

Aldiminium Cations as Countercations to Discrete Main Group Fluoroanions


Abstract
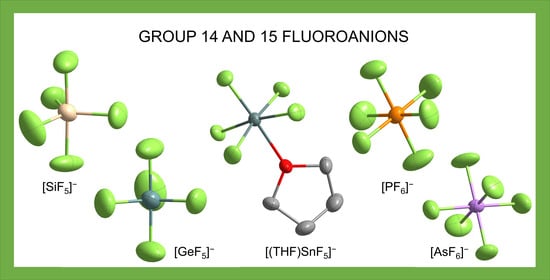
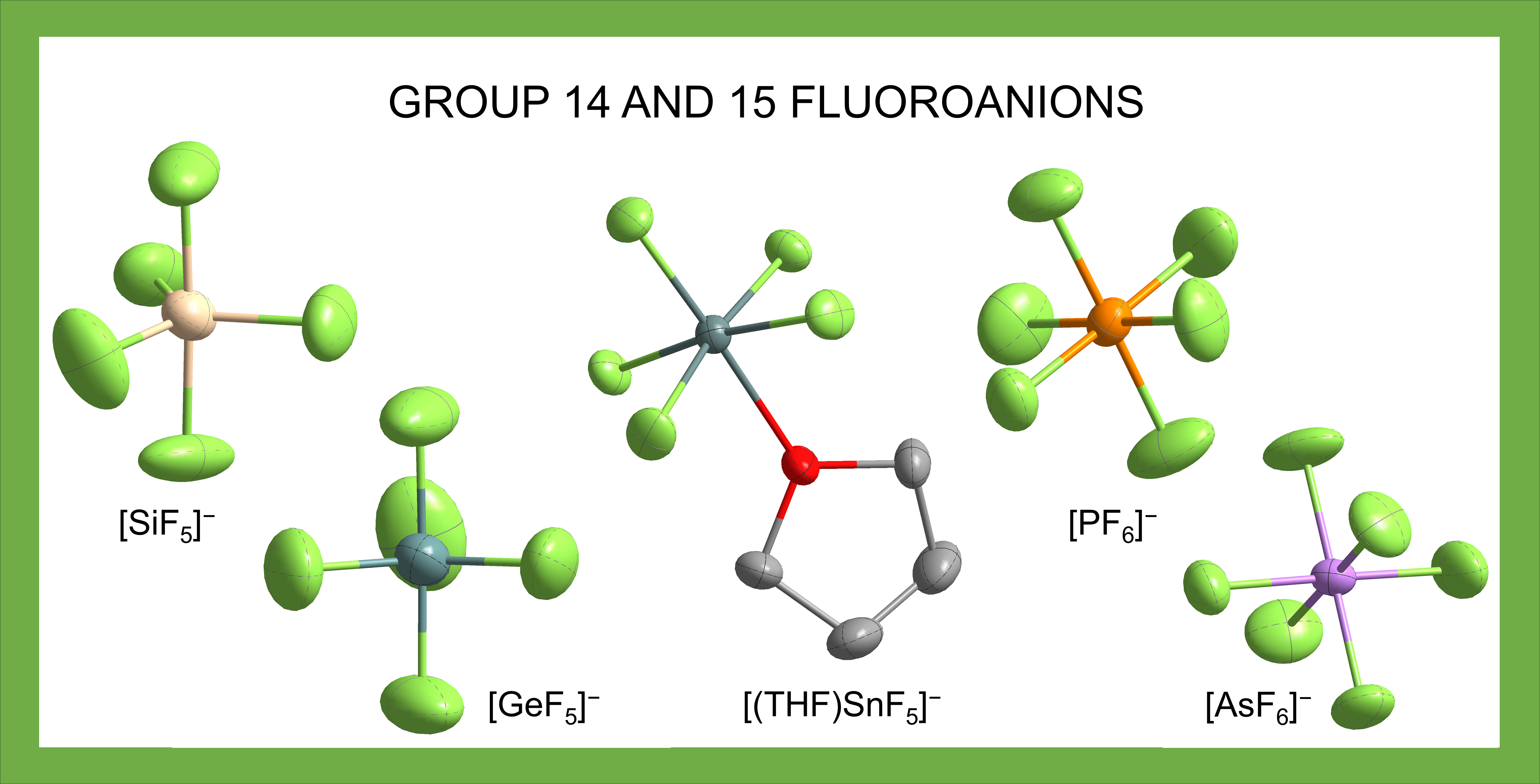
:
1. Introduction
2. Results and Discussion
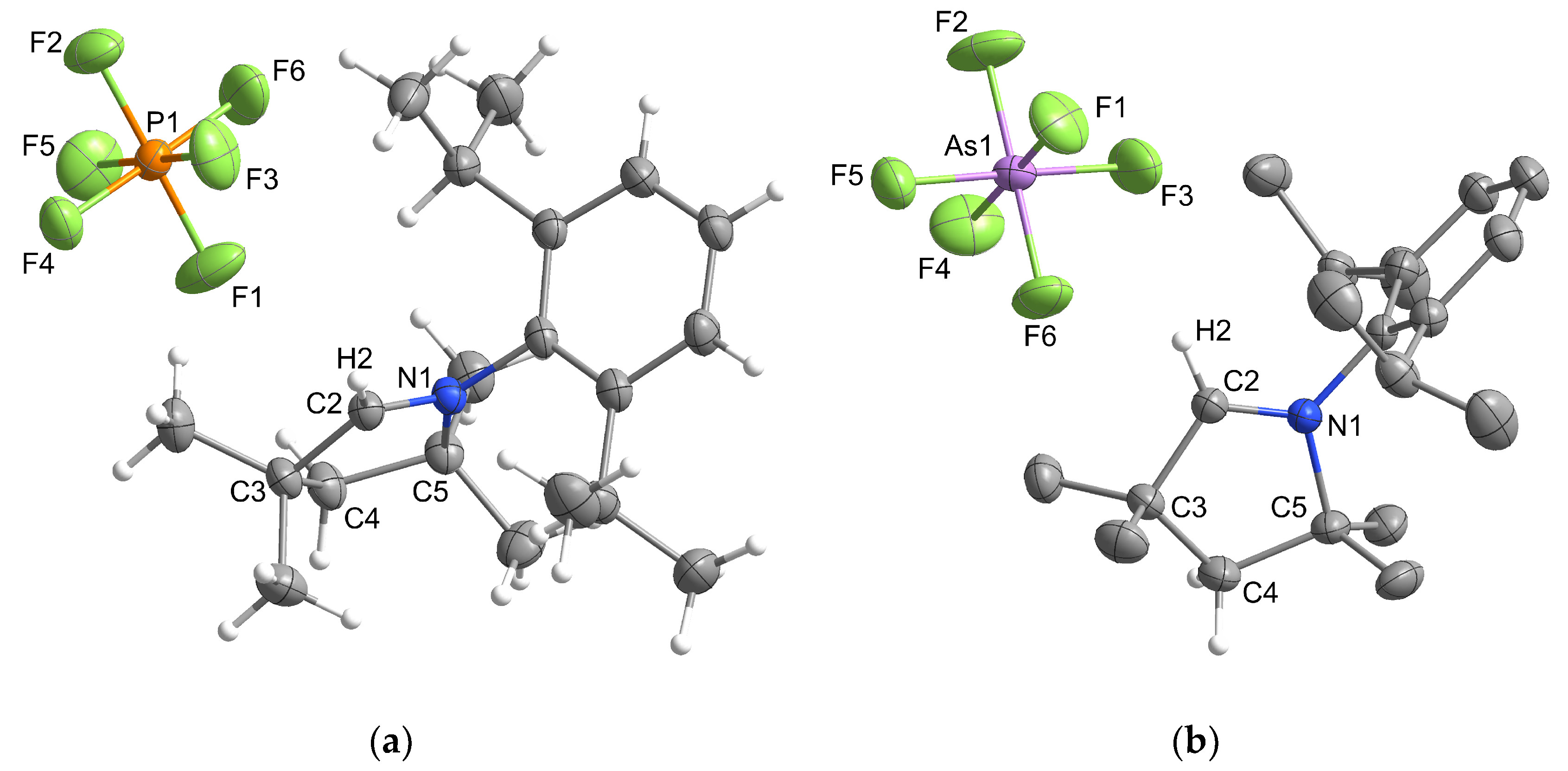
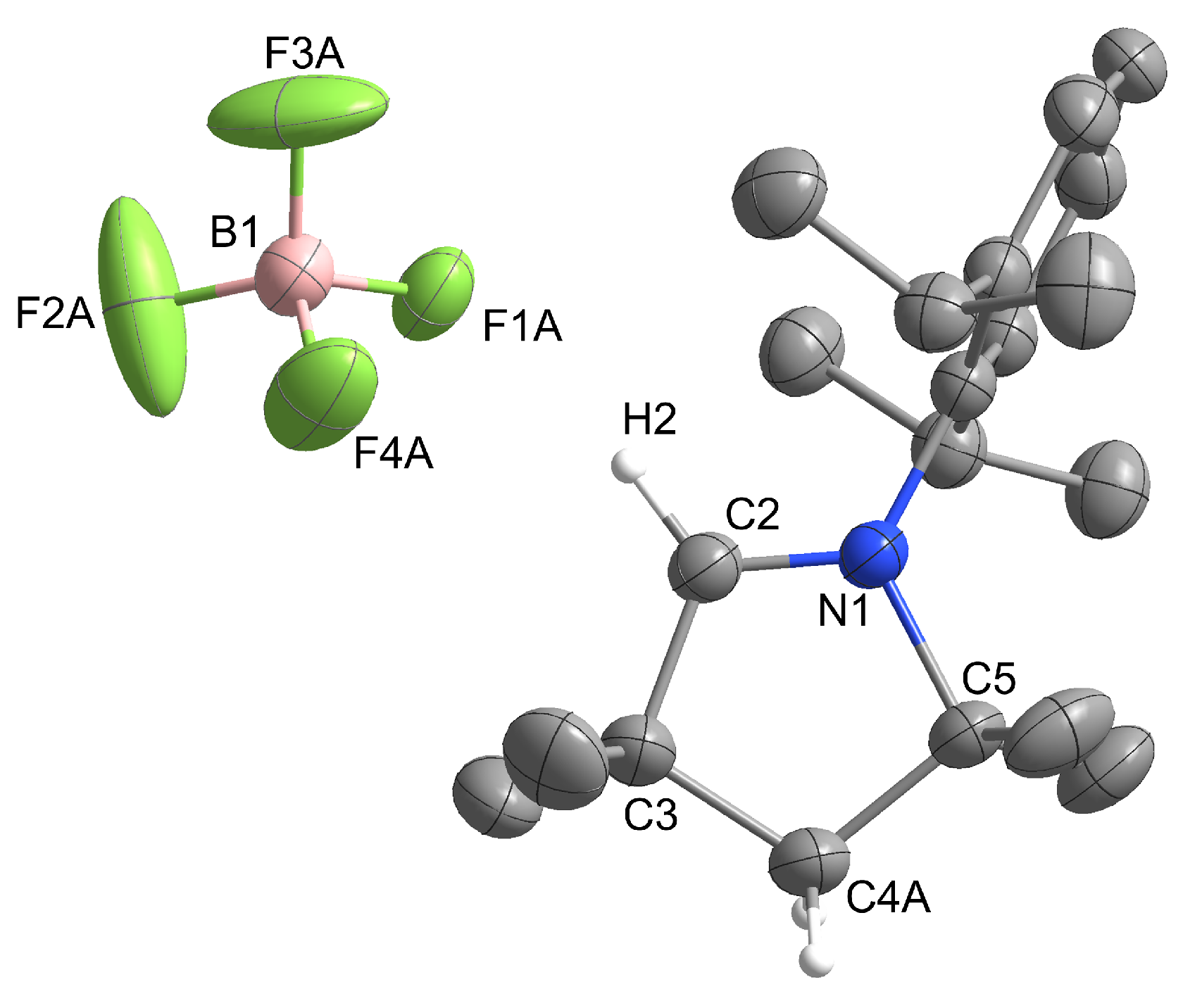
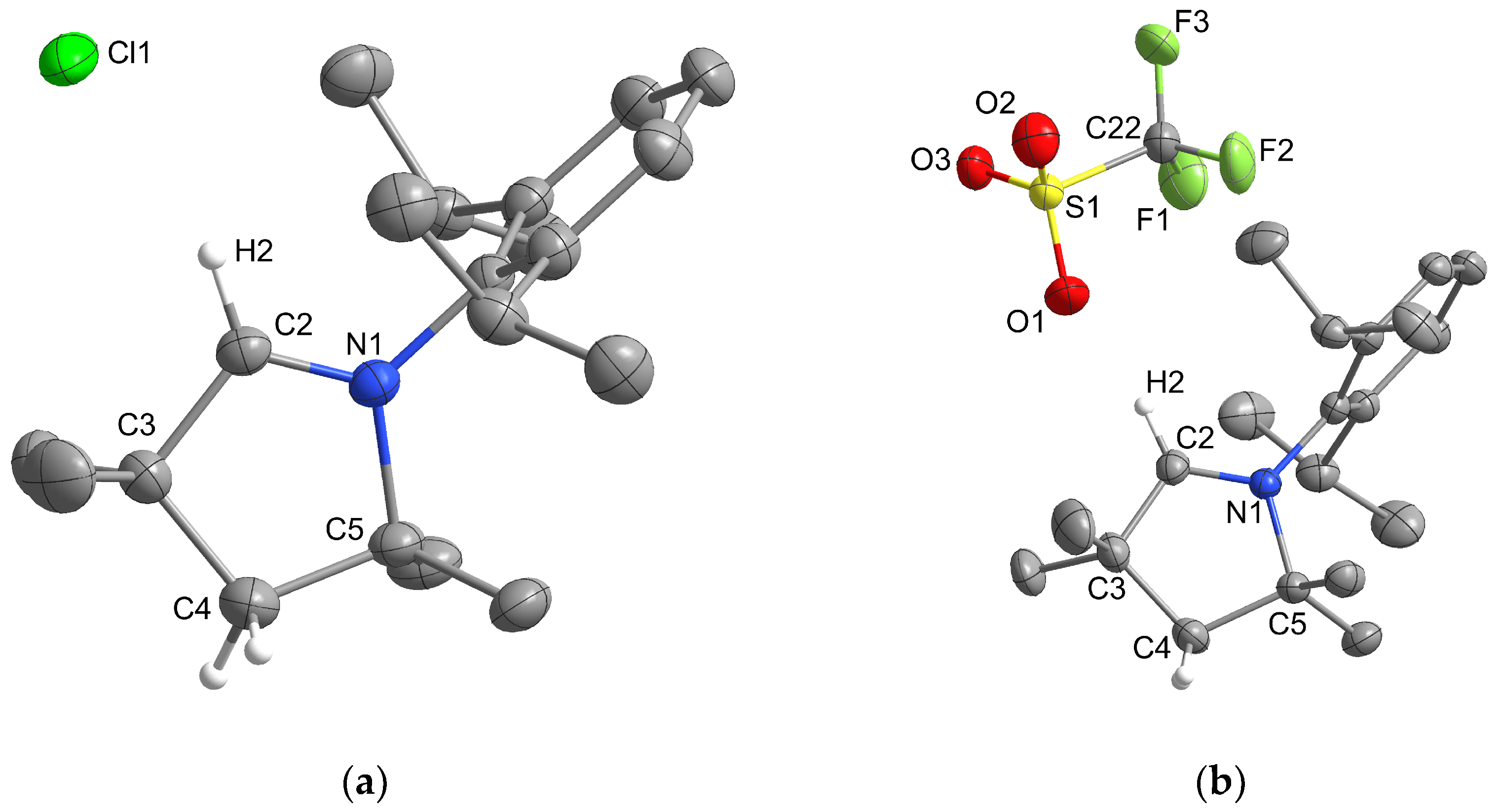
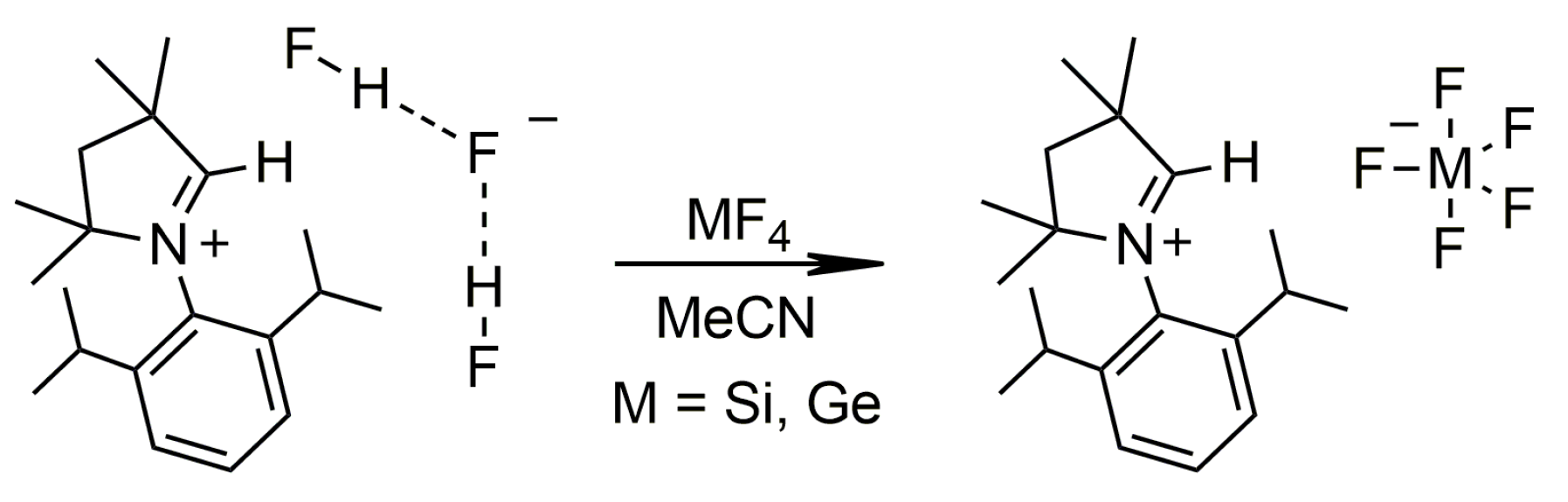
2.1. Synthesis and Structural Characterization
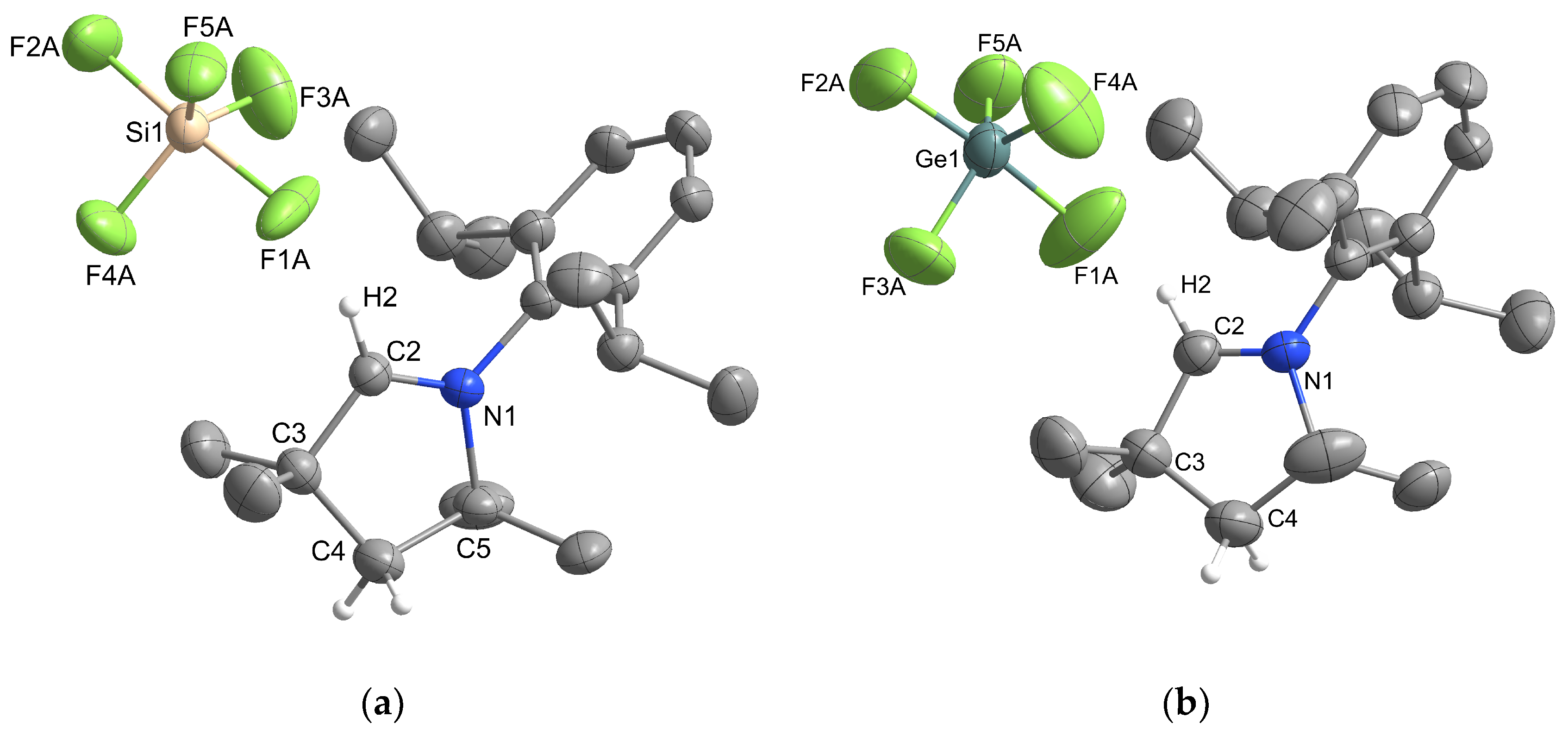
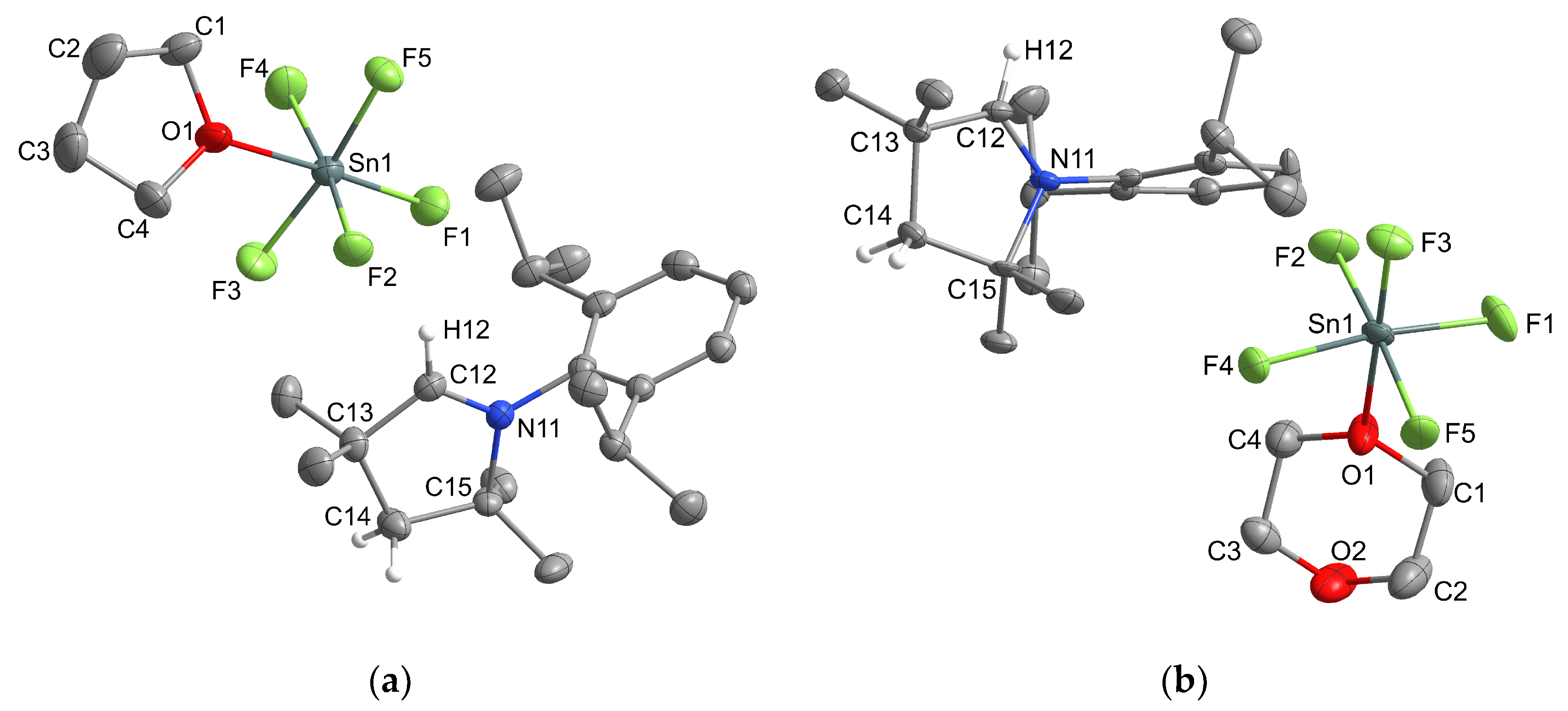
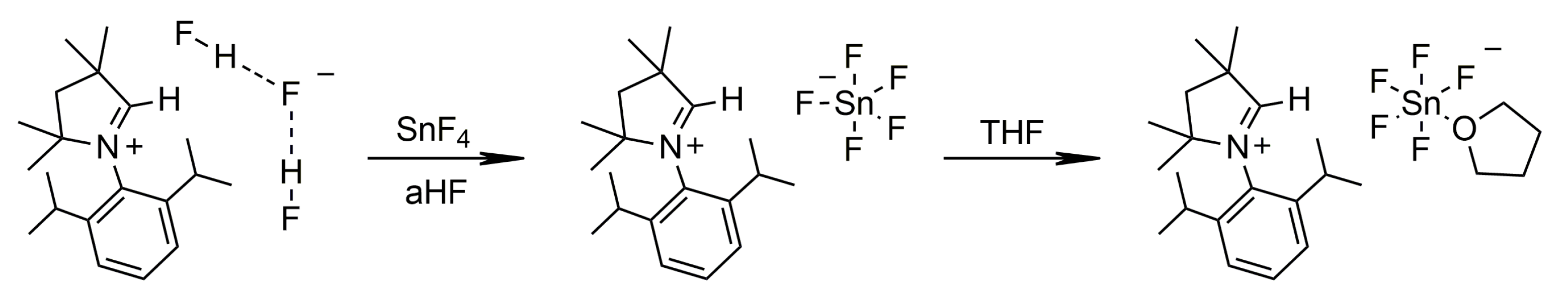
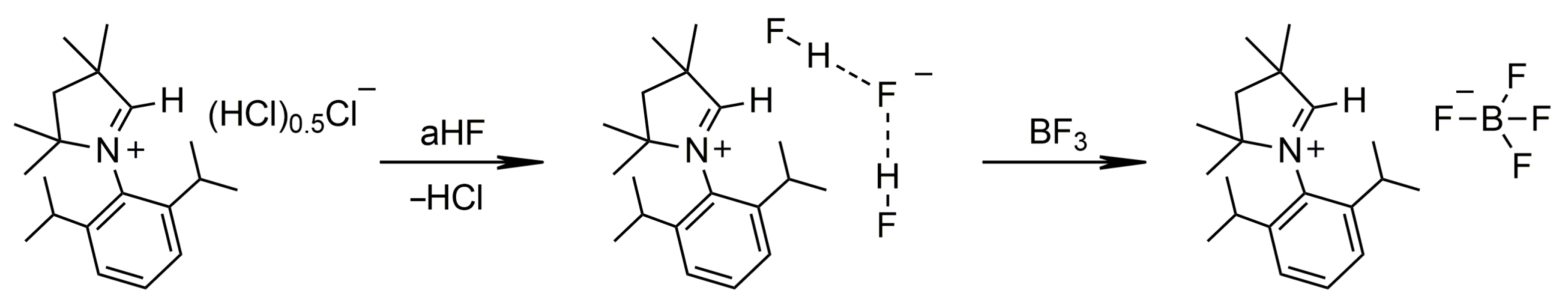
2.1.1. Group 14
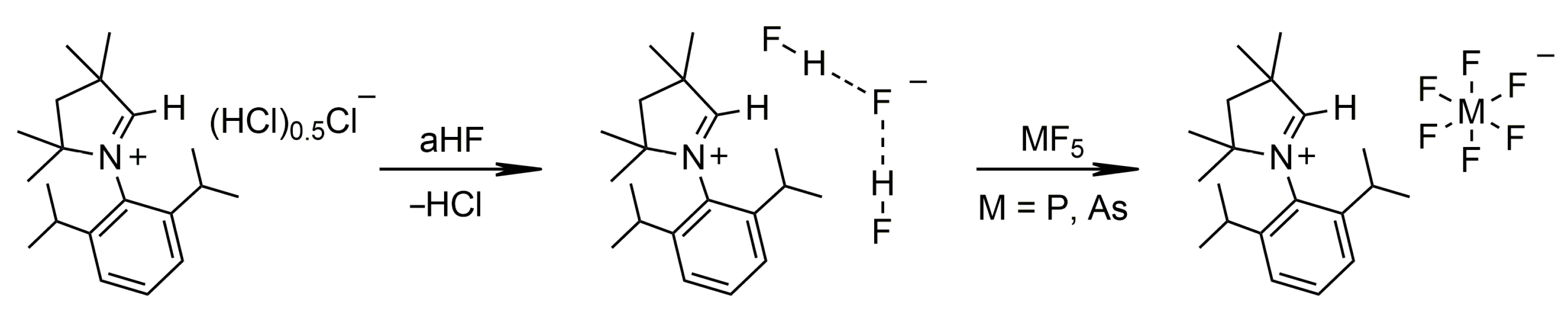
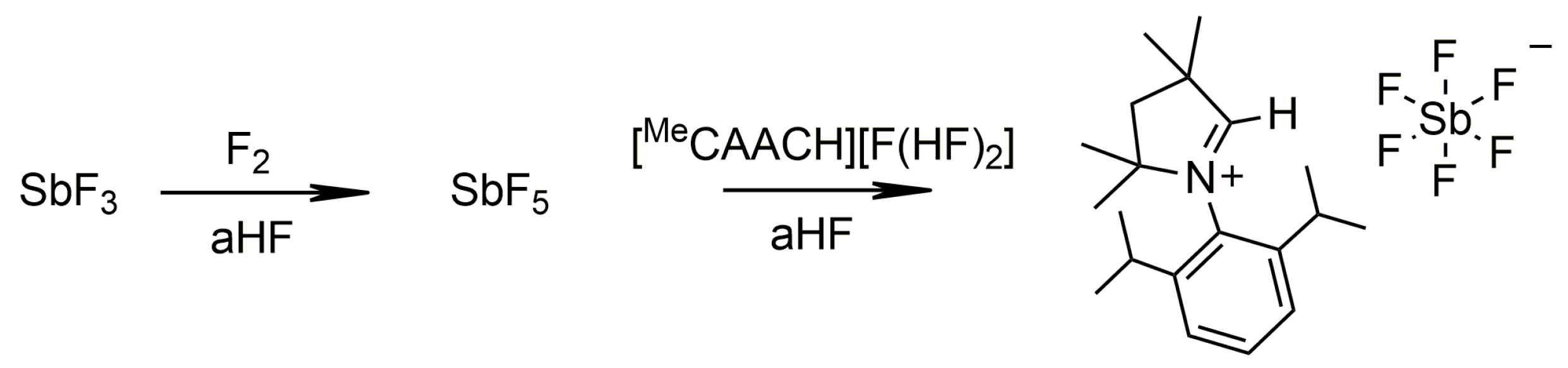
2.1.2. Group 15
2.1.3. CAAC Precursors
2.2. NMR Spectroscopy
2.3. Raman Spectroscopy
3. Materials and Methods
3.1. Reagents
3.2. Caution
3.3. General
3.4. Syntheses
3.4.1. [MeCAACH][SiF5]
3.4.2. [MeCAACH][GeF5]
3.4.3. [MeCAACH][(THF)SnF5]
3.4.4. [MeCAACH][PF6]
3.4.5. [MeCAACH][AsF6]
3.4.6. [MeCAACH][SbF6]
3.4.7. [MeCAACH][BF4]
3.5. NMR Spectroscopy
3.6. Crystal Structure Determination
3.7. Raman Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Stable Cyclic (Alkyl)(Amino)Carbenes as Rigid or Flexible, Bulky, Electron-Rich Ligands for Transition-Metal Catalysts: A Quaternary Carbon Atom Makes the Difference. Angew. Chem. Int. Ed. 2005, 44, 5705–5709. [Google Scholar] [CrossRef] [PubMed]
- Melaimi, M.; Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)(Amino)Carbenes (CAACs): Recent Developments. Angew. Chem. Int. Ed. 2017, 56, 10046–10068. [Google Scholar] [CrossRef] [PubMed]
- Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)(Amino)Carbenes (CAACs): Stable Carbenes on the Rise. Acc. Chem. Res. 2015, 48, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Jazzar, R.; Soleilhavoup, M.; Bertrand, G. Cyclic (Alkyl)- and (Aryl)-(Amino)Carbene Coinage Metal Complexes and Their Applications. Chem. Rev. 2020, 120, 4141–4168. [Google Scholar] [CrossRef] [PubMed]
- Jazzar, R.; Dewhurst, R.D.; Bourg, J.-B.; Donnadieu, B.; Canac, Y.; Bertrand, G. Intramolecular “Hydroiminiumation” of Alkenes: Application to the Synthesis of Conjugate Acids of Cyclic Alkyl Amino Carbenes (CAACs). Angew. Chem. Int. Ed. 2007, 46, 2899–2902. [Google Scholar] [CrossRef]
- Das, A.; Elvers, B.J.; Nayak, M.K.; Chrysochos, N.; Anga, S.; Kumar, A.; Rao, D.K.; Narayanan, T.N.; Schulzke, C.; Yildiz, C.B.; et al. Realizing 1,1-Dehydration of Secondary Alcohols to Carbenes: Pyrrolidin-2-Ols as a Source of Cyclic (Alkyl)(Amino)Carbenes. Angew. Chem. Int. Ed. 2022, 61, e202202637. [Google Scholar] [CrossRef]
- Bissinger, P.; Braunschweig, H.; Damme, A.; Krummenacher, I.; Phukan, A.K.; Radacki, K.; Sugawara, S. Isolation of a Neutral Boron-Containing Radical Stabilized by a Cyclic (Alkyl)(Amino)Carbene. Angew. Chem. Int. Ed. 2014, 53, 7360–7363. [Google Scholar] [CrossRef]
- Vermersch, F.; Oliveira, L.; Hunter, J.; Soleilhavoup, M.; Jazzar, R.; Bertrand, G. Cyclic (Alkyl)(Amino)Carbenes: Synthesis of Iminium Precursors and Structural Properties. J. Org. Chem. 2022, 87, 3511–3518. [Google Scholar] [CrossRef]
- Maiti, A.; Elvers, B.J.; Bera, S.; Lindl, F.; Krummenacher, I.; Ghosh, P.; Braunschweig, H.; Yildiz, C.B.; Schulzke, C.; Jana, A. Disclosing Cyclic(Alkyl)(Amino)Carbenes as One-Electron Reductants: Synthesis of Acyclic(Amino)(Aryl)Carbene-Based Kekulé Diradicaloids. Chem. Eur. J. 2022, 28, e202104567. [Google Scholar] [CrossRef]
- Gruden, E.; Tavčar, G. Synthesis and Characterization of Partially Substituted NHC Supported Alane Adducts Using Triflate or Chloride Salts. Polyhedron 2021, 196, 115009. [Google Scholar] [CrossRef]
- Gruden, E.; Prinčič, G.G.; Hočevar, J.; Iskra, J.; Kvíčala, J.; Tavčar, G. From Cyclic (Alkyl)(Amino)Carbene (CAAC) Precursors to Fluorinating Reagents. Experimental and Theoretical Study. Dalton Trans. 2023, 52, 9562–9572. [Google Scholar] [CrossRef] [PubMed]
- Alič, B.; Tramšek, M.; Kokalj, A.; Tavčar, G. Discrete GeF5– Anion Structurally Characterized with a Readily Synthesized Imidazolium Based Naked Fluoride Reagent. Inorg. Chem. 2017, 56, 10070–10077. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, F.; Lagow, R.J. A Novel Mononuclear Gold(I) Complex: Synthesis and x-Ray Structure of [Au{P(C6H5)3}3][SiF5]∙1.5CH2Cl2. Z. Anorg. Allg. Chem. 1995, 621, 1929–1932. [Google Scholar] [CrossRef]
- Bolli, C.; Gellhaar, J.; Jenne, C.; Keßler, M.; Scherer, H.; Seeger, H.; Uzun, R. Bis(Triphenyl-λ5-Phosphanylidene)Ammonium Fluoride: A Reactive Fluoride Source to Access the Hypervalent Silicates [MenSiF5−n]− (n = 0–3). Dalton Trans. 2014, 43, 4326–4334. [Google Scholar] [CrossRef] [PubMed]
- Petz, W.; Neumüller, B. The Carbodiphosphorane–CS2 Adduct as a Complex Ligand: Crystallographic Characterization of [I2Pt{S2CC(PPh3)2}]·CH2Cl2, [Pt{S2CC(PPh3)2}2][SiF5]2·2CH2Cl2 and [(Me)2PtFI{S2CC(PPh3)2}]·2CH2Cl2. Polyhedron 2008, 27, 2539–2544. [Google Scholar] [CrossRef]
- Keßler, M.; Neumann, B.; Stammler, H.; Hoge, B. Ishikawa’s Reagent—A Valuable Source for Fluoroorganic Iminium Salts. Z. Anorg. Allg. Chem. 2021, 647, 225–230. [Google Scholar] [CrossRef]
- Gorol, M.; Mösch-Zanetti, N.C.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H. Synthesis of a Novel Organoiridium(I) Fluoro Complex. Eur. J. Inorg. Chem. 2004, 2004, 2678–2682. [Google Scholar] [CrossRef]
- Christe, K.O.; Dixon, D.A.; Grant, D.J.; Haiges, R.; Tham, F.S.; Vij, A.; Vij, V.; Wang, T.-H.; Wilson, W.W. Dinitrogen Difluoride Chemistry. Improved Syntheses of cis- and trans-N2F2, Synthesis and Characterization of N2F+Sn2F9−, Ordered Crystal Structure of N2F+Sb2F11−, High-Level Electronic Structure Calculations of cis-N2F2, trans-N2F2, F2N=N, and N2F+, and Mechanism of the trans-cis Isomerization of N2F2. Inorg. Chem. 2010, 49, 6823–6833. [Google Scholar] [CrossRef]
- Wilson, W.W.; Vij, A.; Vij, V.; Bernhardt, E.; Christe, K.O. Polynitrogen Chemistry: Preparation and Characterization of (N5)2SnF6, N5SnF5, and N5B(CF3)4. Chem. Eur. J. 2003, 9, 2840–2844. [Google Scholar] [CrossRef]
- Calov, U.; Schneider, M.; Leibnitz, P. Guanidiniumhexafluorometallate von Titan, Silicium, Germanium Und Zinn. Guanidiniumpentafluorooxoniobat Und Guanidiniumtetrafluorodioxowolframat. Z. Anorg. Allg. Chem. 1991, 604, 77–83. [Google Scholar] [CrossRef]
- Sensharma, D.; Wilson, B.H.; Kumar, N.; O’Hearn, D.J.; Zaworotko, M.J. Pillar Modularity in fsc Topology Hybrid Ultramicroporous Materials Based upon Tetra(4-Pyridyl)Benzene. Cryst. Growth Des. 2022, 22, 5472–5480. [Google Scholar] [CrossRef] [PubMed]
- Lusi, M.; Fechine, P.B.A.; Chen, K.-J.; Perry, J.J.; Zaworotko, M.J. A Rare Cationic Building Block That Generates a New Type of Polyhedral Network with “Cross-Linked” pto Topology. Chem. Commun. 2016, 52, 4160–4162. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, T.; Bassil, B.S.; Röschenthaler, G.-V. Complexes of Ge(IV)- and Sn(IV)-Fluorides with Cyclic and Acyclic Carbenes: Bis(Dialkylamino)-Difluoromethylenes as Carbene Sources. Inorg. Chem. 2012, 51, 763–765. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Mao, L.; Liu, L.-M.; Liu, Y.-C.; Peng, Y.; Hong, X.; Wang, H.-H.; Liu, H.-G.; Liang, H. Potential New Inorganic Antitumour Agents from Combining the Anticancer Traditional Chinese Medicine (TCM) Matrine with Ga(III), Au(III), Sn(IV) Ions, and DNA Binding Studies. J. Inorg. Biochem. 2011, 105, 171–180. [Google Scholar] [CrossRef]
- Lee, S.M.; Lo, K.M.; Tiekink, E.R.T. Crystal Structure of Dichlorido-Bis(4-Chlorophenyl-κC)-Bis(Triphenylarsine Oxide-κO)Tin(IV), C48H38As2Cl4O2Sn. Z. Kristallogr. -N. Crys. Struct. 2021, 236, 1255–1257. [Google Scholar] [CrossRef]
- Beck, S.; Feller, M.; Spies, L.; Dietrich, K.J.; Jessen, C.; Stierstorfer, K.; Kornath, A.J. Protonation of γ-Butyrolactone and γ-Butyrolactam. ChemistryOpen 2021, 10, 8–15. [Google Scholar] [CrossRef]
- Bakulina, O.; Merkt, F.K.; Knedel, T.; Janiak, C.; Müller, T.J.J. Synthesis of Water-Soluble Blue-Emissive Tricyclic 2-Aminopyridinium Salts by Three-Component Coupling-(3+3)-Anellation. Angew. Chem. Int. Ed. 2018, 57, 17240–17244. [Google Scholar] [CrossRef]
- Golding, J.; Hamid, N.; MacFarlane, D.R.; Forsyth, M.; Forsyth, C.; Collins, C.; Huang, J. N-Methyl-N-Alkylpyrrolidinium Hexafluorophosphate Salts: Novel Molten Salts and Plastic Crystal Phases. Chem. Mater. 2001, 13, 558–564. [Google Scholar] [CrossRef]
- Smith, G.L.; Mercier, H.P.A.; Schrobilgen, G.J. F5SN(H)Xe+; a Rare Example of Xenon Bonded to sp3-Hybridized Nitrogen; Synthesis and Structural Characterization of [F5SN(H)Xe][AsF6]. Inorg. Chem. 2008, 47, 4173–4184. [Google Scholar] [CrossRef]
- Matsumoto, K.; Hagiwara, R.; Yoshida, R.; Ito, Y.; Mazej, Z.; Benkič, P.; Žemva, B.; Tamada, O.; Yoshino, H.; Matsubara, S. Syntheses, Structures and Properties of 1-Ethyl-3-Methylimidazolium Salts of Fluorocomplex Anions. Dalton Trans. 2004, 4, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Axhausen, J.; Ritter, C.; Lux, K.; Kornath, A. The Protonation of Acetamide and Thioacetamide in Superacidic Solutions: Crystal Structures of [H3CC(OH)NH2]+AsF6− and [H3CC(SH)NH2]+AsF6−. Z. Anorg. Allg. Chem. 2013, 639, 65–72. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part A: Theory and Applications in Inorganic Chemistry, 5th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1997; ISBN 978-0471163947. [Google Scholar]
- W. Kwasnik Fluor. Handbuch Der Präparativen Anorganischen Chemie; Brauer, G., Ed.; Ferdinand Enke Verlag: Stuttgart, Germany, 1975; Volume 1, pp. 159–287. ISBN 3-432-02328-6. [Google Scholar]
- Mazej, Z.; Žemva, B. Synthesis of Arsenic Pentafluoride by Static Fluorination of As2O3 in a Closed System. J. Fluor. Chem. 2005, 126, 1432–1434. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; De Menezes, S.M.C.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008). Magn. Reson. Chem. 2008, 46, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Rigaku Oxford Diffraction, CrysAlisPro, Software System, Version 1.171.41.120a; (Release Date 26-10-2021); Rigaku Corporation: Wroclaw, Poland, 2021.
- Clark, R.C.; Reid, J.S. The Analytical Calculation of Absorption in Multifaceted Crystals. Acta Crystallogr. Sect. A Found. Crystallogr. 1995, 51, 887–897. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Diamond—Crystal and Molecular Structure Visualization (v4.6.8), Crystal Impact—Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany. Available online: https://www.crystalimpact.com/diamond/ (accessed on 15 May 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | δ(19F) M–F | Isotope (M) | δ(Isotope) M–F |
|---|---|---|---|
| [MeCAACH][BF4] | −151.94 (s) | 11B | −1.23 (s) |
| [MeCAACH][SiF5] | −137.53 (br) | 29Si | −149.86 (s) |
| [MeCAACH][GeF5] | −137.45 (br) | 74Ge | na |
| [MeCAACH][(THF)SnF5] | −160.37 (d), −170.20 (quint) | 119Sn | −789.93 (m) |
| [MeCAACH][PF6] | −72.84 (d) | 31P | −146.09 (sept) |
| [MeCAACH][AsF6] | −65.77 (m) | 75As | 5.45 (sept) |
| [MeCAACH][SbF6] | −123.96 (m) | 121Sb | 86.29 (sept) |
| Compound | Raman ν M–F (cm−1) | Isotope (M) |
|---|---|---|
| [MeCAACH][BF4] | na | 11B |
| [MeCAACH][SiF5] | 712 | 29Si |
| [MeCAACH][GeF5] | 665 | 74Ge |
| [MeCAACH][(THF)SnF5] | 581 | 119Sn |
| [MeCAACH][PF6] | 744 | 31P |
| [MeCAACH][AsF6] | 712 | 75As |
| [MeCAACH][SbF6] | 645 | 121Sb |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruden, E.; Tavčar, G. Aldiminium Cations as Countercations to Discrete Main Group Fluoroanions. Molecules 2023, 28, 6270. https://doi.org/10.3390/molecules28176270
Gruden E, Tavčar G. Aldiminium Cations as Countercations to Discrete Main Group Fluoroanions. Molecules. 2023; 28(17):6270. https://doi.org/10.3390/molecules28176270
Chicago/Turabian StyleGruden, Evelin, and Gašper Tavčar. 2023. "Aldiminium Cations as Countercations to Discrete Main Group Fluoroanions" Molecules 28, no. 17: 6270. https://doi.org/10.3390/molecules28176270
APA StyleGruden, E., & Tavčar, G. (2023). Aldiminium Cations as Countercations to Discrete Main Group Fluoroanions. Molecules, 28(17), 6270. https://doi.org/10.3390/molecules28176270