
Meso-Formyl, Vinyl, and Ethynyl Porphyrins—Multipotent Synthons for Obtaining a Diverse Array of Functional Derivatives



Abstract
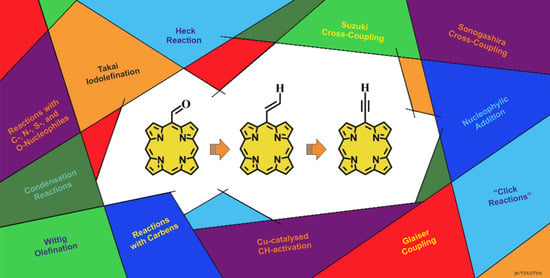
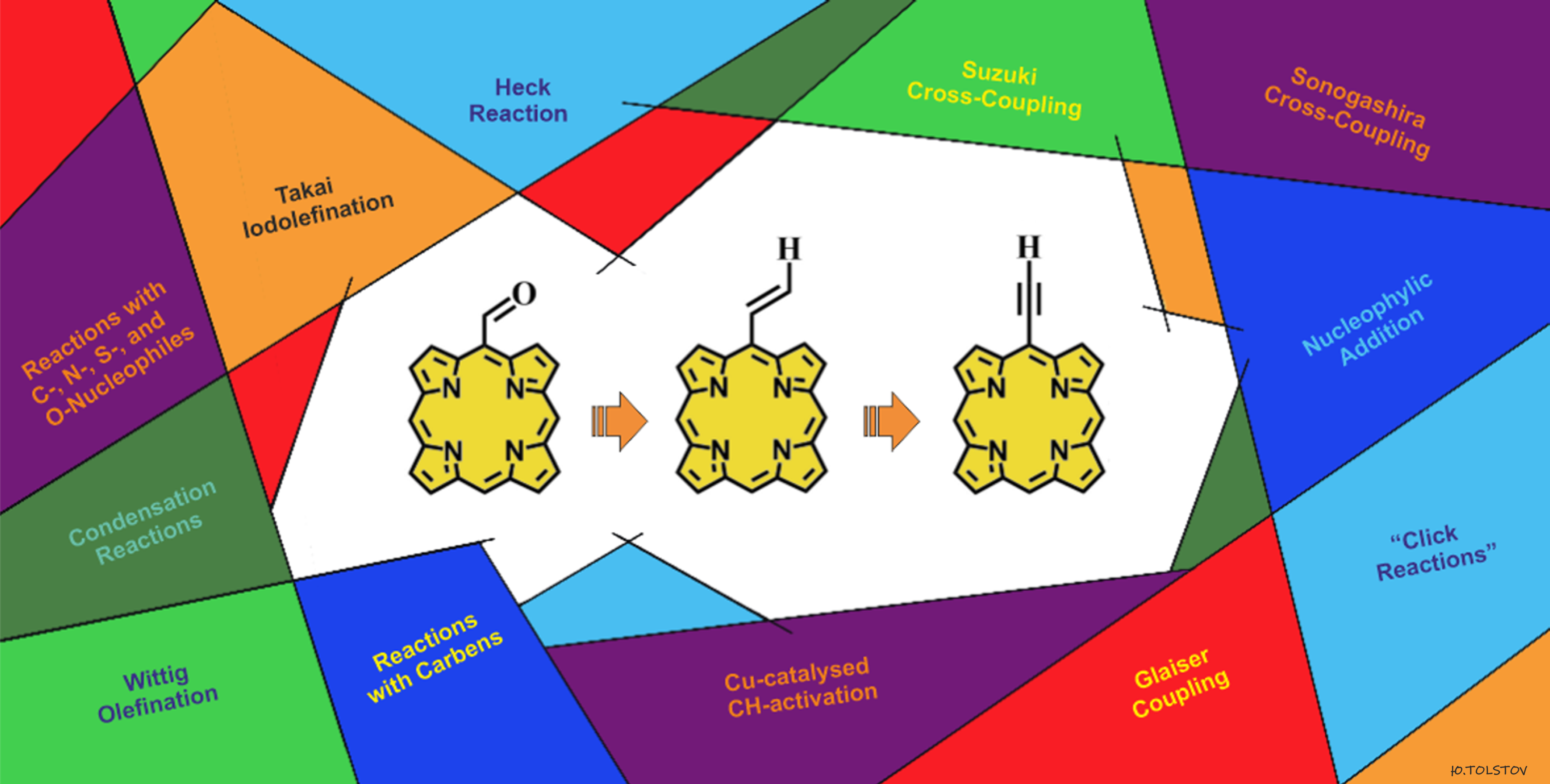
:
{kind=link}
{kind=link}
{kind=link}
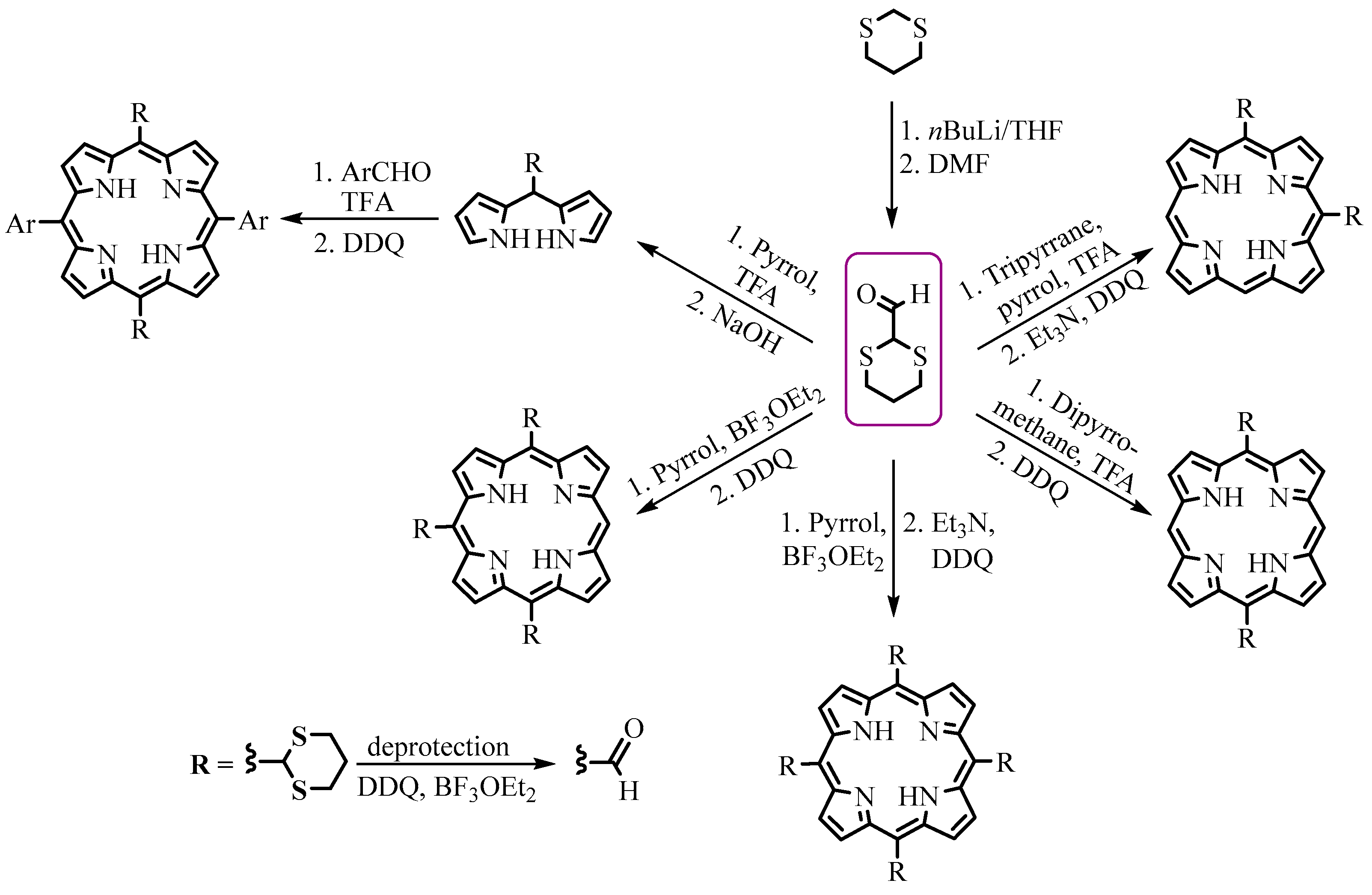{kind=link}
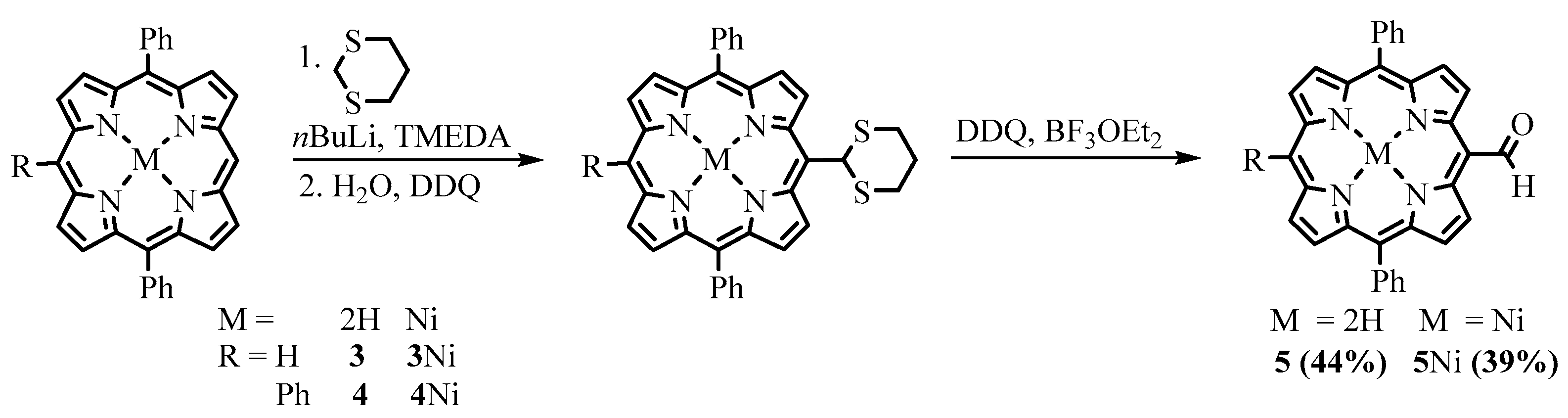{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
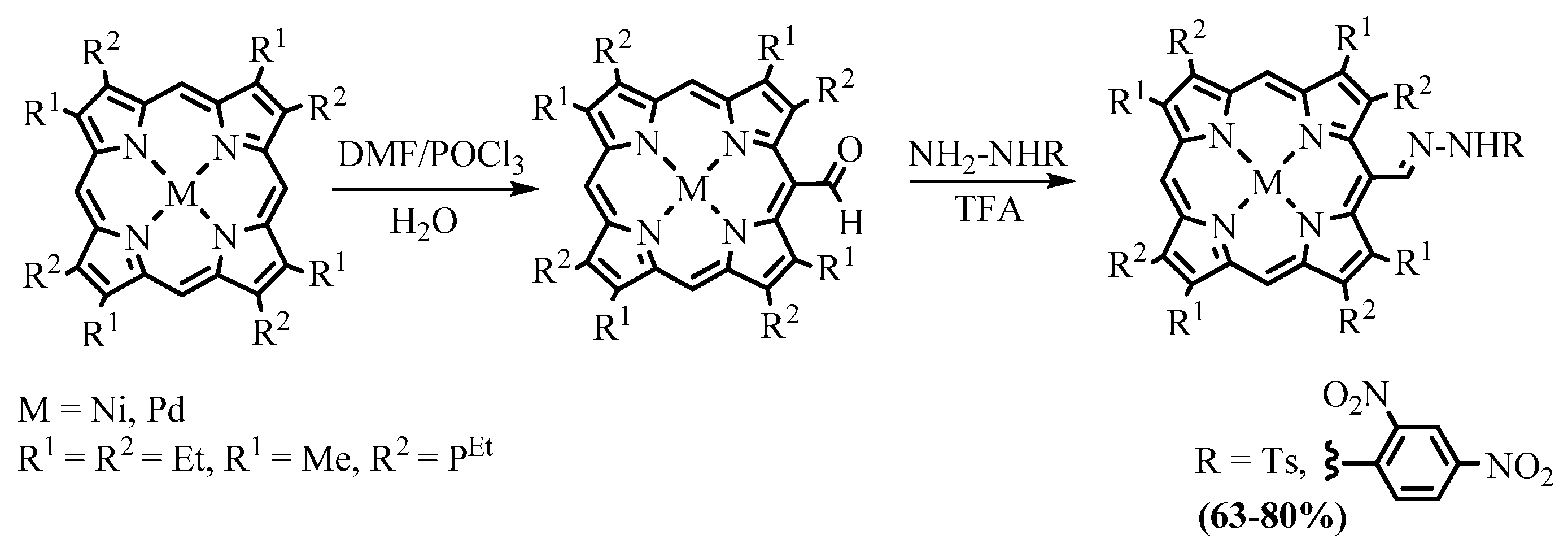{kind=link}
{kind=link}
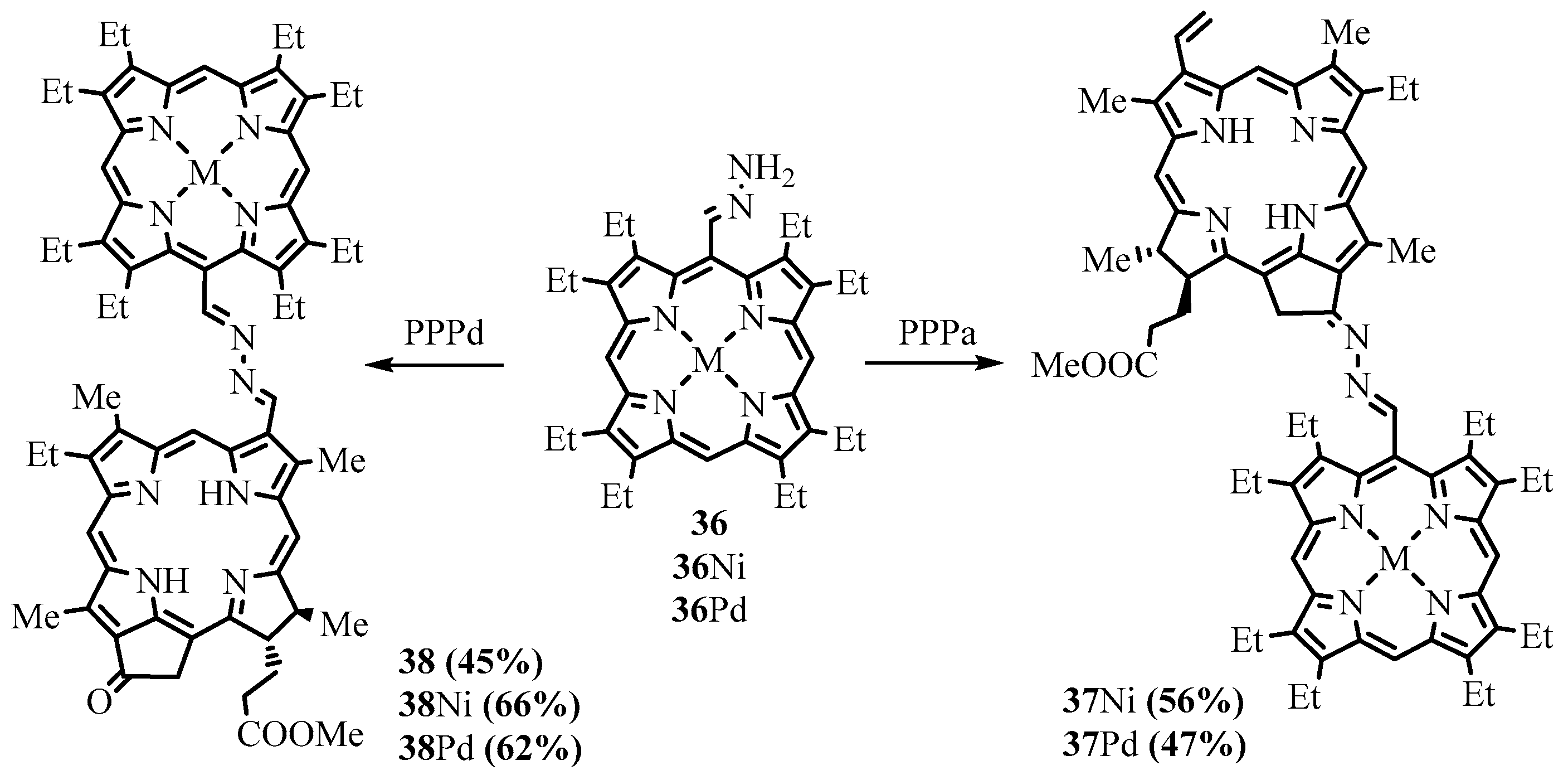{kind=link}
{kind=link}
{kind=link}
{kind=link}
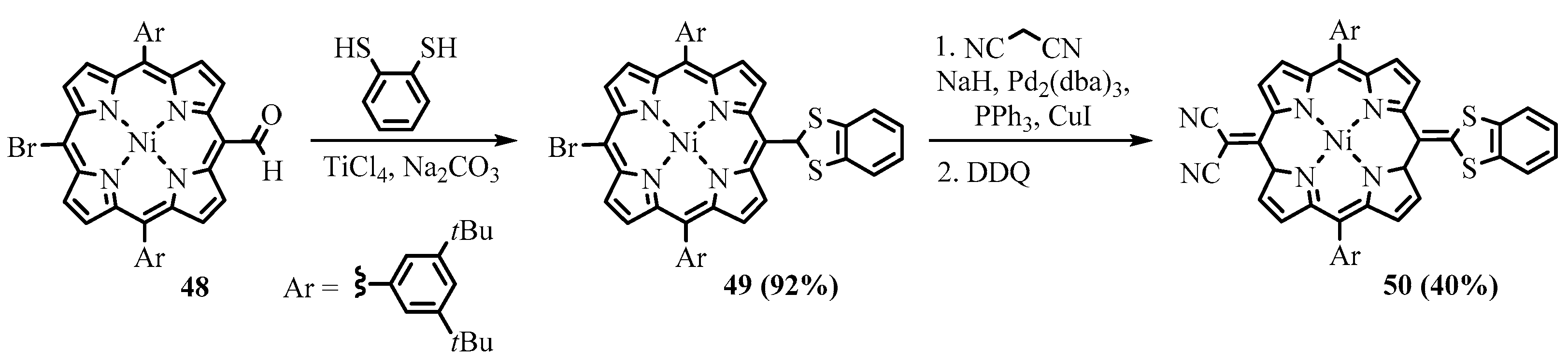{kind=link}
{kind=link}
{kind=link}
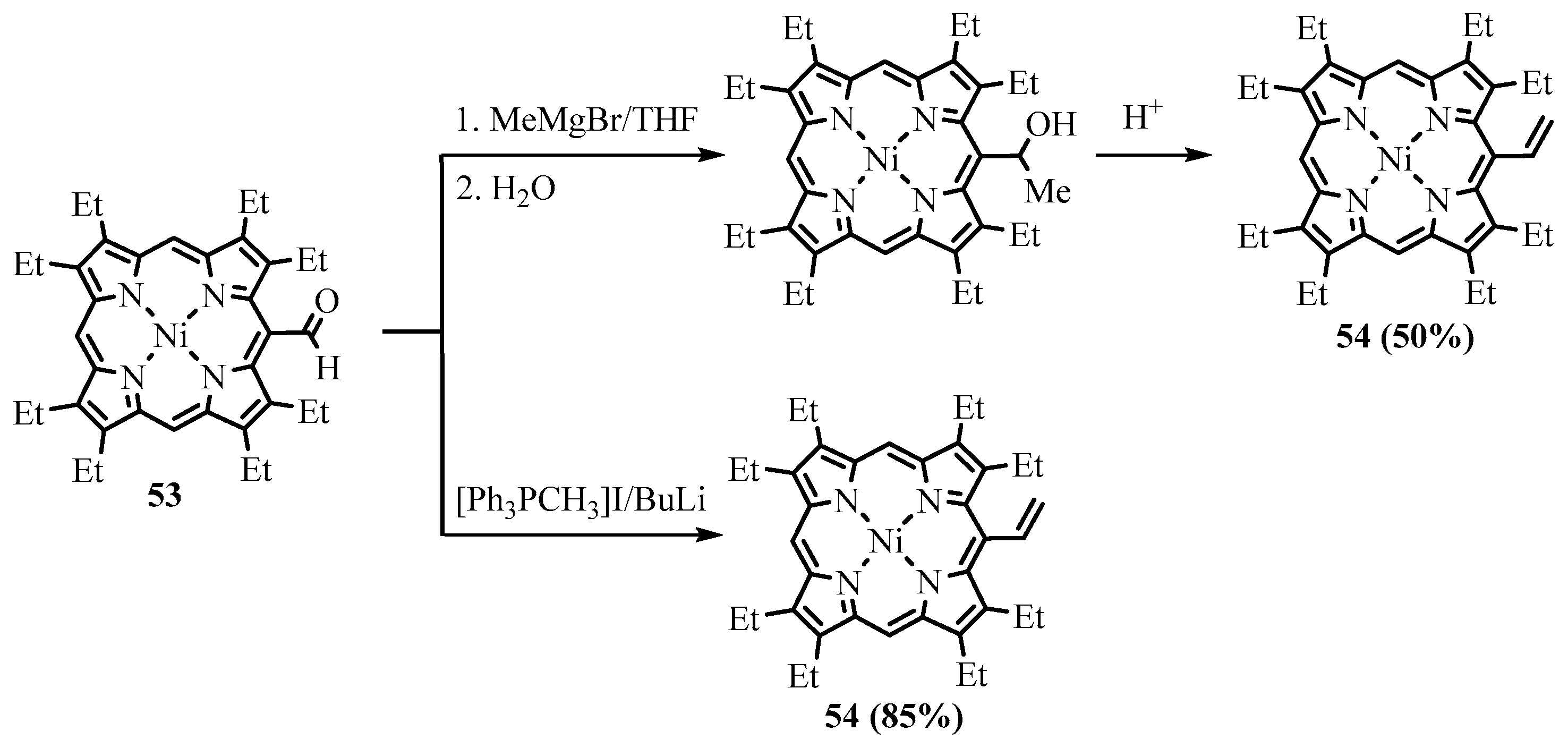{kind=link}
{kind=link}
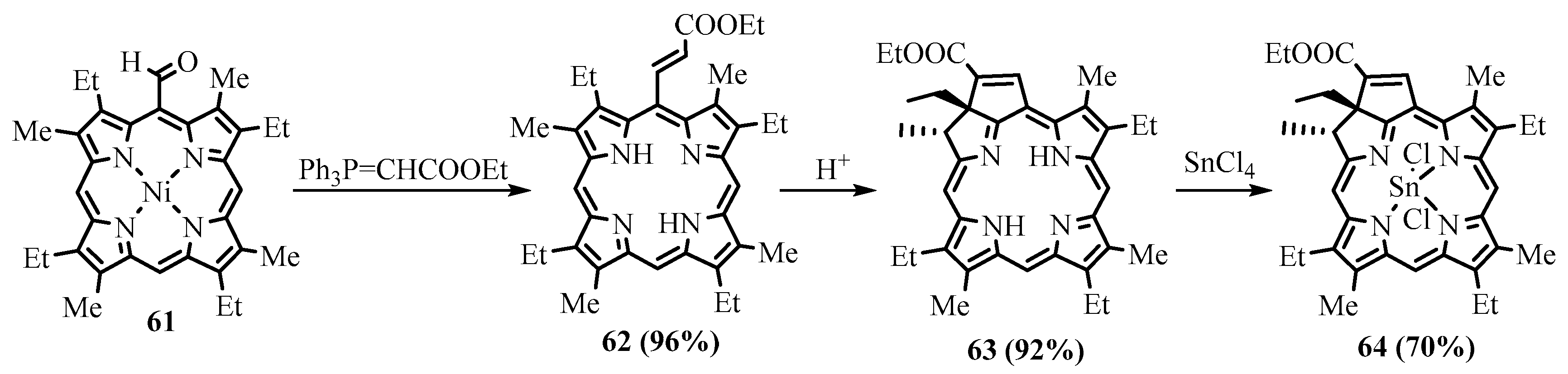{kind=link}
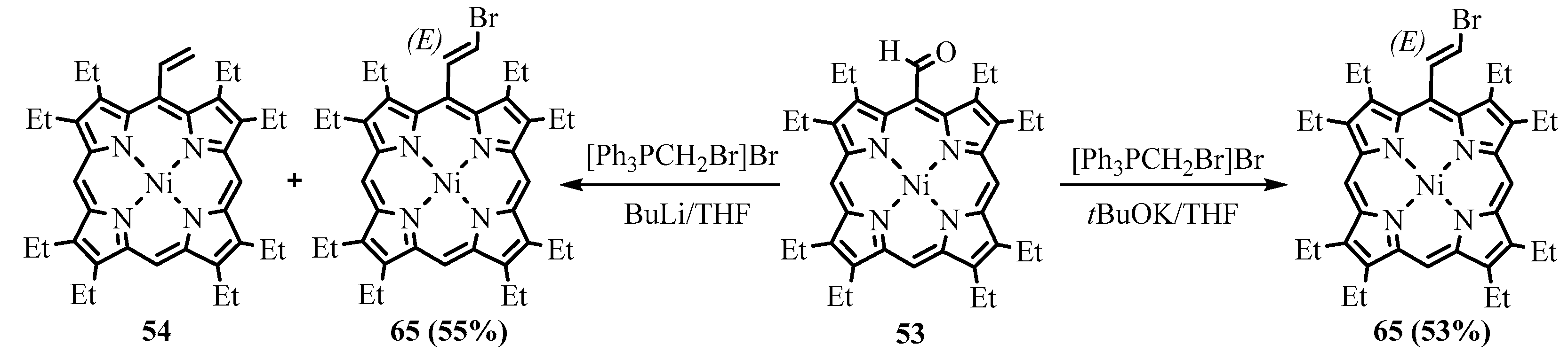{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
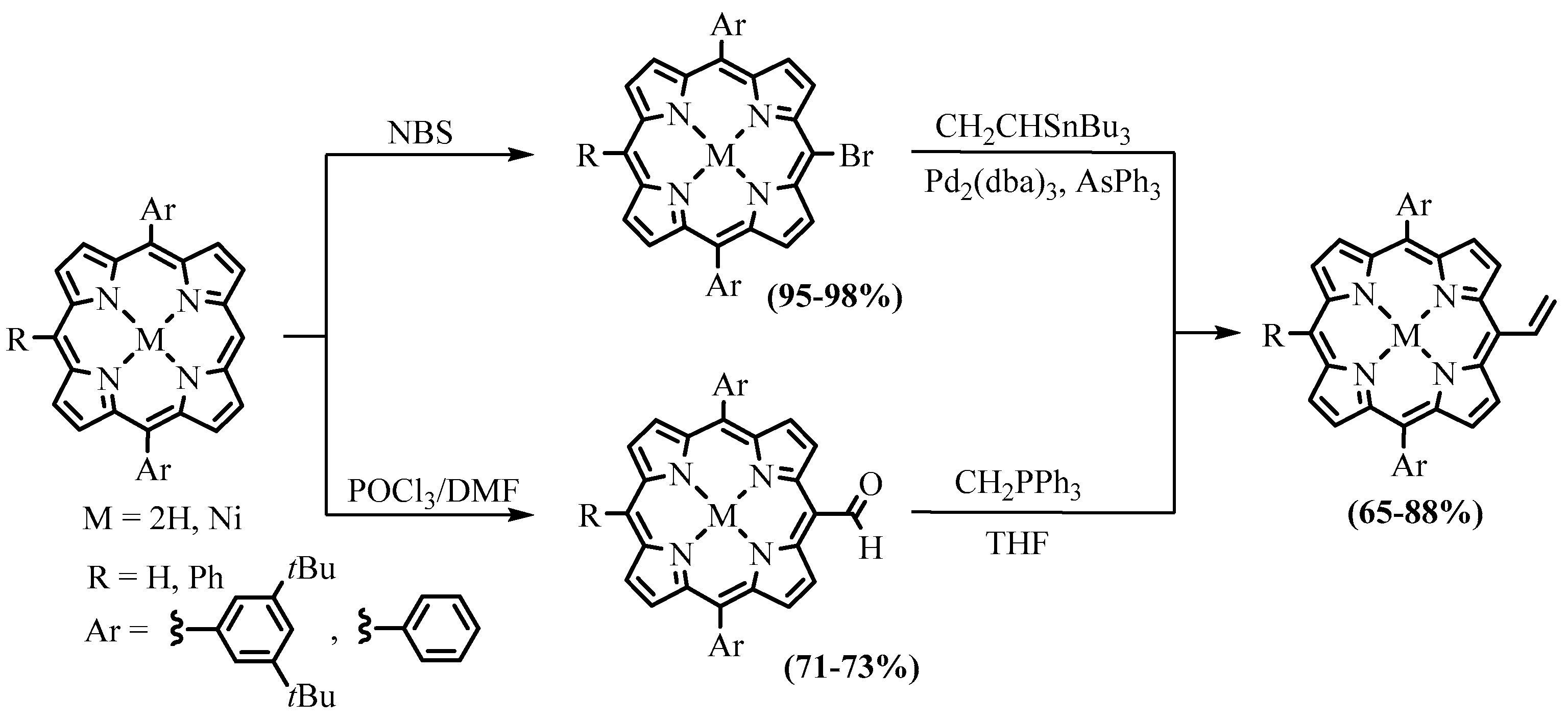{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
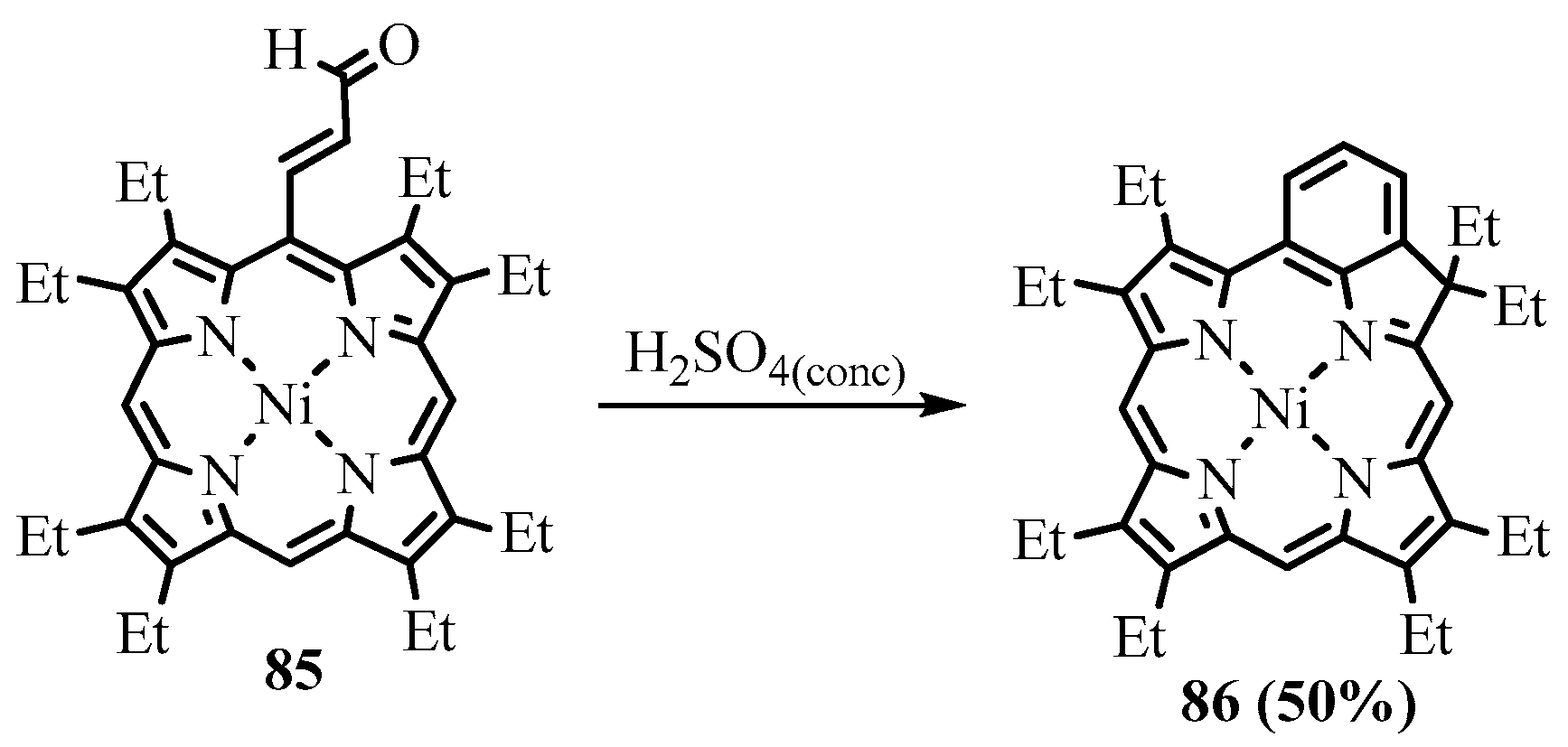{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
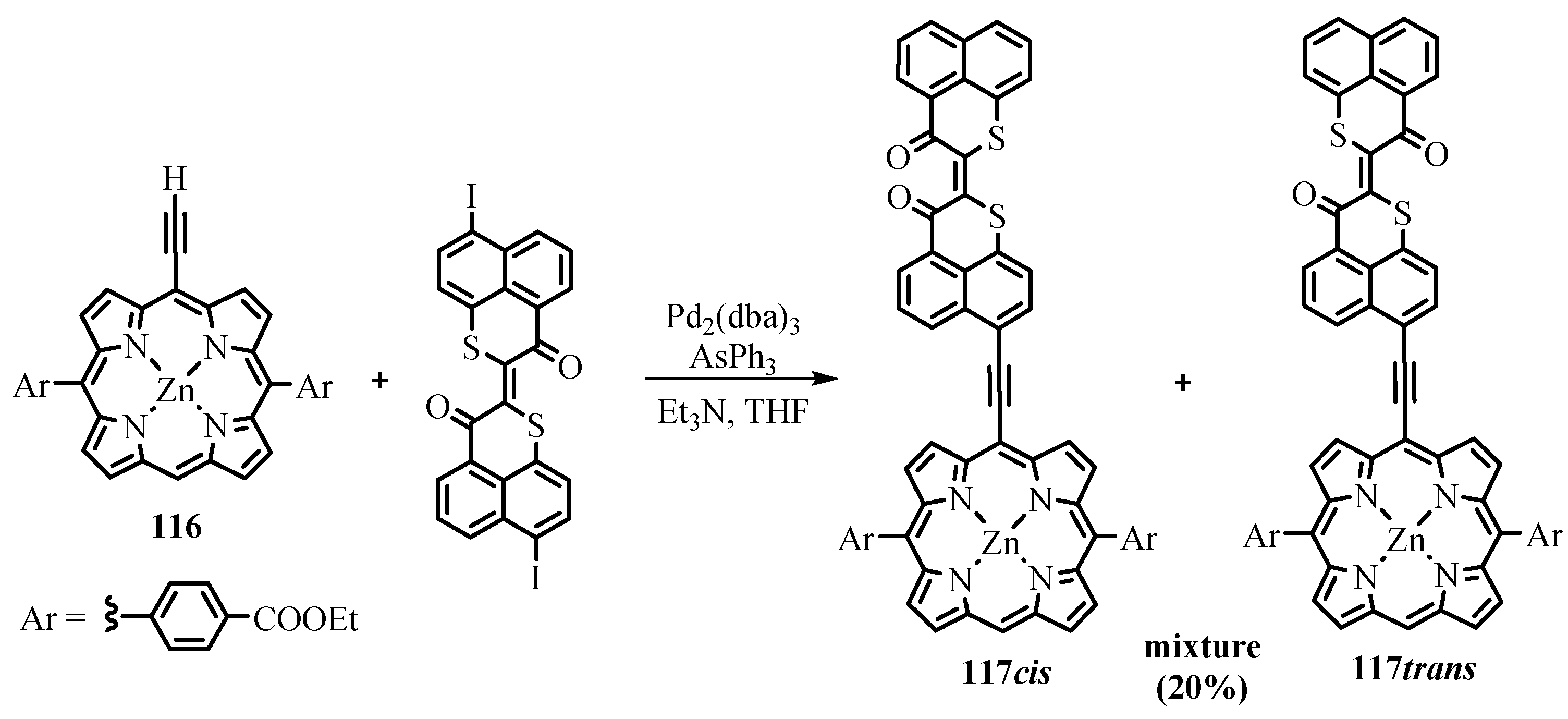{kind=link}
{kind=link}
{kind=link}
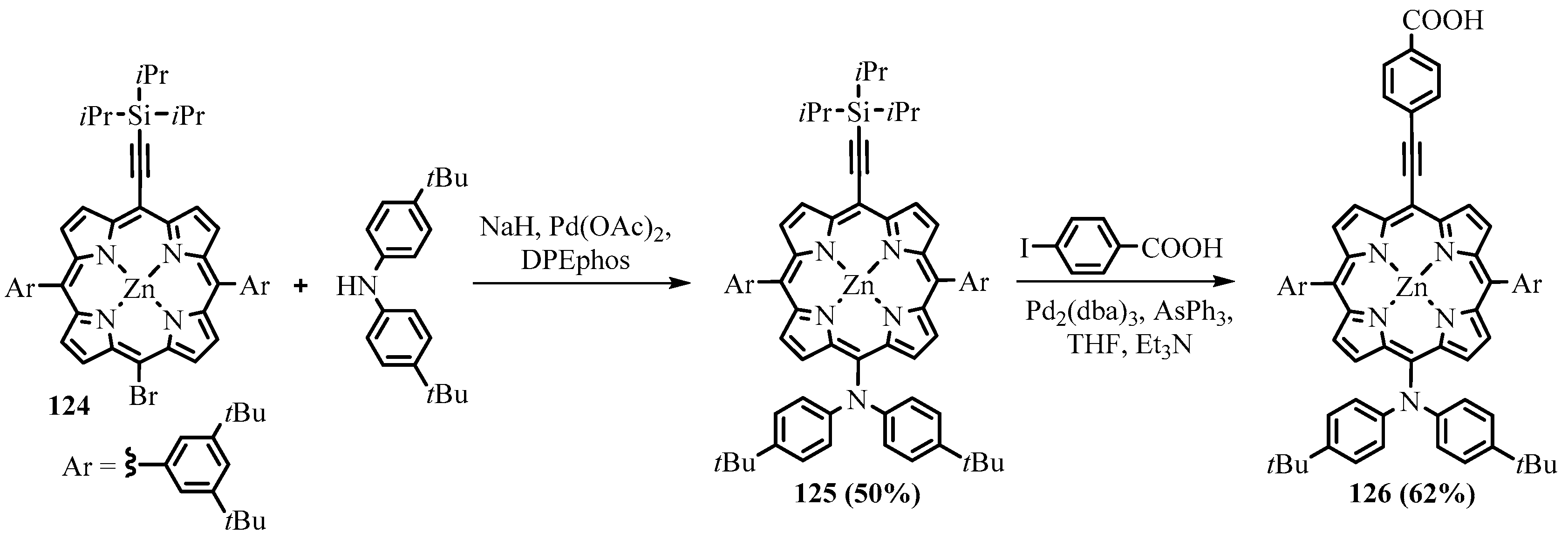{kind=link}
{kind=link}
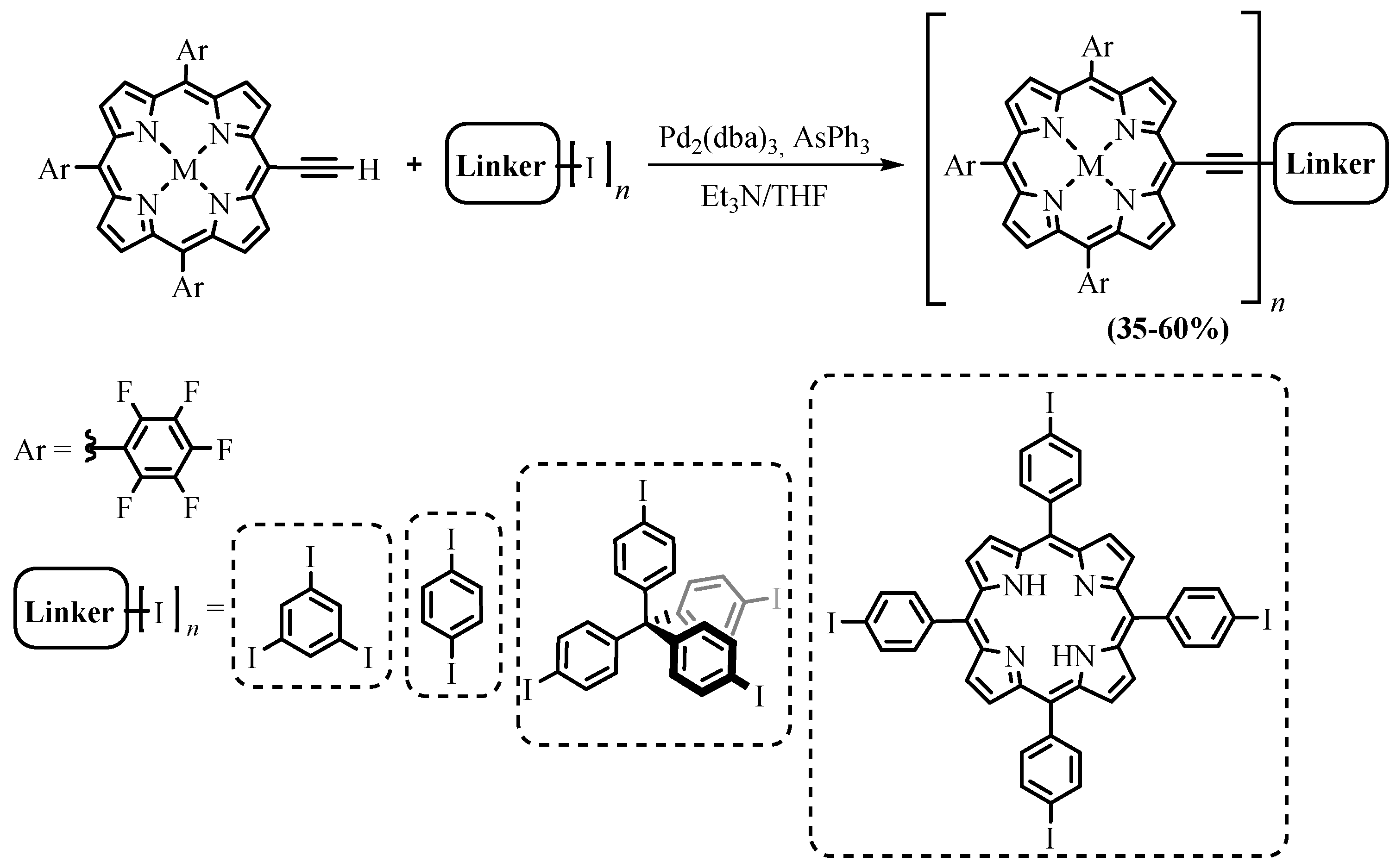
{kind=link}
{kind=link}
{kind=link}
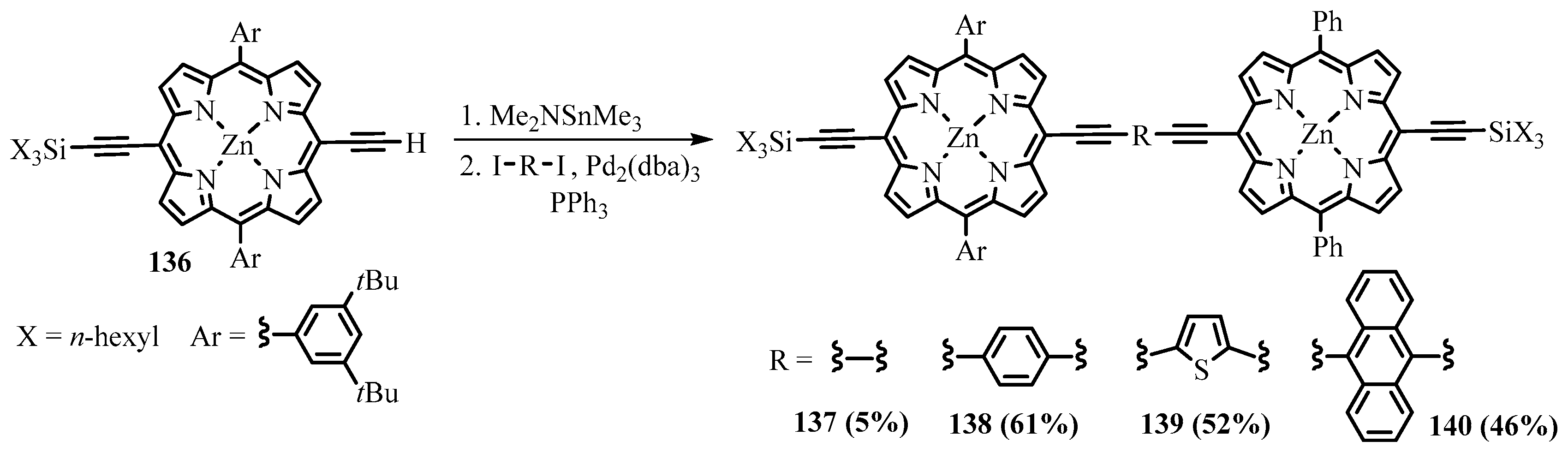{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Meso-Formylporphyrins
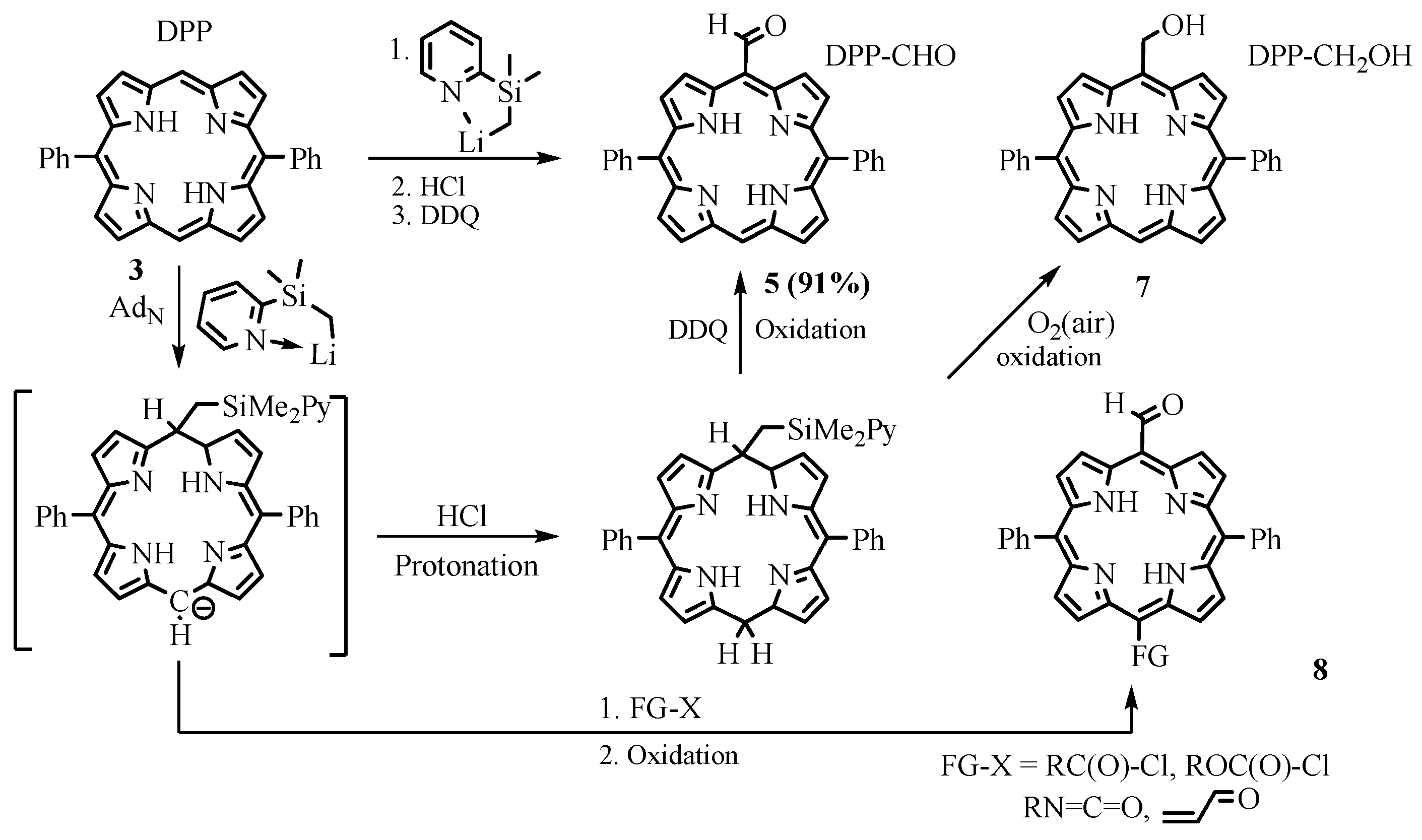
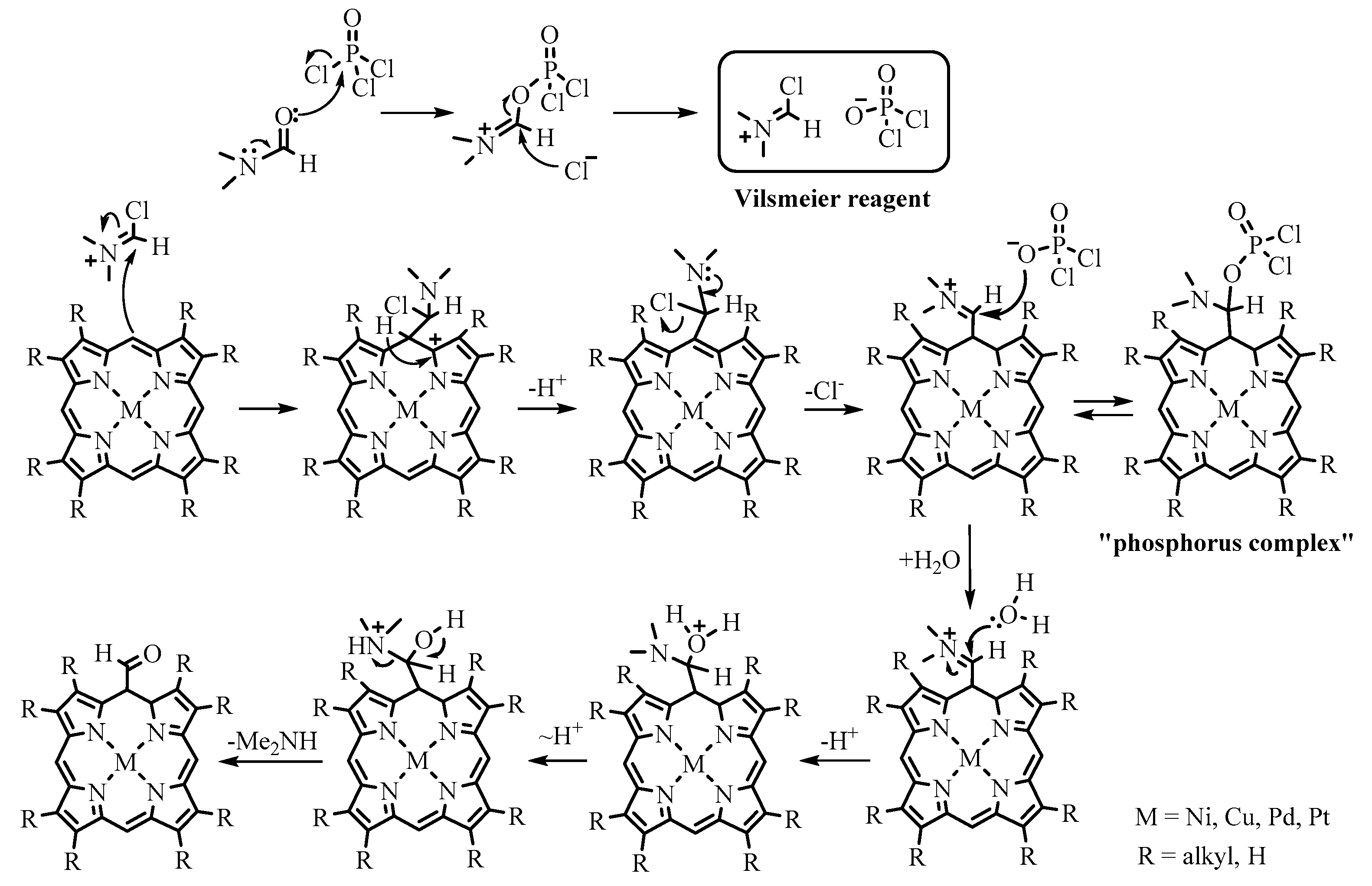
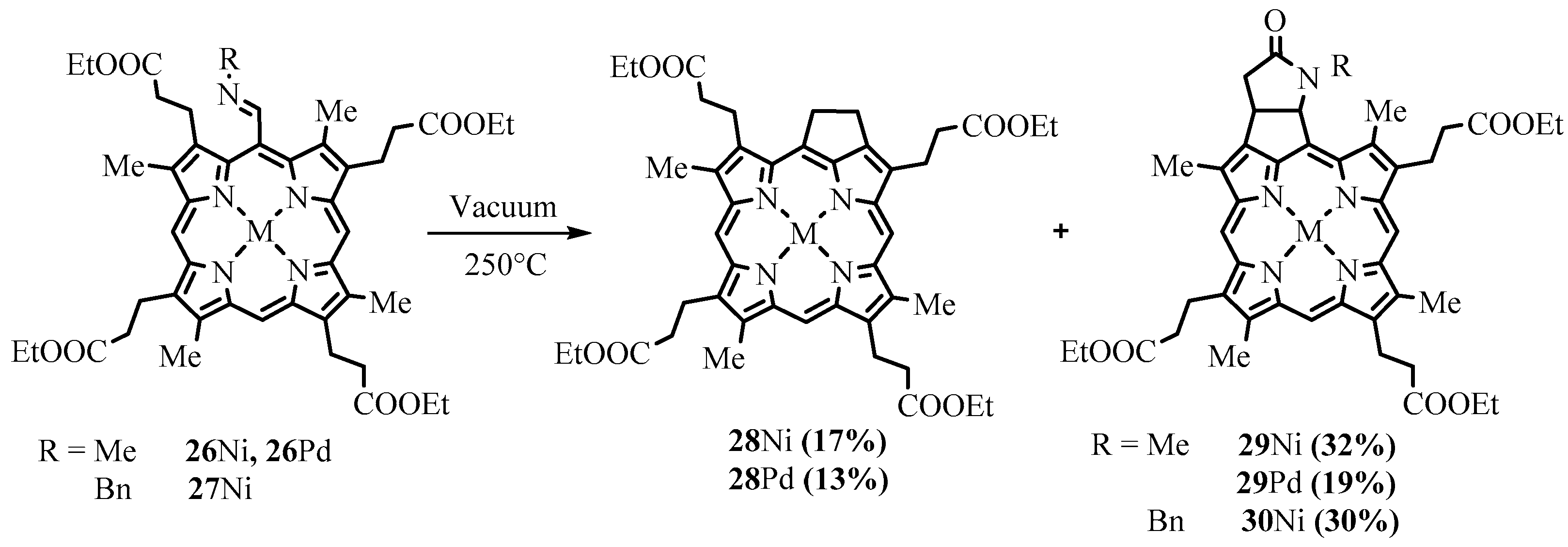
2.1. Preparation of Meso-Formylporphyrins
2.2. Reactions of Meso-Formylporphyrins with Nitrogen Nucleophiles
2.3. Reactions of Meso-Formylporphyrins with Miscellaneous Nucleophiles
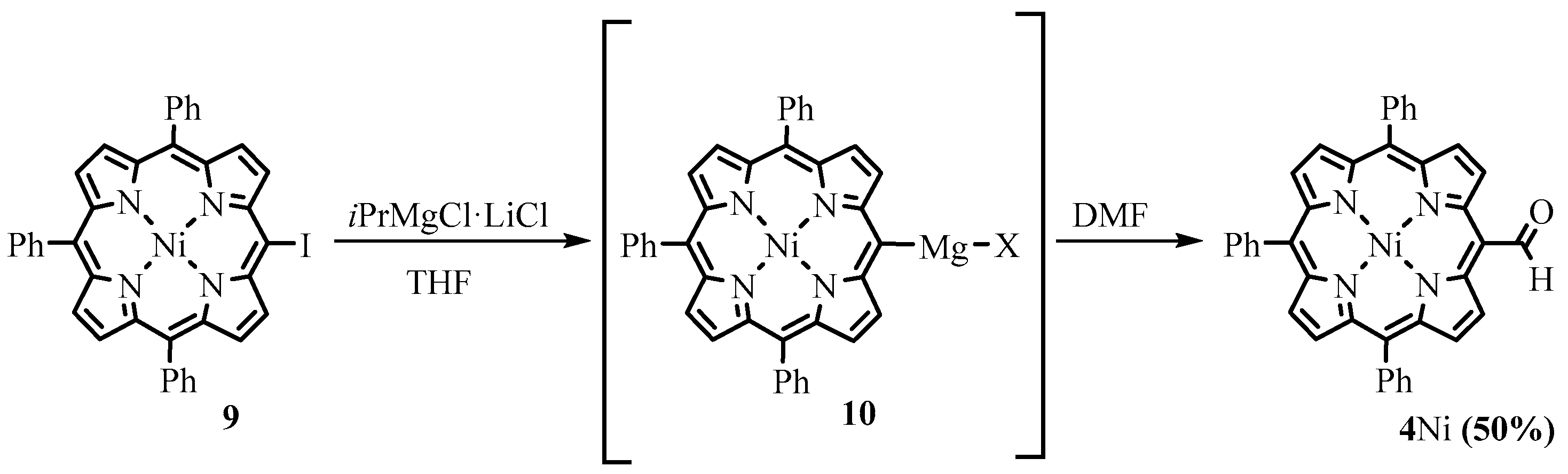
2.4. Reactions of Meso-Formylporphyrins with Organometallic Reagents
2.5. The Reaction of Meso-Formylporphyrins with CH Acids
3. Meso-Vinylporphyrins
3.1. Preparation of Meso-Vinylporphyrins
3.2. Transformations of Meso-Vinylporphyrins
4. Meso-Ethynylporphyrins
Synthesis of Meso-Ethynylporphyrins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CuOEP | Cu(II) β-octaethylporphyrin |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DNA | deoxyribonucleic acid |
| DPP | 5,15-diphenylporphyrin |
| DPP-CHO | 5-formyl-10,20-diphenylporphyrin |
| DSSC | dye sensitized solar cells |
| MOF | metal-organic frameworks |
| NBS | N-bromosuccinimide |
| NiOEP | Ni(II) β-octaethylporphyrin |
| NLO | nonlinear optical |
| OEC | β-octaethylchlorin |
| OEP | β-octaethylporphyrin |
| OEP-CHO | 5-formyl-β-octaethylporphyrin |
| PPPa | methyl pyropheophorbide-a |
| PPPd | methyl pyropheophorbide-d |
| PtOEP | Pt(II) β-octaethylporphyrin |
| STM | scanning tunneling microscope |
| TMEDA | N,N,N,N-tetramethylethylenediamine |
| TPP | 5,10,15,20-tetraphenylporphyrin |
| TrPP | 5,10,15-triphenylporphyrin |
| ZnOEP | Zn(II) β-octaethylporphyrin |
References
- Koifman, O.I.; Ageeva, T.A. Main Strategies for the Synthesis of meso-Arylporphyrins. Russ. J. Org. Chem. 2022, 58, 443–479. [Google Scholar] [CrossRef]
- Tyurin, V.S.; Uglov, A.; Beletskaya, I.P.; Stern, C.; Guilard, R. Survey of Synthetic Routes for Synthesis and Substitution in Porphyrins. In Handbook of Porphyrin Science; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; World Scientific: Singapore, 2012; Volume 23, pp. 81–279. [Google Scholar]
- Vicente, M.D.G.H.; Smith, K.M. Syntheses and Functionalizations of Porphyrin Macrocycles. Curr. Org. Synth. 2014, 11, 3–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senge, M.O. Stirring the porphyrin alphabet soup—Functionalization reactions for porphyrins. Chem. Commun. 2011, 47, 1943–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponomarev, G.V. Formylporphyrins and their derivatives in the chemistry of porphyrins (review). Chem. Heterocycl. Compd. 1994, 30, 1444–1465. [Google Scholar] [CrossRef]
- Paolesse, R.; Nardis, S.; Monti, D.; Stefanelli, M.; Di Natale, C. Porphyrinoids for Chemical Sensor Applications. Chem. Rev. 2017, 117, 2517–2583. [Google Scholar] [CrossRef] [Green Version]
- Nyman, E.S.; Hynninen, P.H. Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2004, 73, 1–28. [Google Scholar] [CrossRef]
- Sternberg, E.D.; Dolphin, D.; Brückner, C. Porphyrin-based photosensitizers for use in photodynamic therapy. Tetrahedron 1998, 54, 4151–4202. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Cerqueira, A.F.R.; Moura, N.M.M.; Serra, V.V.; Faustino, M.A.F.; Tomé, A.C.; Cavaleiro, J.A.S.; Neves, M.G.P.M.S. β-Formyl- and β-Vinylporphyrins: Magic Building Blocks for Novel Porphyrin Derivatives. Molecules 2017, 22, 1269. [Google Scholar] [CrossRef] [Green Version]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.K.; Graetzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Burrell, A.K.; Officer, D.L. Functionalizing Porphyrins via Wittig Reactions: A Building Block Approach. Synlett 1998, 1998, 1297–1307. [Google Scholar] [CrossRef]
- Ando, A.; Yamazaki, M.; Komura, M.; Sano, Y.; Hattori, N.; Omote, M.; Kumadaki, I. An efficient route to formyldeuteroporphyrins and their Wittig reaction. Heterocycles 1999, 50, 913–918. [Google Scholar] [CrossRef]
- Dahms, K.; Senge, M.O.; Bakri Bakar, M. Exploration of meso-substituted formyl porphyrins and their Grignard and Wittig reactions. Eur. J. Org. Chem. 2007, 2007, 3833–3848. [Google Scholar] [CrossRef]
- Runge, S.; Senge, M.O. Reaction of β-formylporphyrins with organometallic reagents—A facile method for the preparation of porphyrins with exocyclic double bonds. Tetrahedron 1999, 55, 10375–10390. [Google Scholar] [CrossRef]
- Arnold, D.P.; Johnson, A.W.; Mahendran, M. Some reactions of meso-formyloctaethylporhyrin. J. Chem. Soc. Perkin Trans. 1 1978, 366–370. [Google Scholar] [CrossRef]
- Smith, K.M. The McMurry Reaction in Porphyrinoid Chemistry. In Synthesis and Modifications of Porphyrinoids; Paolesse, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–34. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Lemmetyinen, H.; Sonoda, J.; Ohkubo, K.; Sato, T.; Imahori, H.; Fukuzumi, S. Ultrafast Photodynamics of Exciplex Formation and Photoinduced Electron Transfer in Porphyrin−Fullerene Dyads Linked at Close Proximity. J. Phys. Chem. A 2003, 107, 8834–8844. [Google Scholar] [CrossRef]
- Fuhrhop, J.-H.; Witte, L.; Sheldrick, W.S. Darstellung, Struktur und Reaktivität hochsubtituierter Porphyrine. Justus Liebigs Ann. Der Chem. 1976, 1976, 1537–1559. [Google Scholar] [CrossRef]
- Ponomarev, G.V. Synthesis and properties of Schiff bases of mesoformylporphyrins (Review). Chem. Heterocycl. Compd. 1996, 32, 1263–1280. [Google Scholar] [CrossRef]
- Hong, S.-K.; Jeoung, E.; Lee, C.-H. Meso-meso linked hybrid porphyrin arrays from meso-formylated porphyrins. J. Porphyr. Phthalocyanines 2005, 9, 285–289. [Google Scholar] [CrossRef]
- Balakumar, A.; Muthukumaran, K.; Lindsey, J.S. A New Route to meso-Formyl Porphyrins. J. Org. Chem. 2004, 69, 5112–5115. [Google Scholar] [CrossRef]
- Senge, M.O.; Hatscher, S.S.; Wiehe, A.; Dahms, K.; Kelling, A. The Dithianyl Group as a Synthon in Porphyrin Chemistry: Condensation Reactions and Preparation of Formylporphyrins under Basic Conditions. J. Am. Chem. Soc. 2004, 126, 13634–13635. [Google Scholar] [CrossRef]
- Seebach, D.; Corey, E.J. Generation and synthetic applications of 2-lithio-1,3-dithianes. J. Org. Chem. 1975, 40, 231–237. [Google Scholar] [CrossRef]
- Takanami, T.; Wakita, A.; Sawaizumi, A.; Iso, K.; Onodera, H.; Suda, K. One-Pot Synthesis of meso-Formylporphyrins by SNAr Reaction of 5,15-Disubstituted Porphyrins with (2-Pyridyldimethylsilyl)methyllithium. Org. Lett. 2008, 10, 685–687. [Google Scholar] [CrossRef] [PubMed]
- Takanami, T.; Wakita, A.; Matsumoto, J.; Sekine, S.; Suda, K. An efficient one-pot procedure for asymmetric bifunctionalization of 5,15-disubstituted porphyrins: A simple preparation of mesoacyl-, alkoxycarbonyl-, and carbamoyl-substituted meso-formylporphyrins. Chem. Commun. 2009, 101–103. [Google Scholar] [CrossRef]
- Takanami, T.; Matsumoto, J.; Kumagai, Y.; Sawaizumi, A.; Suda, K. A facile one-pot preparation of meso-hydroxymethylporphyrins via a sequential SNAr reaction with (2-pyridyldimethylsilyl)methyllithium followed by hydrolysis and aerobic oxidation. Tetrahedron Lett. 2009, 50, 68–70. [Google Scholar] [CrossRef]
- Takanami, T.; Hayashi, S.; Iso, K.; Matsumoto, J.; Hino, F. An efficient one-pot protocol for asymmetric bifunctionalization of 5,15-disubstituted porphyrins: Direct access to meso activated alkenyl-substituted meso-formylporphyrins. Tetrahedron Lett. 2011, 52, 5345–5348. [Google Scholar] [CrossRef]
- Fujimoto, K.; Yorimitsu, H.; Osuka, A. Efficient Synthesis and Versatile Reactivity of Porphyrinyl Grignard Reagents. Eur. J. Org. Chem. 2014, 2014, 4327–4334. [Google Scholar] [CrossRef] [Green Version]
- Inhoffen, H.H.; Fuhrhop, J.-H.; Voigt, H.; Brockmann, H., Jr. Zur weiteren Kenntnis des Chlorophylls und des Hämins, VI. Formylierung der meso-Kohlenstoffatome von Alkyl-substituierten Porphyrinen. Justus Liebigs Ann. Der Chem. 1966, 695, 133–143. [Google Scholar] [CrossRef]
- Johnson, A.W.; Oldfield, D. meso-Substitution products of ætioporphyrin I. J. Chem. Soc. C Org. 1966, 794–798. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Rozynov, B.V. Synthesis of copper and iron complexes of meso-substituted etioporphyrin derivatives. Chem. Heterocycl. Compd. 1973, 9, 1065–1068. [Google Scholar] [CrossRef]
- Volov, A.N.; Zamilatskov, I.A.; Mikhel, I.S.; Erzina, D.R.; Ponomarev, G.V.; Koifman, O.I.; Tsivadze, A.Y. Synthesis of the First Azomethine Derivatives of Pd-II Coproporphyrins I and II. Macroheterocycles 2014, 7, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Tyurin, V.S.; Erzina, D.R.; Zamilatskov, I.A.; Chernyadyev, A.Y.; Ponomarev, G.V.; Yashunskiy, D.V.; Maksimova, A.V.; Krasnovskiy, A.A.; Tsivadze, A.Y. Palladium Complexes of Azomethine Derivatives of Porphyrins as Potential Photosensitizers. Macroheterocycles 2015, 8, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Erzina, D.R.; Zamilatskov, I.A.; Stanetskaya, N.M.; Tyurin, V.S.; Kozhemyakin, G.L.; Ponomarev, G.V.; Chernyshev, V.V.; Fitch, A.N. Transformations of meso-Iminofunctionalized Pd(II) and Ni(II)-Complexes of β-Alkylsubstituted Porphyrins. Eur. J. Org. Chem. 2019, 2019, 1508–1522. [Google Scholar] [CrossRef]
- Papkovsky, D.B.; Ponomarev, G.V.; Wolfbeis, O.S. Protonation of porphyrins in liquid PVC membranes: Effects of anionic additives and application to pH-sensing. J. Photochem. Photobiol. A Chem. 1997, 104, 151–158. [Google Scholar] [CrossRef]
- Borchert, N.B.; Ponomarev, G.V.; Kerry, J.P.; Papkovsky, D.B. O2/pH Multisensor Based on One Phosphorescent Dye. Anal. Chem. 2011, 83, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, A.V.; Li, L.; Yang, P.; Shkirdova, A.O.; Tang, S.; Yashunsky, D.V.; Ponomarev, G.V.; Zamilatskov, I.A.; Papkovsky, D.B. Advanced multi-modal, multi-analyte optochemical sensing platform for cell analysis. Sens. Actuators B Chem. 2022, 355, 131116. [Google Scholar] [CrossRef]
- Harper, S.R.; Pfrunder, M.C.; Esdaile, L.J.; Jensen, P.; McMurtrie, J.C.; Arnold, D.P. Synthetic, Structural, and Spectroscopic Studies of Bis(porphyrinzinc) Complexes Linked by Two-Atom Conjugating Bridges. Eur. J. Org. Chem. 2015, 2015, 2807–2825. [Google Scholar] [CrossRef]
- Dahlstedt, E.; Collins, H.A.; Balaz, M.; Kuimova, M.K.; Khurana, M.; Wilson, B.C.; Phillips, D.; Anderson, H.L. One- and two-photon activated phototoxicity of conjugated porphyrin dimers with high two-photon absorption cross sections. Org. Biomol. Chem. 2009, 7, 897–904. [Google Scholar] [CrossRef] [Green Version]
- Yashunsky, D.V.; Morozova, Y.V.; Ponomarev, G.V. Chemistry ofmeso-formly-porphyrin oximes. An elegant synthesis ofmeso-cyanoporphyrins. Chem. Heterocycl. Compd. 2000, 36, 485–486. [Google Scholar] [CrossRef]
- Yashunsky, D.V.; Morozova, Y.V.; Ponomarev, G.V. Chemistry ofmeso-formylporphyrin oximes. The synthesis of the hetero analog of australochlorin. Chem. Heterocycl. Compd. 2000, 36, 487–488. [Google Scholar] [CrossRef]
- Yashunsky, D.V.; Morozova, Y.V.; Ponomarev, G.V. Chemistry of Metal Complexes of Oximes of meso-Formylporphyrins. Oxidative Cyclization to Metal Complexes of Hydroxy-1,2-oxazinochlorins. Chem. Heterocycl. Compd. 2001, 37, 380–381. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Shul’ga, A.M. Porphyrins. 16. Thermolysis of schiff bases of meso-formylporphyrins—A convenient method for the synthesis of porphyrins with a cyclopentane ring. Chem. Heterocycl. Compd. 1984, 20, 383–388. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Shul’ga, A.M. Porphyrins. 22. Synthesis of porphyrins with two cyclopentane rings. Chem. Heterocycl. Compd. 1987, 23, 757–762. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Shul’ga, A.M.; Rozynov, B.V. Porphyrin. Chem. Heterocycl. Compd. 1993, 29, 155–162. [Google Scholar] [CrossRef]
- Shkirdova, A.O.; Zamilatskov, I.A.; Stanetskaya, N.M.; Tafeenko, V.A.; Tyurin, V.S.; Chernyshev, V.V.; Ponomarev, G.V.; Tsivadze, A.Y. Synthesis and Study of New N-Substituted Hydrazones of Ni(II) Complexes of beta-Octaethylporphyrin and Coproporphyrin I Tetraethyl Ester. Macroheterocycles 2017, 10, 480–486. [Google Scholar] [CrossRef]
- Kozhemyakin, G.L.; Tyurin, V.S.; Shkirdova, A.O.; Belyaev, E.S.; Kirinova, E.S.; Ponomarev, G.V.; Chistov, A.A.; Aralov, A.V.; Tafeenko, V.A.; Zamilatskov, I.A. Carbene functionalization of porphyrinoids through tosylhydrazones. Org. Biomol. Chem. 2021, 19, 9199–9210. [Google Scholar] [CrossRef] [PubMed]
- Belyaev, E.S.; Shkirdova, A.O.; Kozhemyakin, G.L.; Tyurin, V.S.; Emets, V.V.; Grinberg, V.A.; Cheshkov, D.A.; Ponomarev, G.V.; Tafeenko, V.A.; Radchenko, A.S.; et al. Azines of porphyrinoids. Does azine provide conjugation between chromophores? Dye Pigment. 2021, 191, 109354. [Google Scholar] [CrossRef]
- Andreeva, V.D.; Ponomarev, G.V.; Shkirdova, A.O.; Tyurin, V.S.; Zamilatskova, I.A. Modification of β-Octaethylporphyrin via Insertion of Amino and Azino Groups into meso-Positions. Macroheterocycles 2021, 14, 201–207. [Google Scholar] [CrossRef]
- Birin, K.P.; Gorbunova, Y.G.; Tsivadze, A.Y. New approach for post-functionalization of meso-formylporphyrins. RSC Adv. 2015, 5, 67242–67246. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, D.; Zhao, Y.; Wu, F.; Yang, K.; Wang, K. Synthesis, DNA binding mode, singlet oxygen photogeneration and DNA photocleavage activity of ruthenium compounds with porphyrin-imidazo[4,5-f]phenanthroline conjugated ligand. Appl. Organomet. Chem. 2018, 32, e4468. [Google Scholar] [CrossRef]
- Smith, M.J.; Blake, I.M.; Clegg, W.; Anderson, H.L. Push–pull quinoidal porphyrins. Org. Biomol. Chem. 2018, 16, 3648–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhu, Y.-Z.; Fan, Q.-J.; Song, H.-B.; Zheng, J.-Y. Syntheses, Crystal Structure, and Spectroscopic Properties of Meso–Meso-Linked Porphyrin–Corrole Hybrids. Chem. Lett. 2013, 42, 936–938. [Google Scholar] [CrossRef]
- Murugavel, M.; Reddy, R.V.R.; Sankar, J. A new meso–meso directly-linked corrole–porphyrin–corrole hybrid: Synthesis and photophysical properties. RSC Adv. 2014, 4, 13669–13672. [Google Scholar] [CrossRef]
- Higashino, T.; Imahori, H. Development of Efficient Sensitizers Based on Porphyrin Dimers and Fused Porphyrins for Dye-Sensitized Solar Cells. ECS Meet. Abstr. 2021, MA2021-01, 769. [Google Scholar] [CrossRef]
- Ponomarev, G.V.; Sidorov, A.N. Porphyrins. Chem. Heterocycl. Compd. 1977, 13, 742–747. [Google Scholar] [CrossRef]
- Jiang, X.; Nurco, D.J.; Smith, K.M. Direct meso-alkylation of meso-formylporphyrins using Grignard reagents. Chem. Commun. 1996, 1759–1760. [Google Scholar] [CrossRef]
- Locos, O.B.; Arnold, D.P. The Heck reaction for porphyrin functionalisation: Synthesis of meso-alkenyl monoporphyrins and palladium-catalysed formation of unprecedented meso–β ethene-linked diporphyrins. Org. Biomol. Chem. 2006, 004, 902–916. [Google Scholar] [CrossRef] [Green Version]
- van der Haas, R.N.S.; de Jong, R.L.P.; Noushazar, M.; Erkelens, K.; Smijs, T.G.M.; Liu, Y.; Gast, P.; Schuitmaker, H.J.; Lugtenburg, J. The Synthesis of the Dimethyl Ester of Quino[4,4a,5,6-efg]-Annulated 7-Demethyl-8-deethylmesoporphyrin and Three of Its Isomers with Unprecedented peri-Condensed Quinoline Porphyrin Structures. Molecules with Outstanding Properties as Sensitizers for Photodynamic Therapy in the Far-Red Region of the Visible Spectrum. Eur. J. Org. Chem. 2004, 2004, 4024–4038. [Google Scholar] [CrossRef]
- Kessel, D.; Morgan, A. Photosensitization with Etiobenzochlorins and Octaethylbenzochlorins. Photochem. Photobiol. 1993, 58, 521–526. [Google Scholar] [CrossRef]
- Morgan, A.R.; Rampersaud, A.; Garbo, G.M.; Keck, R.W.; Selman, S.H. New sensitizers for photodynamic therapy. Controlled synthesis of purpurins and their effect on normal tissue. J. Med. Chem. 1989, 32, 904–908. [Google Scholar] [CrossRef]
- Li, G.; Graham, A.; Potter, W.; Grossman, Z.D.; Oseroff, A.; Dougherty, T.J.; Pandey, R.K. A Simple and Efficient Approach for the Synthesis of Fluorinated and Nonfluorinated Octaethylporphyrin-Based Benzochlorins with Variable Lipophilicity, Their in Vivo Tumor Uptake, and the Preliminary in Vitro Photosensitizing Efficacy. J. Org. Chem. 2001, 66, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Meunier, I.; Pandey, R.K.; Senge, M.O.; Dougherty, T.J.; Smith, K.M. Benzoporphyrin derivatives: Synthesis, structure and preliminary biological activity. J. Chem. Soc. Perkin Trans. 1 1994, 961–969. [Google Scholar] [CrossRef]
- Li, G.; Pandey, S.K.; Graham, A.; Dobhal, M.P.; Mehta, R.; Chen, Y.; Gryshuk, A.; Rittenhouse-Olson, K.; Oseroff, A.; Pandey, R.K. Functionalization of OEP-Based Benzochlorins To Develop Carbohydrate-Conjugated Photosensitizers. Attempt To Target β-Galactoside-Recognized Proteins. J. Org. Chem. 2004, 69, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Kohli, D.H.; Morgan, A.R. Preparation of substituted chlorins and benzochlorins. Bioorg. Med. Chem. Lett. 1995, 5, 2175–2178. [Google Scholar] [CrossRef]
- Arnold, D.P.; Nitschinsk, L.J. Porphyrin Dimers Linked by Conjugated Butadiynes. Tetrahedron 1992, 48, 8781–8792. [Google Scholar] [CrossRef]
- Arnold, D.P.; Hartnell, R.D. Butadiyne-linked bis(chlorin) and chlorin–porphyrin dyads and an improved synthesis of bis[octaethylporphyrinatonickel(II)-5-yl]butadiyne using the Takai iodoalkenation. Tetrahedron 2001, 57, 1335–1345. [Google Scholar] [CrossRef]
- Vicente, M.G.H.; Smith, K.M. Vilsmeier reactions of porphyrins and chlorins with 3-(dimethylamino)acrolein to give meso-(2-formylvinyl)porphyrins: New syntheses of benzochlorins, benzoisobacteriochlorins, and benzobacteriochlorins and reductive coupling of porphyrins and chlorins using low-valent titanium complexes. J. Org. Chem. 1991, 56, 4407–4418. [Google Scholar] [CrossRef]
- Rong, J.; Wu, Y.; Ji, X.; Zhao, T.; Yin, B.; Rao, Y.; Zhou, M.; Osuka, A.; Xu, L.; Song, J. Porphyrinatonickel(II)–Cyclopentene and Porphyrinatonickel(II)–Cyclopentadiene Hybrids: Zirconacyclopentadiene-Mediated Syntheses, Structures, and Mechanistic Study. Org. Lett. 2022, 24, 6128–6132. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, F.; Tang, W.; Zhao, S.; Li, C.; Xie, Y. A novel nitroethylene-based porphyrin as a NIR fluorescence turn-on probe for biothiols based on the Michael addition reaction. Dye Pigm. 2018, 148, 437–443. [Google Scholar] [CrossRef]
- Muthiah, C.; Taniguchi, M.; Kim, H.-J.; Schmidt, I.; Kee, H.L.; Holten, D.; Bocian, D.F.; Lindsey, J.S. Synthesis and Photophysical Characterization of Porphyrin, Chlorin and Bacteriochlorin Molecules Bearing Tethers for Surface Attachment. Photochem. Photobiol. 2007, 83, 1513–1528. [Google Scholar] [CrossRef]
- Chen, B.; Ding, Y.; Li, X.; Zhu, W.; Hill, J.P.; Ariga, K.; Xie, Y. Steric hindrance-enforced distortion as a general strategy for the design of fluorescence “turn-on” cyanide probes. Chem. Commun. 2013, 49, 10136–10138. [Google Scholar] [CrossRef] [PubMed]
- Shkirdova, A.O.; Orlova, E.A.; Ponomarev, G.V.; Tyurin, V.S.; Zamilatskov, I.A.; Buryak, A.K. Synthesis of β-Octaethylporphyrin Conjugates with Nitrogen and Sulfur Containing Heterocycles. Macroheterocycles 2021, 14, 208–212. [Google Scholar] [CrossRef]
- DiMagno, S.G.; Lin, V.S.Y.; Therien, M.J. Facile elaboration of porphyrins via metal-mediated cross-coupling. J. Org. Chem. 1993, 58, 5983–5993. [Google Scholar] [CrossRef]
- Kato, A.; Hartnell, R.D.; Yamashita, M.; Miyasaka, H.; Sugiura, K.-I.; Arnold, D.P. Selective meso-monobromination of 5,15-diarylporphyrins via organopalladium porphyrins. J. Porphyr. Phthalocyanines 2004, 8, 1222–1227. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Amin, S.R.; Liebeskind, L.S. 3-Cyclobutenyl-1,2-dione-Substituted Porphyrins. A General and Efficient Entry to Porphyrin−Quinone and Quinone−Porphyrin−Quinone Architectures. J. Org. Chem. 2000, 65, 1650–1664. [Google Scholar] [CrossRef]
- Locos, O.; Bašić, B.; McMurtrie, J.C.; Jensen, P.; Arnold, D.P. Homo- and Heteronuclear meso,meso-(E)-Ethene-1,2-diyl-Linked Diporphyrins: Preparation, X-ray Crystal Structure, Electronic Absorption and Emission Spectra and Density Functional Theory Calculations. Chem.—A Eur. J. 2012, 18, 5574–5588. [Google Scholar] [CrossRef]
- Arnold, D.P.; Gaete-Holmes, R.; Johnson, A.W.; Smith, A.R.P.; Williams, G.A. Wittig condensation products from nickel meso-formyl-octaethyl-porphyrin and -aetioporphyrin I and some cyclisation reactions. J. Chem. Soc. Perkin Trans. 1 1978, 1660–1670. [Google Scholar] [CrossRef]
- Glowacka-Sobotta, A.; Wrotynski, M.; Kryjewski, M.; Sobotta, L.; Mielcarek, J. Porphyrinoids in photodynamic diagnosis and therapy of oral diseases. J. Porphyr. Phthalocyanines 2019, 23, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Moreira, L.M.; Vieira dos Santos, F.; Lyon, J.P.; Maftoum-Costa, M.; Pacheco-Soares, C.; Soares da Silva, N. Photodynamic Therapy: Porphyrins and Phthalocyanines as Photosensitizers. Aust. J. Chem. 2008, 61, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.; Garbo, G.; Rampersaud, A.; Skalkos, D.; Keck, R.; Selman, S. Photodynamic Action Of Benzochlorins; SPIE: Bellingham, WA, USA, 1989; Volume 1065. [Google Scholar]
- Frampton, M.J.; Akdas, H.; Cowley, A.R.; Rogers, J.E.; Slagle, J.E.; Fleitz, P.A.; Drobizhev, M.; Rebane, A.; Anderson, H.L. Synthesis, Crystal Structure, and Nonlinear Optical Behavior of β-Unsubstituted meso-meso E-Vinylene-Linked Porphyrin Dimers. Org. Lett. 2005, 7, 5365–5368. [Google Scholar] [CrossRef]
- Tokuji, S.; Awane, H.; Yorimitsu, H.; Osuka, A. Direct Arylation of meso-Formyl Porphyrin. Chem.—A Eur. J. 2013, 19, 64–68. [Google Scholar] [CrossRef]
- Belyaev, E.S.; Kozhemyakin, G.L.; Tyurin, V.S.; Frolova, V.V.; Lonin, I.S.; Ponomarev, G.V.; Buryak, A.K.; Zamilatskov, I.A. Direct C–H borylation of vinylporphyrins via copper catalysis. Org. Biomol. Chem. 2022, 20, 1926–1932. [Google Scholar] [CrossRef]
- Orlova, E.A.; Romanenko, Y.V.; Tyurin, V.S.; Shkirdova, A.O.; Belyaev, E.S.; Grigoriev, M.S.; Koifman, O.I.; Zamilatskov, I.A. Dimer of Pd(II) β-octaethylporphyrin bound by a 1,3-butadiene bridge. Macroheterocycles 2022, 15, 139–146. [Google Scholar] [CrossRef]
- Tanaka, T.; Osuka, A. Conjugated porphyrin arrays: Synthesis, properties and applications for functional materials. Chem. Soc. Rev. 2015, 44, 943–969. [Google Scholar] [CrossRef] [PubMed]
- Chachisvilis, M.; Chirvony, V.S.; Shulga, A.M.; Källebring, B.; Larsson, S.; Sundström, V. Spectral and Photophysical Properties of Ethylene-Bridged Side-to-Side Porphyrin Dimers. 1. Ground-State Absorption and Fluorescence Study and Calculation of Electronic Structure of trans-1,2-Bis(meso-octaethylporphyrinyl)ethene. J. Phys. Chem. 1996, 100, 13857–13866. [Google Scholar] [CrossRef]
- Screen, T.E.O.; Blake, I.M.; Rees, L.H.; Clegg, W.; Borwick, S.J.; Anderson, H.L. Making conjugated connections to porphyrins: A comparison of alkyne, alkene, imine and azo links. J. Chem. Soc. Perkin Trans. 1 2002, 320–329. [Google Scholar] [CrossRef]
- Rintoul, L.; Harper, S.R.; Arnold, D.P. A systematic theoretical study of the electronic structures of porphyrin dimers: DFT and TD-DFT calculations on diporphyrins linked by ethane, ethene, ethyne, imine, and azo bridges. Phys. Chem. Chem. Phys. 2013, 15, 18951–18964. [Google Scholar] [CrossRef]
- Yang, S.I.; Lammi, R.K.; Seth, J.; Riggs, J.A.; Arai, T.; Kim, D.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Excited-State Energy Transfer and Ground-State Hole/Electron Hopping in p-Phenylene-Linked Porphyrin Dimers. J. Phys. Chem. B 1998, 102, 9426–9436. [Google Scholar] [CrossRef]
- Lin, V.S.-Y.; DiMagno, S.G.; Therien, M.J. Highly Conjugated, Acetylenyl Bridged Porphyrins: New Models for Light-Harvesting Antenna Systems. Science 1994, 264, 1105–1111. [Google Scholar] [CrossRef]
- Taylor, P.N.; Anderson, H.L. Cooperative Self-Assembly of Double-Strand Conjugated Porphyrin Ladders. J. Am. Chem. Soc. 1999, 121, 11538–11545. [Google Scholar] [CrossRef]
- Nakamura, K.; Fujimoto, T.; Takara, S.; Sugiura, K.-I.; Miyasaka, H.; Ishii, T.; Yamashita, M.; Sakata, Y. Systematic synthesis of porphyrin dimers linked by conjugated oligoacetylene bridges. Chem. Lett. 2003, 32, 694–695. [Google Scholar] [CrossRef]
- Hayashi, N.; Sato, M.; Miyabayashi, K.; Miyake, M.; Higuchi, H. Synthesis and electronic properties of trimeric octaethylporphyrin (OEP) derivative connected with diacetylene linkage. A comparative study with vinylene-group connected OEP trimer. Sci. Technol. Adv. Mater. 2006, 7, 237–242. [Google Scholar] [CrossRef]
- Yamada, H.; Kushibe, K.; Mitsuogi, S.; Okujima, T.; Uno, H.; Ono, N. Selective synthesis of 5-alkenyl-15-alkynyl-porphyrin and 5,15-dialkynyl-porphyrin by 2+2 acid-catalyzed condensation of dipyrrylmethane and TMS propynal. Tetrahedron Lett. 2008, 49, 4731–4733. [Google Scholar] [CrossRef]
- Hiroyuki, H.; Takashi, I.; Kazumine, M.; Yukari, T.; Koji, Y.; Keita, T.; Keiko, M.; Mikio, M. Synthesis and Properties of Head-to-head, Head-to-tail, and Tail-to-tail Orientational Isomers of Extended Dihexylbithiophene–Octaethylporphyrin System [OEP–(DHBT)n–OEP] Connected with 1,3-Butadiyne Linkages. Bull. Chem. Soc. Jpn. 2001, 74, 889–906. [Google Scholar] [CrossRef]
- Kanwal, I.; Mujahid, A.; Rasool, N.; Rizwan, K.; Malik, A.; Ahmad, G.; Shah, S.A.A.; Rashid, U.; Nasir, N.M. Palladium and Copper Catalyzed Sonogashira cross Coupling an Excellent Methodology for C-C Bond Formation over 17 Years: A Review. Catalysts 2020, 10, 443. [Google Scholar] [CrossRef] [Green Version]
- Gou, F.; Jiang, X.; Fang, R.; Jing, H.; Zhu, Z. Strategy to Improve Photovoltaic Performance of DSSC Sensitized by Zinc Prophyrin Using Salicylic Acid as a Tridentate Anchoring Group. ACS Appl. Mater. Interfaces 2014, 6, 6697–6703. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Lee, G.-H.; Peng, S.-M.; Yeh, C.-Y. Unexpected formation of porphyrinic enyne under Sonogashira conditions. Tetrahedron Lett. 2005, 46, 1541–1544. [Google Scholar] [CrossRef]
- Chang, J.-C.; Ma, C.-J.; Lee, G.-H.; Peng, S.-M.; Yeh, C.-Y. Porphyrin–triarylamine conjugates: Strong electronic communication between triarylamine redox centers via the porphyrin dication. Dalton Trans. 2005, 1504–1508. [Google Scholar] [CrossRef]
- Dy, J.T.; Maeda, R.; Nagatsuka, Y.; Ogawa, K.; Kamada, K.; Ohta, K.; Kobuke, Y. A photochromic porphyrin–perinaphthothioindigo conjugate and its two-photon absorption properties. Chem. Commun. 2007, 1504–1508. [Google Scholar] [CrossRef]
- Wada, T.; Tachi, Y.; Toyota, K.; Kozaki, M. Platinum octaethylporphyrin-diphenylanthracene dyad with an ethynylene linker. Tetrahedron Lett. 2022, 108, 154131. [Google Scholar] [CrossRef]
- Zhang, T.-G.; Zhao, Y.; Asselberghs, I.; Persoons, A.; Clays, K.; Therien, M.J. Design, Synthesis, Linear, and Nonlinear Optical Properties of Conjugated (Porphinato)zinc(II)-Based Donor−Acceptor Chromophores Featuring Nitrothiophenyl and Nitrooligothiophenyl Electron-Accepting Moieties. J. Am. Chem. Soc. 2005, 127, 9710–9720. [Google Scholar] [CrossRef]
- Zhang, T.-G.; Zhao, Y.; Song, K.; Asselberghs, I.; Persoons, A.; Clays, K.; Therien, M.J. Electronic Modulation of Hyperpolarizable (Porphinato)zinc(II) Chromophores Featuring Ethynylphenyl-, Ethynylthiophenyl-, Ethynylthiazolyl-, and Ethynylbenzothiazolyl-Based Electron-Donating and -Accepting Moieties. Inorg. Chem. 2006, 45, 9703–9712. [Google Scholar] [CrossRef] [PubMed]
- Bessho, T.; Zakeeruddin, S.M.; Yeh, C.-Y.; Diau, E.W.-G.; Grätzel, M. Highly Efficient Mesoscopic Dye-Sensitized Solar Cells Based on Donor–Acceptor-Substituted Porphyrins. Angew. Chem. Int. Ed. 2010, 49, 6646–6649. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Lu, H.-P.; Lan, C.-M.; Huang, Y.-L.; Liang, Y.-R.; Yen, W.-N.; Liu, Y.-C.; Lin, Y.-S.; Diau, E.W.-G.; Yeh, C.-Y. Novel Zinc Porphyrin Sensitizers for Dye-Sensitized Solar Cells: Synthesis and Spectral, Electrochemical, and Photovoltaic Properties. Chem.—A Eur. J. 2009, 15, 1403–1412. [Google Scholar] [CrossRef]
- Marschner, S.M.; Haldar, R.; Fuhr, O.; Wöll, C.; Bräse, S. Modular Synthesis of trans-A2B2-Porphyrins with Terminal Esters: Systematically Extending the Scope of Linear Linkers for Porphyrin-Based MOFs. Chem.—A Eur. J. 2021, 27, 1390–1401. [Google Scholar] [CrossRef]
- Felix, L.; Sezer, U.; Arndt, M.; Mayor, M. Synthesis of Highly Fluoroalkyl-Functionalized Oligoporphyrin Systems. Eur. J. Org. Chem. 2014, 2014, 6884–6895. [Google Scholar] [CrossRef]
- Wagner, R.W.; Johnson, T.E.; Lindsey, J.S. Soluble Synthetic Multiporphyrin Arrays. 1. Modular Design and Synthesis. J. Am. Chem. Soc. 1996, 118, 11166–11180. [Google Scholar] [CrossRef]
- Wagner, R.W.; Johnson, T.E.; Li, F.; Lindsey, J.S. Synthesis of Ethyne-Linked or Butadiyne-Linked Porphyrin Arrays Using Mild, Copper-Free, Pd-Mediated Coupling Reactions. J. Org. Chem. 1995, 60, 5266–5273. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kato, K.; Shimizu, D.; Tanaka, T.; Osuka, A. Phenylene-bridged Porphyrin meso-Oxy Radical Dimers. Chem.—Asian J. 2019, 14, 4031–4034. [Google Scholar] [CrossRef]
- Okuno, Y.; Kamikado, T.; Yokoyama, S.; Mashiko, S. Determining the conformation of a phenylene-linked porphyrin dimer by NMR spectroscopy and quantum chemical calculations. J. Mol. Struct. THEOCHEM 2003, 631, 13–20. [Google Scholar] [CrossRef]
- Taylor, P.N.; Wylie, A.P.; Huuskonen, J.; Anderson, H.L. Enhanced Electronic Conjugation in Anthracene-Linked Porphyrins. Angew. Chem. Int. Ed. 1998, 37, 986–989. [Google Scholar] [CrossRef]
- Drobizhev, M.; Stepanenko, Y.; Dzenis, Y.; Karotki, A.; Rebane, A.; Taylor, P.N.; Anderson, H.L. Understanding Strong Two-Photon Absorption in π-Conjugated Porphyrin Dimers via Double-Resonance Enhancement in a Three-Level Model. J. Am. Chem. Soc. 2004, 126, 15352–15353. [Google Scholar] [CrossRef] [PubMed]
- Balaz, M.; Collins, H.A.; Dahlstedt, E.; Anderson, H.L. Synthesis of hydrophilic conjugated porphyrin dimers for one-photon and two-photon photodynamic therapy at NIR wavelengths. Org. Biomol. Chem. 2009, 7, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.-I.; Fujimoto, Y.; Sakata, Y. A porphyrin square: Synthesis of a square-shaped π-conjugated porphyrin tetramer connected by diacetylene linkages. Chem. Commun. 2000, 1105–1106. [Google Scholar] [CrossRef]
- Taylor, P.N.; Huuskonen, J.; Aplin, R.T.; Anderson, H.L.; Huuskonen, J.; Rumbles, G.; Williams, E. Conjugated porphyrin oligomers from monomer to hexamer. Chem. Commun. 1998, 909–910. [Google Scholar] [CrossRef]
- Aiko, K.; Ken-ichi, S.; Hitoshi, M.; Hiroyuki, T.; Tomoji, K.; Manabu, S.; Masahiro, Y. A Square Cyclic Porphyrin Dodecamer: Synthesis and Single-Molecule Characterization. Chem. Lett. 2004, 33, 578–579. [Google Scholar] [CrossRef]
- Hoffmann, M.; Wilson, C.J.; Odell, B.; Anderson, H.L. Template-Directed Synthesis of a π-Conjugated Porphyrin Nanoring. Angew. Chem. Int. Ed. 2007, 46, 3122. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kärnbratt, J.; Chang, M.-H.; Herz, L.M.; Albinsson, B.; Anderson, H.L. Enhanced π Conjugation around a Porphyrin[6] Nanoring. Angew. Chem. Int. Ed. 2008, 47, 4993–4996. [Google Scholar] [CrossRef]
- Nakamura, Y.; Aratani, N.; Osuka, A. Cyclic porphyrin arrays as artificial photosynthetic antenna: Synthesis and excitation energy transfer. Chem. Soc. Rev. 2007, 36, 831–845. [Google Scholar] [CrossRef]
- Wang, S.-P.; Shen, Y.-F.; Zhu, B.-Y.; Wu, J.; Li, S. Recent advances in the template-directed synthesis of porphyrin nanorings. Chem. Commun. 2016, 52, 10205–10216. [Google Scholar] [CrossRef]
- Ryan, A.; Gehrold, A.; Perusitti, R.; Pintea, M.; Fazekas, M.; Locos, O.B.; Blaikie, F.; Senge, M.O. Porphyrin Dimers and Arrays. Eur. J. Org. Chem. 2011, 2011, 5817–5844. [Google Scholar] [CrossRef]
- Zawadzka, M.; Wang, J.; Blau, W.J.; Senge, M.O. Correlation studies on structurally diverse porphyrin monomers, dimers and trimers and their nonlinear optical responses. Chem. Phys. Lett. 2009, 477, 330–335. [Google Scholar] [CrossRef]
- Maeda, C.; Yamaguchi, S.; Ikeda, C.; Shinokubo, H.; Osuka, A. Dimeric Assemblies from 1,2,3-Triazole-Appended Zn(II) Porphyrins with Control of NH-Tautomerism in 1,2,3-Triazole. Org. Lett. 2008, 10, 549–552. [Google Scholar] [CrossRef]
- Séverac, M.; Pleux, L.L.; Scarpaci, A.; Blart, E.; Odobel, F. Synthesis of new azido porphyrins and their reactivity in copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition reaction with alkynes. Tetrahedron Lett. 2007, 48, 6518–6522. [Google Scholar] [CrossRef]
- Shen, D.-M.; Liu, C.; Chen, Q.-Y. Synthesis and Versatile Reactions of β-Azidotetraarylporphyrins. Eur. J. Org. Chem. 2007, 2007, 1419–1422. [Google Scholar] [CrossRef]
- Thorley, K.J.; Anderson, H.L. Extending conjugation in porphyrin dimer carbocations. Org. Biomol. Chem. 2010, 8, 3472–3479. [Google Scholar] [CrossRef]


































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyurin, V.S.; Shkirdova, A.O.; Koifman, O.I.; Zamilatskov, I.A. Meso-Formyl, Vinyl, and Ethynyl Porphyrins—Multipotent Synthons for Obtaining a Diverse Array of Functional Derivatives. Molecules 2023, 28, 5782. https://doi.org/10.3390/molecules28155782
Tyurin VS, Shkirdova AO, Koifman OI, Zamilatskov IA. Meso-Formyl, Vinyl, and Ethynyl Porphyrins—Multipotent Synthons for Obtaining a Diverse Array of Functional Derivatives. Molecules. 2023; 28(15):5782. https://doi.org/10.3390/molecules28155782
Chicago/Turabian StyleTyurin, Vladimir S., Alena O. Shkirdova, Oscar I. Koifman, and Ilya A. Zamilatskov. 2023. "Meso-Formyl, Vinyl, and Ethynyl Porphyrins—Multipotent Synthons for Obtaining a Diverse Array of Functional Derivatives" Molecules 28, no. 15: 5782. https://doi.org/10.3390/molecules28155782
APA StyleTyurin, V. S., Shkirdova, A. O., Koifman, O. I., & Zamilatskov, I. A. (2023). Meso-Formyl, Vinyl, and Ethynyl Porphyrins—Multipotent Synthons for Obtaining a Diverse Array of Functional Derivatives. Molecules, 28(15), 5782. https://doi.org/10.3390/molecules28155782