
A UPLC-MS/MS Based Rapid, Sensitive, and Non-Enzymatic Methodology for Quantitation of Dietary Isoflavones in Biological Fluids
Abstract
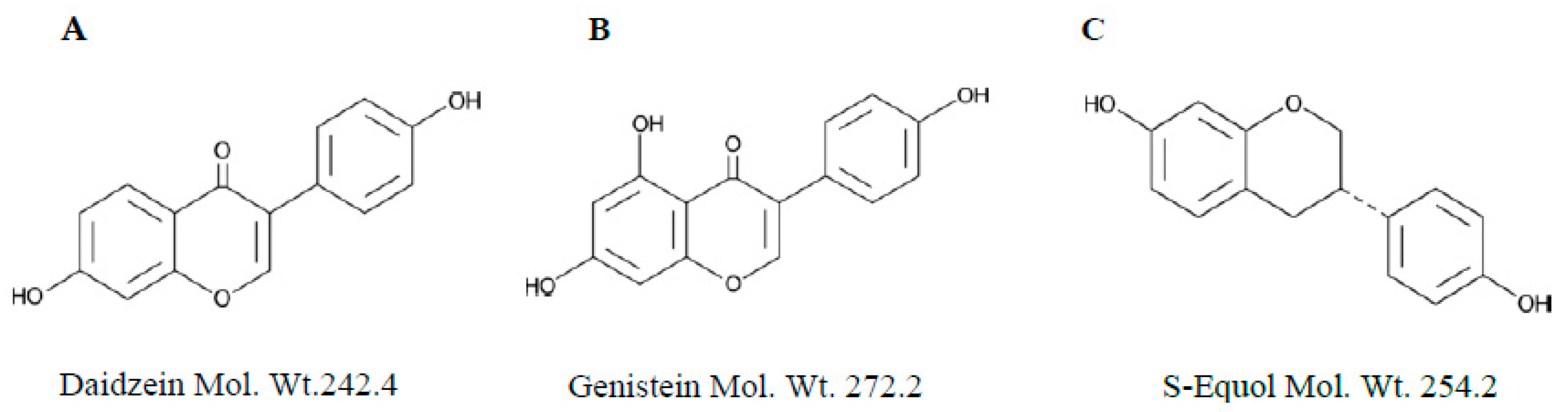
:1. Introduction
2. Results and Discussion
2.1. Optimization of Mass Spectrometry and Chromatography Parameters
2.2. Optimization of Chromatography Parameters for Rapid and Sensitive Detection
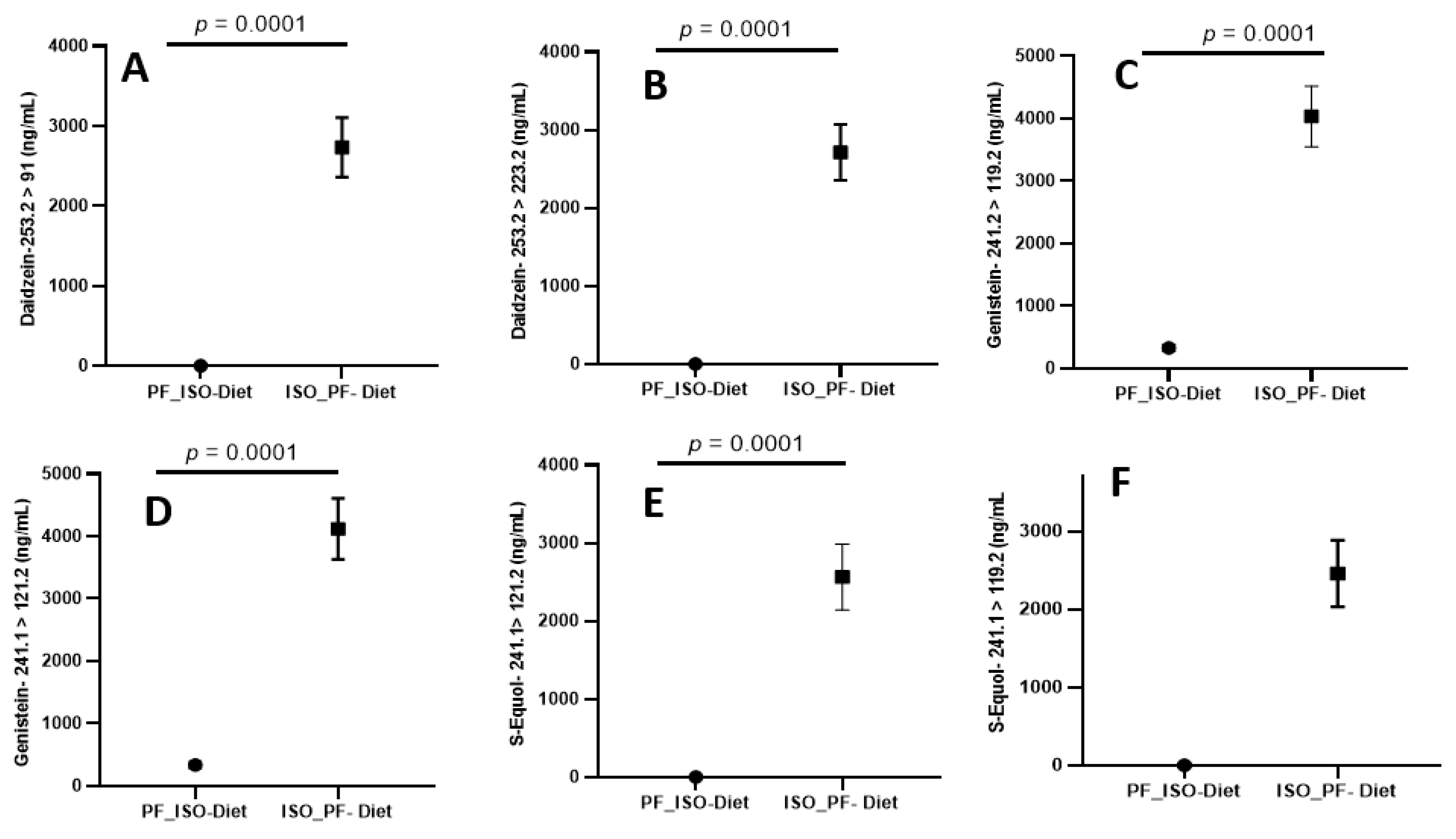
2.3. Extraction of Isoflavones from Urine
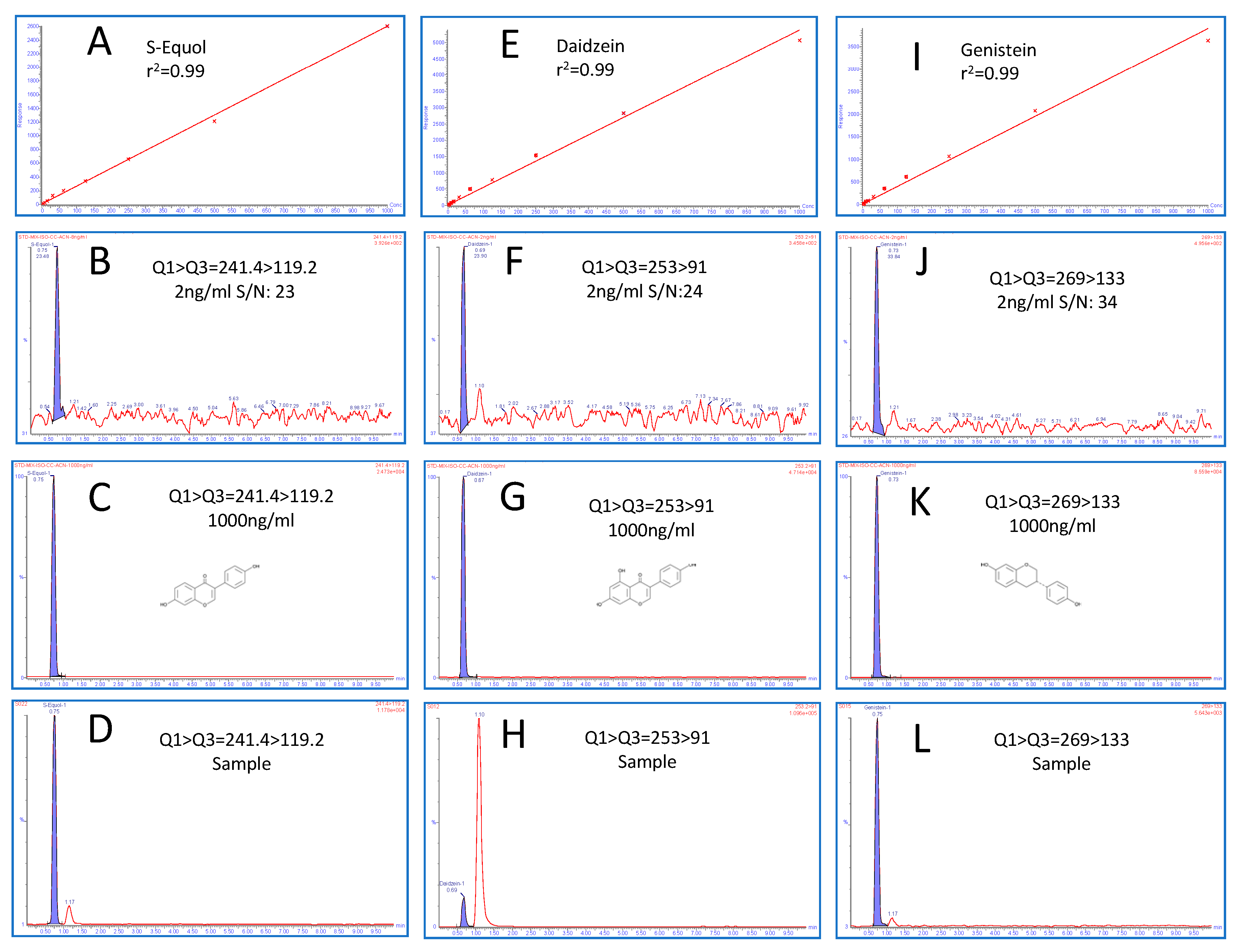
2.4. Method Validation
2.5. Linear Dynamic Range and Sensitivity
2.6. Precision Accuracy and Recovery
2.7. Extraction Recovery, Matrix Effect, and Carryover
2.8. Data Analysis
2.9. Metabolites Stability
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Stock Preparation
3.3. Sample Preparation and Isoflavones Extraction from Urine
3.4. LC-MS/MS Instrumentation and Conditions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kao, T.H.; Huang, R.F.; Chen, B.H. Antiproliferation of hepatoma cell and progression of the cell cycle as affected by isoflavone extracts from soybean cake. Int. J. Mol. Sci. 2007, 8, 1095–1110. [Google Scholar] [CrossRef]
- Lee, C.H.; Yang, L.; Xu, J.Z.; Yeung, S.Y.V.; Huang, Y.; Chen, Z.Y. Relative antioxidant activity of soybean isoflavones and their glycosides. Food Chem. 2005, 90, 735–741. [Google Scholar] [CrossRef]
- Ghimire, S.; Cady, N.M.; Lehman, P.; Peterson, S.R.; Shahi, S.K.; Rashid, F.; Giri, S.; Mangalam, A.K. Dietary Isoflavones Alter Gut Microbiota and Lipopolysaccharide Biosynthesis to Reduce Inflammation. Gut Microbes 2022, 14, 2127446. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M. Isoflavone and bone metabolism: It is cellular mechanism and preventive role in bone loss. J. Health Sci. 2002, 48, 209–222. [Google Scholar] [CrossRef]
- Migliaccio, S.; Anderson, J.J. Isoflavones and skeletal health: Are these molecules ready for clinical application. Osteoporos. Int. 2003, 14, 361–368. [Google Scholar]
- Sathyapalan, T.; Aye, M.; Rigby, A.S.; Thatcher, N.J.; Dargham, S.R.; Kilpatrick, E.S.; Atkin, S.L. Soy isoflavones improve cardiovascular disease risk markers in women during the early menopause. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 691–697. [Google Scholar] [CrossRef]
- Kim, I.S. Current Perspectives on the Beneficial Effects of Soybean Isoflavones and Their Metabolites for Humans. Antioxidants 2021, 10, 1064. [Google Scholar] [CrossRef]
- Mayo, B.; Vázquez, L.; Flórez, A.B. S-Equol: A Bacterial Metabolite from The Daidzein Isoflavone and Its Presumed Beneficial Health Effects. Nutrients 2019, 11, 2231. [Google Scholar] [CrossRef]
- Pabich, M.; Materska, M. Biological effect of soy isoflavones in the prevention of civilization diseases. Nutrients 2019, 11, 1660. [Google Scholar] [CrossRef]
- Szeja, W.; Grynkiewicz, G.; Rusin, A. Isoflavones, their glycosides and glycoconjugates. Synthesis and biological activity. Curr. Org. Chem. 2017, 21, 218–235. [Google Scholar] [CrossRef]
- Ningtyas, D.W.; Hati, S.; Prakash, S. Bioconversion and bio accessibility of isoflavones from sogurt during in vitro digestion. Food Chem. 2021, 343, 128553. [Google Scholar] [CrossRef] [PubMed]
- Vitale, D.C.; Piazza, C.; Melilli, B.; Drago, F.; Salomone, S. Isoflavones: Estrogenic activity, biological effect and bioavailability. Eur. J. Drug Metab. Pharmacokinet. 2013, 38, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C. Flavonoids and isoflavones: Absorption, metabolism, and bioactivity. Free Radic. Biol. Med. 2004, 36, 827–828. [Google Scholar] [CrossRef] [PubMed]
- Yoshikata, R.; Myint, K.Z.Y.; Ohta, H. Effects of S-Equol supplement on bone and cardiovascular parameters in middle-aged Japanese women: A prospective observational study. J. Altern. Complement. Med. 2018, 24, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, Z.; Huang, W.; Omedi, J.O.; Wang, F.; Zou, Q.; Zheng, J. Isoflavone aglycones enrichment in soybean sourdough bread fermented by lactic acid bacteria strains isolated from traditional Qu starters: Effects on in vitro gastrointestinal digestion, nutritional, and baking properties. Cereal Chem. 2019, 96, 129–141. [Google Scholar] [CrossRef]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to S-Equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Brown, J.E.; Hawdon, A.; Faughnan, M.A.; King, L.J.; Millward, J. Factors affecting the bioavailability of soy isoflavones in humans after ingestion of physiologically relevant levels from different soy foods. J. Nutr. 2006, 136, 45–51. [Google Scholar] [CrossRef]
- Available online: https://www.cdc.gov/biomonitoring/Phytoestrogens_BiomonitoringSummary.html (accessed on 23 June 2023).
- Saha, S.; Kroon, P.A. A Simple and Rapid LC-MS/MS Method for Quantification of Total Daidzein, Genistein, and S-Equol in Human Urine. J. Anal. Methods Chem. 2020, 2020, 2359397. [Google Scholar] [CrossRef]
- Bustamante-Rangel, M.; Delgado-Zamarreño, M.M.; Pérez-Martín, L.; Rodríguez-Gonzalo, E.; Domínguez-Álvarez, J. Analysis of Isoflavones in Foods. Compr. Rev. Food Sci. Food Saf. 2018, 17, 391–411. [Google Scholar] [CrossRef]
- Hsu, B.-Y.; Inbaraj, B.S.; Chen, B.-H. Analysis of soy isoflavones in foods and biological fluids: An overview. J. Food Drug Anal. 2010, 18, 8. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Maskarinec, G.; Oshiro, C.; Morimoto, Y.; Hebshi, S.; Novotny, R.; Franke, A.A. Urinary isoflavone excretion as a compliance measure in a soy intervention among young girls: A pilot study. Eur. J. Clin. Nutr. 2005, 59, 369–375. [Google Scholar] [CrossRef]
- Rashid, F.; Baghla, R.; Kale, P.; Shah, M.; Malakar, D.; Pillai, M. Absolute Quantification of Follicle Stimulating Hormone (FSH) Using Its Signature Peptides and Enzymatic Digestion in Human Serum by UPLC/LC–MS/MS. Chromatographia 2021, 84, 793–802. [Google Scholar] [CrossRef]
- Wilson, L.; Arabshahi, A.; Simons, B.; Prasain, J.K.; Barnes, S. Improved high sensitivity analysis of polyphenols and their metabolites by nano-liquid chromatography-mass spectrometry. Arch. Biochem. Biophys. 2014, 559, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhao, W.; Wang, C.; Wang, Z.; Wang, Z.; Zhang, J. A Comprehensive Screening and Identification of Genistin Metabolites in Rats Based on Multiple Metabolite Templates Combined with UHPLC-HRMS Analysis. Molecules 2018, 23, 1862. [Google Scholar] [CrossRef] [PubMed]
- Soukup, S.T.; Al-Maharik, N.; Botting, N.; Kulling, S.E. Quantification of soy isoflavones and their conjugative metabolites in plasma and urine: An automated and validated UHPLC-MS/MS method for use in large-scale studies. Anal. Bioanal. Chem. 2014, 406, 6007–6020. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.B.; Mistry, N.S.; Carter, M.H.; Leathem, A.J.; Teale, P. High throughput quantification of phytoestrogens in human urine and serum using liquid chromatography/tandem mass spectrometry (LC–MS/MS). J. Chromatogr. B 2007, 853, 138–146. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Metabolites | Calibration Range (ng/mL) | r2 | Limit of Detection (ng/mL) | Limit of Quantitation (ng/mL) | Signal to Noise (s/n) | Retention Time (min) |
|---|---|---|---|---|---|---|
| Daidzein-P | 2.0–1000 | 0.9973 | 0.5 | 2.0 | 23.90 | 0.69 ± 0.04 |
| Daidzein-C | 2.0–1000 | 0.9964 | 0.5 | 2.0 | 24.45 | 0.69 ± 0.04 |
| Genistein-P | 4.0–1000 | 0.9973 | 1.0 | 4.0 | 33.84 | 0.73 ± 0.03 |
| Genistein-C | 4.0–1000 | 0.9899 | 1.0 | 4.0 | 38.56 | 0.73 ± 0.03 |
| S-Equol-P | 2.0–1000 | 0.9970 | 0.25 | 2.0 | 23.34 | 0.75 ± 0.05 |
| S-Equol-C | 2.0–1000 | 0.9925 | 0.25 | 2.0 | 29.89 | 0.75 ± 0.05 |
| Metabolites | Selected MRM Precursor > Fragment | Collision Energy | Cove Voltage | Confirmative Ions | r2 |
|---|---|---|---|---|---|
| Daidzein-P | 253.2 > 91 | 10 | 32 | Primary | 0.997 |
| Daidzein-C | 253.2 > 223.2 | 12 | 32 | Confirmatory | 0.996 |
| Genistein-P | 269 > 133 | 10 | 45 | Primary | 0.997 |
| Genistein-C | 269 > 135 | 12 | 48 | Confirmatory | 0.989 |
| S-Equol-P | 241.1 > 121.2 | 12 | 45 | Primary | 0.997 |
| S-Equol-C | 241.1 > 119.2 | 10 | 42 | Confirmatory | 0.992 |
| Metabolites | Low-10 ng/mL Spike | Medium-25 ng/mL Spike | High-100 ng/mL Spike | Intraday Variation | Interday Variation | |||
|---|---|---|---|---|---|---|---|---|
| Recovery | RSD | Recovery | RSD | Recovery | RSD | RSD | RSD | |
| Daidzein (P) | 93 ± 2 | 5.2 | 98 ± 6 | 5.3 | 96 ± 7 | 2.1 | 1.3 | 4.8 |
| Genistein (P) | 96 ± 5 | 6.1 | 97 ± 4 | 6.3 | 99 ± 3 | 3.9 | 2.1 | 5.0 |
| S-Equol (P) | 92 ± 3 | 5.3 | 101 ± 5 | 4.2 | 91 ± 7 | 2.3 | 3.6 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, F.; Ghimire, S.; Mangalam, A.K.; Giri, S. A UPLC-MS/MS Based Rapid, Sensitive, and Non-Enzymatic Methodology for Quantitation of Dietary Isoflavones in Biological Fluids. Molecules 2023, 28, 6729. https://doi.org/10.3390/molecules28186729
Rashid F, Ghimire S, Mangalam AK, Giri S. A UPLC-MS/MS Based Rapid, Sensitive, and Non-Enzymatic Methodology for Quantitation of Dietary Isoflavones in Biological Fluids. Molecules. 2023; 28(18):6729. https://doi.org/10.3390/molecules28186729
Chicago/Turabian StyleRashid, Faraz, Sudeep Ghimire, Ashutosh K. Mangalam, and Shailendra Giri. 2023. "A UPLC-MS/MS Based Rapid, Sensitive, and Non-Enzymatic Methodology for Quantitation of Dietary Isoflavones in Biological Fluids" Molecules 28, no. 18: 6729. https://doi.org/10.3390/molecules28186729
APA StyleRashid, F., Ghimire, S., Mangalam, A. K., & Giri, S. (2023). A UPLC-MS/MS Based Rapid, Sensitive, and Non-Enzymatic Methodology for Quantitation of Dietary Isoflavones in Biological Fluids. Molecules, 28(18), 6729. https://doi.org/10.3390/molecules28186729