
Oxidation of the Platinum(II) Anticancer Agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] to Platinum(IV) Complexes by Hydrogen Peroxide

Abstract
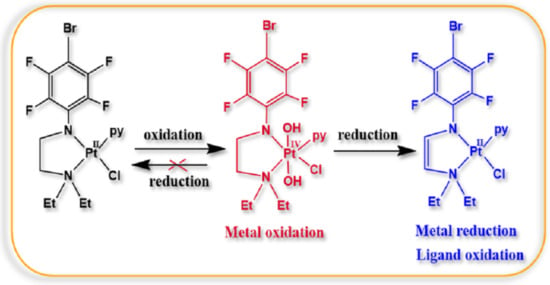
:
1. Introduction
2. Results and Discussion
2.1. X-ray Crystal Structures
2.2. Powder X-ray Diffraction (PXRD) Study
2.3. Isolation of PtIV from the Solution of an Aged Bulk Sample
2.4. NMR Spectroscopy
Variable-Temperature NMR Spectra
2.5. Electrospray MS Measurements
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation/Analytical Procedure
3.3. X-ray Crystallography
3.4. Experimental Section
3.4.1. Oxidation of 1 with H2O2
3.4.2. Oxidation of 1 with 30% H2O2 in the Presence of LiCl
3.4.3. Oxidation of 1 with Excess H2O2 in the Presence of Tetrabutylammonium Chloride (TBACl)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of cell division in escherichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Wong, E.; Giandomenico, C.M. Current status of platinum-based antitumor drugs. Chem. Rev. 1999, 99, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Fricker, S.P. Metal based drugs: From serendipity to design. Dalton Trans. 2007, 4903–4917. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef]
- Farrell, N.P. Platinum formulations as anticancer drugs clinical and pre-clinical studies. Curr. Top. Med. Chem. 2011, 11, 2623–2631. [Google Scholar] [CrossRef]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Weiss, R.B.; Christian, M.C. New cisplatin analogs in development—A Review. Drugs 1993, 46, 360–377. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.T.; Lipp, H.P. Toxicity of platinum compounds. Expert Opin. Pharmacother. 2003, 4, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Abu-Surrah, A.S.; Kettunen, M. Platinum group antitumor chemistry: Design and development of new anticancer drugs complementary to cisplatin. Curr. Med. Chem. 2006, 13, 1337–1357. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef]
- Klein, A.V.; Hambley, T.W. Platinum drug distribution in cancer cells and tumors. Chem. Rev. 2009, 109, 4911–4920. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.; Lamb, H.; Vermorken, J.B. Current and future potential roles of the platinum drugs in the treatment of ovarian cancer. Ann. Oncol. 2001, 12, 1195–1203. [Google Scholar]
- Hambley, T.W. The influence of structure on the activity and toxicity of Pt anticancer drugs. Coord. Chem. Rev. 1997, 166, 181–223. [Google Scholar] [CrossRef]
- Gibson, D. Platinum(iv) anticancer prodrugs—Hypotheses and facts. Dalton Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef]
- Lee, Y.-A.; Lee, S.S.; Kim, K.M.; Lee, C.O.; Sohn, Y.S. Synthesis and oral antitumor activity of tetrakis(carboxylato)platinum(IV) complexes. J. Med. Chem. 2000, 43, 1409–1412. [Google Scholar] [CrossRef]
- Barbanente, A.; Gandin, V.; Ceresa, C.; Marzano, C.; Ditaranto, N.; Hoeschele, J.D.; Natile, G.; Arnesano, F.; Pacifico, C.; Intini, F.P.; et al. Improvement of Kiteplatin efficacy by a benzoato Pt(IV) prodrug suitable for oral administration. Int. J. Mol. Sci. 2022, 23, 7081. [Google Scholar] [CrossRef]
- Galanski, M.; Keppler, B.K. Searching for the magic bullet: Anticancer platinum drugs which can be accumulated or activated in the tumor tissue. Anticancer Agents Med. Chem. 2007, 7, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Harrap, K.R.; Kelland, L.R.; Jones, M.; Goddard, P.M.; Orr, R.M.; Morgan, S.E.; Murrer, B.A.; Abrams, M.J.; Giandomenico, C.M.; Cobbleigh, T. Platinum coordination complexes which circumvent cisplatin resistance. Adv. Enzym. Res. 1991, 31, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Albert, A. Chemical aspects of selective toxicity. Nature 1958, 182, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Song, H.; Yang, Q.; Cai, H.; Qi, R.; Yan, L.; Liu, S.; Zheng, Y.; Huang, Y.; Liu, T.; et al. A prodrug strategy to deliver cisplatin(IV) and paclitaxel in nanomicelles to improve efficacy and tolerance. Biomaterials 2012, 33, 6507–6519. [Google Scholar] [CrossRef]
- Hall, M.D.; Hambley, T.W. Platinum(IV) antitumour compounds: Their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Yao, H.; Wang, Z.; Wang, N.; Deng, Z.; Liu, G.; Zhou, J.; Chen, S.; Shi, J.; Zhu, G. Enhancing circulation and tumor accumulation of carboplatin via an erythrocyte-anchored prodrug strategy. Angew. Chem. Int. Ed. 2022, 61, e202203838. [Google Scholar] [CrossRef]
- Ellis, L.T.; Er, H.M.; Hambley, T.W. The influence of the axial ligands of a series of platinum(IV) anticancer complexes on their reduction to platinum(II) and reaction with DNA. Aust. J. Chem. 1995, 48, 793–806. [Google Scholar] [CrossRef]
- Chaney, S.G.; Wyrick, S.; Till, G.K. In vitro biotransformations of tetrachloro(d,l-trans)-1,2-diaminocyclohexaneplatinum(IV) (tetraplatin) in rat plasma. Cancer Res. 1990, 50, 4539–4545. [Google Scholar]
- Poon, G.K.; Raynaud, F.I.; Mistry, P.; Odell, D.E.; Kelland, L.R.; Harrap, K.R.; Barnard, C.F.; Murrer, B.A. Metabolic studies of an orally active platinum anticancer drug by liquid chromatography-electrospray ionization-mass spectrometry. J. Chromatogr. 1995, 712, 61–66. [Google Scholar] [CrossRef]
- Choi, S.; Filloto, C.; Bisanzo, M.; Delaney, S. Reduction and anticancer activity of Platinum(IV) complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar] [CrossRef]
- Huang, J.; Ding, W.; Zhu, X.; Li, B.; Zeng, F.; Wu, K.; Wu, X.; Wang, F. Ligand evolution in the photoactivatable platinum(IV) anticancer prodrugs. Front. Chem. 2022, 10, 876410. [Google Scholar] [CrossRef] [PubMed]
- Spector, D.; Pavlov, K.; Beloglazkina, E.; Krasnovskaya, O. Recent advances in light-controlled activation of Pt(IV) prodrugs. Int. J. Mol. Sci. 2022, 23, 14511. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, Y.; Lin, X.; Ma, W.; Chen, G.; Li, W.; Wang, X.; Yu, Z. Platinum(iv) prodrugs with long lipid chains for drug delivery and overcoming cisplatin resistance. Chem. Comm. 2018, 54, 5369–5372. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Jin, Q.; Li, Y.; Zhang, J.; Kang, X.; Zhu, J.; Mao, X.; Cao, P.; Liu, C. Nanoparticle delivery of a triple-action Pt(IV) prodrug to overcome cisplatin resistance via synergistic effect. Biomater. Sci. 2022, 10, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D. Platinum(IV) anticancer agents; are we en route to the holy grail or to a dead end? J. Inorg. Biochem. 2021, 217, 111353. [Google Scholar] [CrossRef]
- Sinisi, M.; Intini, F.P.; Natile, G. Dependence of the reduction products of platinum(IV) prodrugs upon the configuration of the substrate, bulk of the carrier ligands, and nature of the reducing agent. Inorg. Chem. 2012, 51, 9694–9704. [Google Scholar] [CrossRef]
- Nemirovski, A.; Vinograd, I.; Takrouri, K.; Mijovilovich, A.; Rompel, A.; Gibson, D. New reduction pathways for ctc-[PtCl2(CH3CO2)2(NH3)(Am)] anticancer prodrugs. Chem. Commun. 2010, 46, 1842–1844. [Google Scholar] [CrossRef]
- Hambley, T.W. New approaches to platinum anti-cancer drugs. Chem. Aust. 1991, 58, 154–156. [Google Scholar]
- Lovejoy, K.S.; Lippard, S.J. Non-traditional platinum compounds for improved accumulation, oral bioavailability, and tumor targeting. Dalton Trans. 2009, 10651–10659. [Google Scholar] [CrossRef]
- Cleare, M.J.; Hoeschele, J.D. Studies on the antitumor activity of group VIII transition metal complexes. Part I. Platinum (II) complexes. Bioinorg. Chem. 1973, 2, 187–210. [Google Scholar] [CrossRef]
- Farrell, N. Metal Ions in Biological Systems, 1st ed.; CRC Press: Boca Raton, FL, USA, 2004; Volume 42, p. 46. [Google Scholar]
- Wheate, N.J.; Collins, J.G. Multi-nuclear platinum drugs: A new paradigm in chemotherapy. Curr. Med. Chem. Anticancer Agents 2005, 5, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Collins, J.G. Multi-nuclear platinum complexes as anticancer drugs. Coord. Chem. Rev. 2003, 241, 133–145. [Google Scholar] [CrossRef]
- Webster, L.K.; Deacon, G.B.; Buxton, D.P.; Hillcoat, B.L.; James, A.M.; Roos, I.A.G.; Thomson, R.J.; Wakelin, L.P.G.; Williams, T.L. Cis-bis(pyridine)platinum(II) organoamides with unexpected growth-inhibition properties and antitumor-activity. J. Med. Chem. 1992, 35, 3349–3353. [Google Scholar] [CrossRef] [PubMed]
- Talarico, T.; Phillips, D.R.; Deacon, G.B.; Rainone, S.; Webster, L.K. Activity and DNA binding of new organoamidoplatinum (II) complexes. Investig. New Drugs 1999, 17, 1–15. [Google Scholar] [CrossRef]
- Ojha, R.; Mason, D.; Forsyth, C.M.; Deacon, G.B.; Junk, P.C.; Bond, A.M. Diverse and unexpected outcomes from oxidation of the platinum(II) anticancer agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] by hydrogen peroxide. J. Inorg. Biochem. 2021, 218, 111360. [Google Scholar] [CrossRef]
- Masztafiak, J.; Nogueira, J.; Lipiec, L.; Kwiatek, W.; Wood, B.; Deacon, G.B.; Kayser, Y.; Fernandes, D.; Pavliuk, M.; Szlachetko, J.; et al. Direct determination of metal complexes interation with DNA by atomic telemetry and multiscale molecular dynamics. J. Phys. Chem. Lett. 2017, 8, 805–811. [Google Scholar] [CrossRef]
- Shaw, P.A.; Clarkson, G.J.; Rourke, J.P. Long-lived five-coordinate platinum(IV) intermediates: Regiospecific c–c coupling. Organometallics 2016, 35, 3751–3762. [Google Scholar] [CrossRef]
- Twigg, M.V. Higher Oxidation State Organopalladium and Platinum Chemistry. Platin. Met. Rev. 2012, 56, 104–109. [Google Scholar] [CrossRef]
- Canty, A.J.; Hoare, J.L.; Davies, N.W.; Traill, P.R. Synthesis and decomposition behavior of Pallada(IV)cyclopentane complexes. Organometallics 1998, 17, 2046–2051. [Google Scholar] [CrossRef]
- Canty, A.J. Development of organopalladium(IV) chemistry: Fundamental aspects and systems for studies of mechanism in organometallic chemistry and catalysis. Acc. Chem. Res. 1992, 25, 83–90. [Google Scholar] [CrossRef]
- Topczewski, J.J.; Sanford, M.S. Carbon–hydrogen (C–H) bond activation at Pd(IV): A frontier in c–h functionalization catalysis. Chem. Sci. 2015, 6, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-X.; Mason, D.N.; Turland, S.A.; Lawrenz, E.T.; Kelly, L.C.; Fallon, G.D.; Gatehouse, B.M.; Bond, A.M.; Deacon, G.B.; Battle, A.R.; et al. Systematic differences in electrochemical reduction of the structurally characterized anticancer platinum(IV) complexes [Pt{((p-HC6F4)NCH2)2}-(pyridine)2Cl2], [Pt{((p-HC6F4)NCH2)2}(pyridine)2(OH)2], and [Pt{((p-HC6F4)NCH2)2}(pyridine)2(OH)Cl]. J. Inorg. Biochem. 2012, 115, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Law, D.J.; Sunley, G.J.; White, A.J.P.; Britovsek, G.J.P. Hydrogen bonding directs the H2O2 oxidation of platinum(ii) to a cis-dihydroxo platinum(iv) complex. Chem. Commun. 2008, 2800–2802. [Google Scholar] [CrossRef]
- Shamsuddin, S.; Santillan, C.C.; Stark, J.L.; Whitmire, K.H.; Siddik, Z.H.; Khokhar, A.R. Synthesis, characterization, and antitumor activity of new platinum(IV) trans-carboxylate complexes: Crystal structure of [Pt(cis-1,4-DACH)trans-(acetate)2Cl2]. J. Inorg. Biochem. 1998, 71, 29–35. [Google Scholar] [CrossRef]
- Lee, Y.-A.; Ho Yoo, K.; Jung, O.-S. Oxidation of Pt(II) to Pt(IV) complex with hydrogen peroxide in glycols. Inorg. Chem. Commun. 2003, 6, 249–251. [Google Scholar] [CrossRef]
- Canil, G.; Braccini, S.; Marzo, T.; Marchetti, L.; Pratesi, A.; Biver, T.; Funaioli, T.; Chiellini, F.; Hoeschele, J.D.; Gabbiani, C. Photocytotoxic Pt(iv) complexes as prospective anticancer agents. Dalton Trans. 2019, 48, 10933–10944. [Google Scholar] [CrossRef]
- Abrams, M.J.; Giandomenico, C.M.; Murrer, B.A.; Vollano, J.F. Pt(IV) Complexes as Anti-Tumor Agents. U.S. Patent Application No. 5,244,919, 1 July 1991. [Google Scholar]
- Giandomenico, C.M.; Abrams, M.J.; Murrer, B.A.; Vollano, J.F.; Rheinheimer, M.I.; Wyer, S.B.; Bossard, G.E.; Higgins, J.D. Carboxylation of kinetically inert platinum(iv) hydroxy complexes. an entree into orally active platinum(iv) antitumor agents. Inorg. Chem. 1995, 34, 1015–1021. [Google Scholar] [CrossRef]
- Ojha, R.; Boas, J.F.; Deacon, G.B.; Junk, P.C.; Bond, A.M. EPR spectroscopic characterization of a monomeric PtIII species produced via electrochemical oxidation of the anticancer compound trans-[PtII{(p-HC6F4)NCH2CH2NEt2}Cl(py)]. J. Inorg. Biochem. 2016, 162, 194–200. [Google Scholar] [CrossRef]
- Ojha, R.; Nafady, A.; Shiddiky, M.J.A.; Mason, D.; Boas, J.F.; Torriero, A.A.J.; Bond, A.M.; Deacon, G.B.; Junk, P.C. Conditions Favoring the Formation of Monomeric PtIII Derivatives in the Electrochemical Oxidation of trans-[PtII{(p-BrC6F4)NCH2CH2NEt2}Cl(py)]. ChemElectroChem 2015, 2, 1048–1061. [Google Scholar] [CrossRef]
- Kauffman, G.B.; Cowan, D.O.; Slusarczuk, G.; Kirschner, S. cis- and trans-Dichlorodiammineplatinum(II). In Inorganic Syntheses; McGraw-Hill Book Company, Inc.: New York, NY, USA, 1963; Volume 7, pp. 239–245. [Google Scholar]
- Wilson, J.J.; Lippard, S.J. Synthetic methods for the preparation of platinum anticancer complexes. Chem. Rev. 2014, 114, 4470–4495. [Google Scholar] [CrossRef]
- Cox, L.E.; Peters, D.G.; Wehry, E.L. Photoaquation of hexachloroplatinate (IV). J. Inorg. Nucl. Chem. 1972, 34, 297–305. [Google Scholar] [CrossRef]
- Dunham, S.O.; Larsen, R.D.; Abbott, E.H. Nuclear Magnetic Resonance investigation of the hydrogen peroxide oxidation of Platinum(II) complexes. crystal and molecular structures of sodium írans-dihydroxobis(malonato)plátinate(iv) hexahydrate and sodium frans-dihydroxobis(oxalato)platinate(iv) hexahydrate. Inorg. Chem. 1993, 32, 2049–2055. [Google Scholar]
- Murray, P.; Koch, K.R.; van Eldik, R. Mechanism of tetrachloroplatinate(ii) oxidation by hydrogen peroxide in hydrochloric acid solution. Dalton Trans. 2014, 43, 6308–6314. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1997. [Google Scholar]
- Deacon, G.B.; Patrick, J.M.; Skelton, B.W.; Thomas, N.C.; White, A.H. Ruthenium carbonyl complexes. III. Peparations, properties and structures of Dicarbonyl- and Monocarbonyl-(2,2′:6′,2″-terpyridyl)ruthenium(II) complexes. Aust. J. Chem. 1984, 37, 929–945. [Google Scholar] [CrossRef]
- Ojha, R.; Junk, P.C.; Deacon, G.B.; Bond, A.M. A supramolecular approach to the examination of the structures of some anticancer organoamidoplatinum(II) complexes. Supramol. Chem. 2018, 30, 418–424. [Google Scholar] [CrossRef]
- Nakamoto, K. Infra-Red and Raman Spectra of Inorganic Coordination Compounds, 4th ed.; John Wiley and Sons: New York, NY, USA, 1986. [Google Scholar]
- McPhillips, T.M.; McPhillips, S.E.; Chiu, H.J.; Cohen, A.E.; Deacon, A.M.; Ellis, P.J.; Garman, E.; Gonzalez, A.; Sauter, N.K.; Phizackerley, R.P.; et al. Blu-Ice and the Distributed Control System: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synch. Rad. 2002, 9, 401–406. [Google Scholar] [CrossRef]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Bruker AXS; APEX2 v 2.0. Bruker AXS Inc.: Madison, WI, USA, 2005.
- Barbour, L.J. X-Seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Broughton, D.B.; Wentworth, R.L.; Laing, M.E. Mechanism of decomposition of hydrogen peroxide solutions with manganese dioxide II. J. Am. Chem. Soc. 1947, 69, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Kukushkin, V.Y.; Belskii, V.; Aleksandrova, E.; Pankova, E.; Konovalov, V.; Yakovlev, V.; Moiseev, A. Unusual Redox-Pairing of Acetoxime Ligands in Platinum Complex. Zhurnal Obs. Khimii 1991, 61, 318–328. [Google Scholar]
- Melanson, R.; Rochon, F.D. The crystal structure of potassium trichloro(2,6-lutidine)platinum(II). Can. J. Chem. 1976, 54, 1002–1006. [Google Scholar] [CrossRef]
- Cingi Biagini, M.; Ferrari, M.; Lanfranchi, M.; Marchiò, L.; Angela Pellinghelli, M. Chirality in mononuclear square planar complexes. J. Chem. Soc. Dalton Trans. 1999, 1575–1580. [Google Scholar] [CrossRef]
- Flack, H.D. On Enantiomorph-Polarity Estimation. Acta Cryst. 1983, A39, 876–881. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron. Radiat. 2015, 22, 187–190. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cl Source | H2O2 | Solvent | Time | Products with Yields |
|---|---|---|---|---|---|
| 1 (1.0 mmol) | none | 2.0 mmol | CH3COCH3 | 12 d | 1H = 1.4%, 2·H2O(gold) = 25%; 1H0.251Br0.75 = 7% |
| 1 (0.5 mmol) | LiCl (0.5 mmol) | 1.0 mmol | * CH2Cl2 | 7 d | 2·H2O(red) = 50%, 2·H2O(gold) = 14% |
| 1 (0.48 mmol) | NBu4Cl (0.48 mmol) | 1.0 mmol | * CH2Cl2 | 4 d | 3·0.5CH2Cl2 = 49%, 2·H2O(gold) = 10% |
| 2·H2O(gold) | 2·H2O(red) | 3·0.5CH2Cl2 | |
|---|---|---|---|
| Empirical formula | C17H19BrClF4N3O3Pt | C17H19BrClF4N3O3Pt | C17.5H22BrCl2F4N3O2Pt |
| Formula weight | 703.83 | 703.83 | 728.28 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | Cc | C2/c |
| a (Å) | 19.719(4) | 19.6389(16) | 14.948(3) |
| b (Å) | 13.357(3) | 13.3360(11) | 14.302(3) |
| c (Å) | 16.946(3) | 16.9327(13) | 21.743(4) |
| α (°) | 90 | 90 | 90 |
| β (°) | 105.29(3) | 104.857(2) | 104.53(3) |
| γ (°) | 90 | 90 | 90 |
| vol (Å3) | 4305.4(16) | 4286.5(6) | 4499.7(17) |
| Z | 8 | 8 | 8 |
| ρ (calcd) (g/cm3) | 2.172 | 2.169 | 2.150 |
| µ (mm−1) | 8.557 | 8.594 | 8.303 |
| F (000) | 2688.0 | 2656.0 | 2776.0 |
| Reflections collected/unique | 31,692/5071 | 35,311/12,158 | 40,998/6383 |
| Rint | 0.1016 | 0.0537 | 0.0504 |
| 2θmax (°) | 56.33 | 61.2 | 63.5 |
| Goodness-of-fit on F2 | 1.287 | 1.044 | 1.060 |
| R1 indices [I ≥ 2σ (I)] | 0.0712 | 0.0387 | 0.0342 |
| wR2 indices [I ≥ 2σ (I)] | 0.1431 | 0.0634 | 0.0841 |
| Flack parameter | n/a | 0.398(5) | n/a |
| Bond | 2·H2O(gold) C17H23BrClF4N3O3Pt (Å) | 2·H2O(red) C17H23BrClF4N3O3Pt (Molecules A and B in Asymmetric Unit) (Å) | 3·0.5CH2Cl2 C17.5H22BrCl2F4N3O2Pt (Å) | 103(OH)2 C24H18F8N4O2Pt [54] (Å) | |
|---|---|---|---|---|---|
| Pt-O1 | 2.029(9) | 2.015(6) | 2.028(7) | 2.061(3) | 2.017(3) |
| Pt-O2 | 2.003(8) | 1.999(6) | 2.016(6) | 2.018(3) | 2.008(3) |
| Pt-Cl | 2.349(3) | 2.359(3) | 2.345(3) | 2.3662 (11) | n/a |
| Pt-N1(amide) | 2.031(9) | 2.026(9) | 2.037(9) | 2.047(3) | 2.033(3) |
| Pt-N2(amine) | 2.119(7) | 2.119(8) | 2.132(9) | 2.124(3) | n/a |
| Pt-N3(py) | 2.078(7) | 2.087(8) | 2.066(9) | 2.061(3) | 2.083(3) |
| N1(amide)-C6F4 | 1.395(13) | 1.387(15) | 1.380(14) | 1.391(5) | 1.392(5) |
| C7(deen)-C8(deen) | 1.492(16) | 1.507(15) | 1.497(15) | 1.506(6) | 1.473(5) |
| N1(amide)-C7(deen) | 1.459(12) | 1.456(13) | 1.469(13) | 1.467 (5) | 1.470(5) |
| N2(amine)-C8(deen) | 1.508(14) | 1.497(13) | 1.504(13) | 1.510(6) | n/a |
| N2(amine)-C9(Et) N2(amine)-C11(Et) | 1.510(14) 1.502(13) | 1.510(13) 1.508(14) | 1.514(14) 1.508(13) | 1.497(5) 1.521(5) | n/a |
| 2·H2O(gold) | 3·0.5CH2Cl2 | ||
|---|---|---|---|
| Interactions | Distance | Interactions | Distance |
| F1⋯HO2 | 2.412(7) | O2⋯ H(CH2CH3) | 2.3561(8) |
| F4⋯ HO1 | 2.438(7) | F4⋯HO2 | 2.7065(7) |
| O1⋯ H(py) | 2.322(7) | Cl⋯H(CH3) | 2.7529(7) |
| O2⋯ H(py) | 2.227(7) | (CH2Cl2)Cl⋯H(CH2N(p-BrC6F4) | 3.0946(7) |
| O1⋯ H(CH2CH3) | 2.380(7) | ||
| Cl⋯H(CH3) | 2.738(3) | Inter (CH2Cl2)Cl⋯H(CH2CH3) | 2.8282(4) |
| Inter O3H⋯O1 | 2.198(7) | ||
| 1H NMR Chemical Shifts Assignment | 1H * (ppm) | 2·H2O(red) (ppm) | 3·0.5CH2Cl2 (ppm) |
|---|---|---|---|
| -NCH2CH3 | 1.56, td | 1.23, t | 1.26, t |
| -CH2NEt2 | - | 2.30, m | 2.30, m |
| =CHNEt2 | 3.75, d | - | - |
| -NCHAHBCH3 | 2.30, m | 3.00, m | 3.00, m |
| -NCHAHBCH3 | 3.43, m | 3.13, m | 3.00, m |
| -CH2N(p-BrC6F4) | - | 3.33, m | 3.47, m |
| =CHN(p-BrC6F4) | 6.07, m | - | - |
| Pt-OH | - | 4.03, m | 4.03, m |
| H 3, 5 (py) | 7.19, m | 7.33, t | 7.19, t |
| H 4 (py) | 7.74, tt | 7.86, t | 7.72, t |
| H 2,6 (py) | 8.42, d | 8.98, d | 8.98, d |
| 19F NMR chemical shifts Assignment | 1H * (ppm) | 2·H2O(red) (ppm) | 3·0.5CH2Cl2 (ppm) |
| (p-BrC6F4) F 3,5 | −138.20, m | −138.34, m | −137.63, m |
| (p-BrC6F4) F 2,6 | −148.20, m | −140.64, m | −142.25, m |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojha, R.; Junk, P.C.; Bond, A.M.; Deacon, G.B. Oxidation of the Platinum(II) Anticancer Agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] to Platinum(IV) Complexes by Hydrogen Peroxide. Molecules 2023, 28, 6402. https://doi.org/10.3390/molecules28176402
Ojha R, Junk PC, Bond AM, Deacon GB. Oxidation of the Platinum(II) Anticancer Agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] to Platinum(IV) Complexes by Hydrogen Peroxide. Molecules. 2023; 28(17):6402. https://doi.org/10.3390/molecules28176402
Chicago/Turabian StyleOjha, Ruchika, Peter C. Junk, Alan M. Bond, and Glen B. Deacon. 2023. "Oxidation of the Platinum(II) Anticancer Agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] to Platinum(IV) Complexes by Hydrogen Peroxide" Molecules 28, no. 17: 6402. https://doi.org/10.3390/molecules28176402
APA StyleOjha, R., Junk, P. C., Bond, A. M., & Deacon, G. B. (2023). Oxidation of the Platinum(II) Anticancer Agent [Pt{(p-BrC6F4)NCH2CH2NEt2}Cl(py)] to Platinum(IV) Complexes by Hydrogen Peroxide. Molecules, 28(17), 6402. https://doi.org/10.3390/molecules28176402