1. Introduction
Following the discovery that the gamma-emitting radionuclide gallium-67 (
67Ga) is taken up specifically in tumours (particularly lymphoma) [
1],
67Ga has become valuable in nuclear medicine as an imaging agent for diagnosing lymphoma, inflammation, and infection using scintigraphy or single-photon emission computed tomography (SPECT) [
2,
3]. It is also being evaluated as a therapeutic radionuclide by virtue of its Auger–Meitner electron emissions [
2,
3,
4]. The positron-emitting isotope
68Ga has also found widespread application in positron emission tomography (PET) [
2]. The discovery also sparked interest in the potential of nonradioactive gallium as the basis of drugs for treatment of cancer and other disorders. The development of gallium-based drugs, primarily as anticancer agents, began with simple salts including gallium(III) nitrate (marketed as Ganite
TM) [
5], gallium(III) chloride [
6], and gallium(III) citrate [
7]. There is little chemical basis to distinguish between these forms from a pharmacology perspective—all contain hydrated and hydrolysed Ga
3+ ions, and despite its formal description as gallium nitrate, the approved formulation of Ganite
TM contains citrate. Ganite
TM showed particular promise in clinical trials in patients with non-Hodgkin’s lymphoma [
8] and bladder cancer [
7,
9] and is approved for treatment of malignancy-related hypercalcemia [
9,
10,
11]. It is administered as an intravenous infusion. Gallium citrate (Panaecin
TM) [
12] is being evaluated in clinical trials as an inhaled formulation for the treatment of a variety of lung infections. Gallium chloride has been evaluated clinically and preclinically for treatment of various cancers [
13,
14,
15]. A range of mechanisms of anticancer and other actions have been suggested, but most mechanistic investigations focus on the downstream consequences of interference with iron transport and metabolism related to the chemical similarity in ligand-binding characteristics between Ga(III) and Fe(III) [
4,
7,
15,
16,
17,
18].
These promising examples of therapy with gallium salts involved primarily intravenous administration. Oral administration would make the drugs more acceptable and would be expected to reduce toxic side effects. However, bioavailability of gallium in the blood after oral administration of gallium nitrate [
11], citrate [
19], and chloride [
11,
13,
20] in animal models was very poor. The search for orally administered forms of gallium led to the evaluation of second-generation compounds: gallium tris(8-hydroxyquinolinate) (known as KP46 or AP002,
Figure 1) and gallium tris(maltolate), both designed as more lipophilic compounds in the expectation of improved absorption [
18]. In clinical trials, KP46 showed promise against renal cancer [
21]; furthermore, as AP-002, it is currently in phase I–II clinical trials (national clinical trial identifier (NCT) 04143789) for patients with breast, lung, and prostate cancer and bone metastases. However, in vivo preclinical data suggest that it, too, has poor bioavailability [
21,
22], possibly related to its very low water solubility [
23,
24]. Ga-maltolate has higher water solubility than KP46, and its oral administration leads to levels of gallium in serum, mainly bound to transferrin, comparable to those achieved during Ganite
TM infusion. Gallium maltolate is currently the subject of a clinical trial in glioblastoma (NCT04319276).
Despite numerous studies both in vitro in cancer cell lines and in vivo in rodent models, and despite the compilation of detailed and critical reviews [
25], the absorption, speciation, pharmacokinetics, and trafficking of gallium after administration of KP46 and gallium maltolate remain poorly understood. KP46 shows cytotoxic activity in some cell lines when added directly to cultured cells [
16,
18,
19,
25,
26,
27,
28,
29,
30], but since the speciation of gallium in vivo en route to the tumours is not fully elucidated, it is unclear whether direct treatment of cultured cells with KP46 is relevant to the in vivo and clinical context. Investigations of transchelation and binding of gallium to serum proteins, particularly transferrin and albumin, have given conflicting results: some works suggest transchelation of gallium from KP46 to transferrin and other data suggest hydrophobic association of the intact complex with apo-transferrin and albumin [
31,
32,
33].
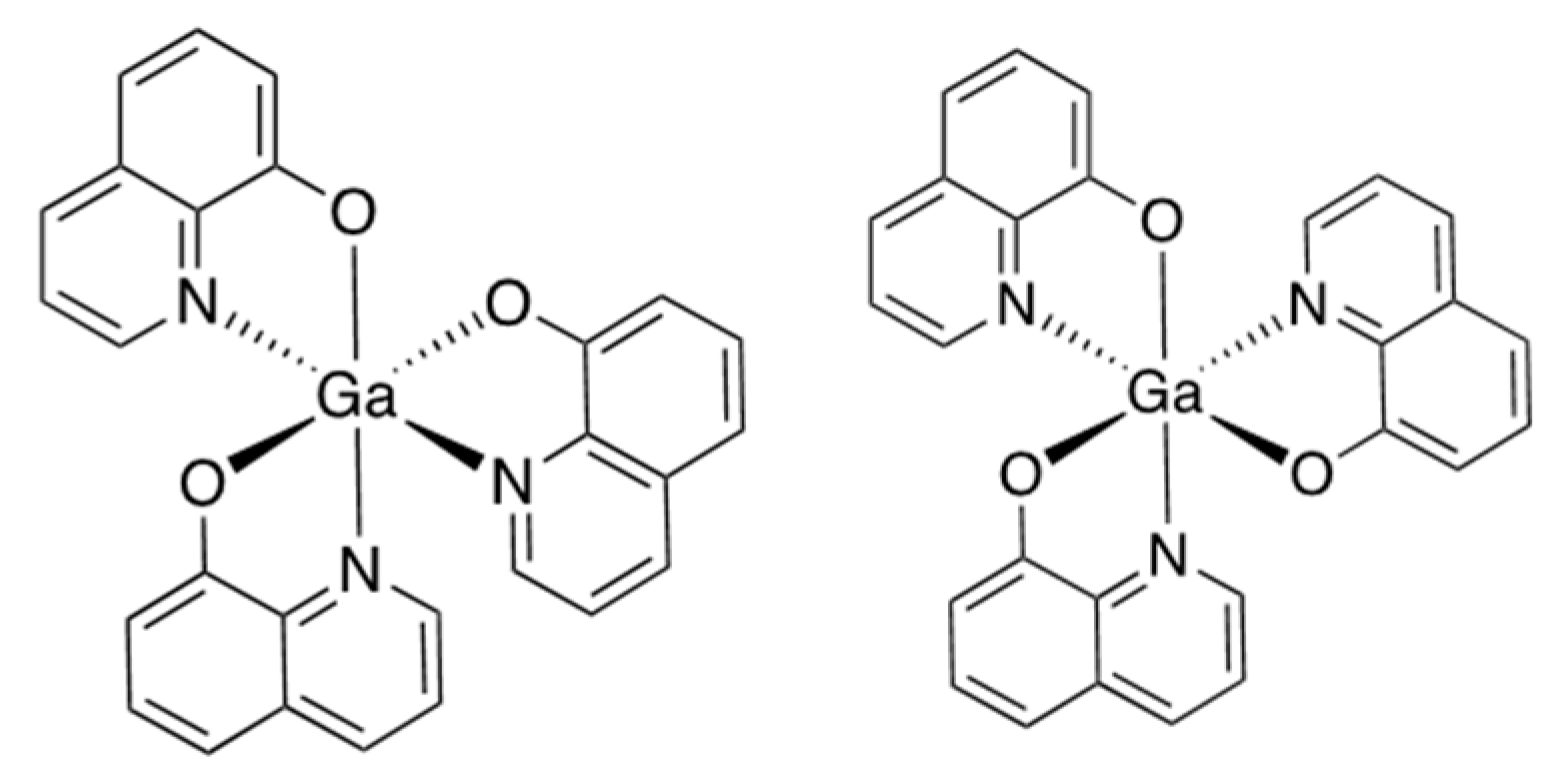
Figure 1.
Structure of tris(8-quinolinolato)gallium (III) (KP46) [
34].
Left:
mer-isomer;
right:
fac-isomer.
Figure 1.
Structure of tris(8-quinolinolato)gallium (III) (KP46) [
34].
Left:
mer-isomer;
right:
fac-isomer.
Thus, despite clinical trials being in progress and some being completed, understanding of the absorption, speciation, and pharmacokinetics of KP46 and gallium maltolate in vivo and of the trafficking of gallium to tumours after their administration remains poor and must be improved if enhanced design and delivery of gallium drugs is to be achieved. It was briefly pointed out [
25] that radionuclides such as
68Ga, mentioned above, could, in principle, be used to illuminate gallium trafficking in this context, but such studies have not been reported despite increased interest recently in the use of radionuclide imaging to study the biology of trace metals and metallodrugs [
35,
36,
37]. Here, we describe for the first time the use of radionuclides
67Ga and
68Ga to help understand the speciation and trafficking of gallium administered in the form of KP46 in vitro and in vivo in a tumour-bearing mouse model using PET and SPECT imaging. The tumour cell line used here is the A375 human melanoma cell line, chosen because KP46 has been shown to be active against human melanomas [
28] and because in our laboratory, A375 tumours in mice have shown significant avidity for i.v. injected
68Ga in the form of gallium chloride or acetate [
38,
39].
3. Discussion
A comparison of the chromatographic and radiochromatographic properties (iTLC and HPLC) of 68Ga- and 67Ga-labelled KP46 indicates that the labelled species can be expected to be a reliable tracer for the bulk KP46 drug. The in vivo experiments, where biodistributions of the radionuclides (using radioactivity measurements) and the nonradioactive gallium-69 were compared, indicated that the radionuclides were also reliable tracers for gallium administered as the KP46 drug in vivo. The additional detail of gallium distribution provided by radionuclide imaging, over and above the direct analysis of nonradioactive gallium in tissue samples, is therefore potentially highly informative. For example, using radionuclide imaging, the distribution of gallium can be studied at a range of different time points without adding to the number of animals, and additional insight is accessible into the biodistribution in tissues that would not typically be sampled ex vivo or that would be hard to sample ex vivo. For example, the SPECT images show that uptake in the bones is primarily in the joints. Thus, we conclude that 68Ga PET imaging and 67Ga SPECT imaging represent useful, and hitherto unexploited, tools with which to study the pharmacokinetics of gallium-based drugs. Subject to satisfactory dosimetric evaluation and regulatory approvals, this utility potentially extends to human studies, since these radionuclides are widely used in nuclear medicine, and human PET and SPECT scanners are widely available in hospitals.
It is acknowledged that the ability of a drug compound to be absorbed across the gastrointestinal barrier can usefully be predicted by measurement of its lipophilicity, e.g., by use of octanol extraction to determine the distribution/partition coefficient [
5,
23,
39]. KP46 was first introduced as a lipophilic form in which to deliver gallium orally, and measurement of the octanol–water partition coefficient of the bulk drug spectrophotometrically, confirming its lipophilicity (log
P(octanol/water) = 0.88), is described in the literature [
23,
25]. In the present work, the octanol–water partition coefficient was determined using radioactivity measurements at the tracer level, again confirming its lipophilicity but showing a much higher log
P value (2.33 ± 0.28). We suggest that the origin of the difference lies in the different methods and conditions: bulk KP46 contains Ga
3+ and 8-hydroxyquinolinate in a 1:3 ratio and is subject to dissociative equilibria in solution to form complexes with a higher Ga:8HQ ratio and which would consequently be positively charged and less lipophilic than the 1:3 complex. The apparent log
P value determined spectrophotometrically using the bulk drug is therefore likely to be an underestimate of the true value for the 1:3 complex itself. The
68Ga measurement, on the other hand, was performed on a solution containing tracer (<nanomolar) levels of
68Ga but a large excess—more than a millionfold—of 8-hydroxyquinoline (a necessary consequence of the radiochemical synthesis). This excess is likely to suppress dissociation, ensuring that virtually all of the
68Ga is in the form of the 1:3 complex and hence more efficiently extracted into octanol, giving a higher log
P that more closely reflects the lipophilicity of the 1:3 complex. This dissociative equilibrium is also suggested by other aspects of the physicochemical characterisation of KP46, including HPLC and mass spectrometry. It is likely to be an important consideration in understanding the absorption of the drug and its interaction with serum proteins. Reverse-phase gradient HPLC of KP46 with UV detection (
Supplementary Materials, Figure S5) shows an unusual line shape suggestive of on-column dissociative equilibria: an unusually broad peak from 14 to 16 min merging into a sharp peak at 16.2 min. Mass spectrometry of the late (16.2 min), sharp part of this peak suggests that it represents primarily the 1:3 complex, (M + H
+ 503.77 for the
71Ga isotope). By contrast, mass spectrometry of the earlier broad peak (14.1 min) shows no peak representing the 1:3 complex; instead, it shows predominantly uncomplexed 8HQ (M + H
+ 146). Other minor peaks observed in mass spectra were consistent with the presence of complexes with lower Ga:8HQ ratios (e.g., 1:2, 2:5, see
Supplementary Materials, Figure S3). Although MS alone cannot determine whether these species were present in solution rather than being formed during the electrospray ionisation process, its combination with the HPLC peak shape and the partition coefficient data strongly suggests that dissociative equilibria are important.
Radiogallium-labelled KP46 (
68Ga initially, for short incubations, and
67Ga when extension to longer time points was required) proved useful in understanding interactions of the drug with serum proteins. These become relevant if KP46 is administered intravenously or is absorbed intact from the GI tract. Apo-transferrin (apo-Tf) and human serum albumin (HSA) are likely candidate proteins for binding to KP46 or gallium released from it: apo-Tf because, as an iron(III)-binding protein, it is known to bind with high affinity to Ga
3+ in the presence of bicarbonate ions [
15,
33,
40,
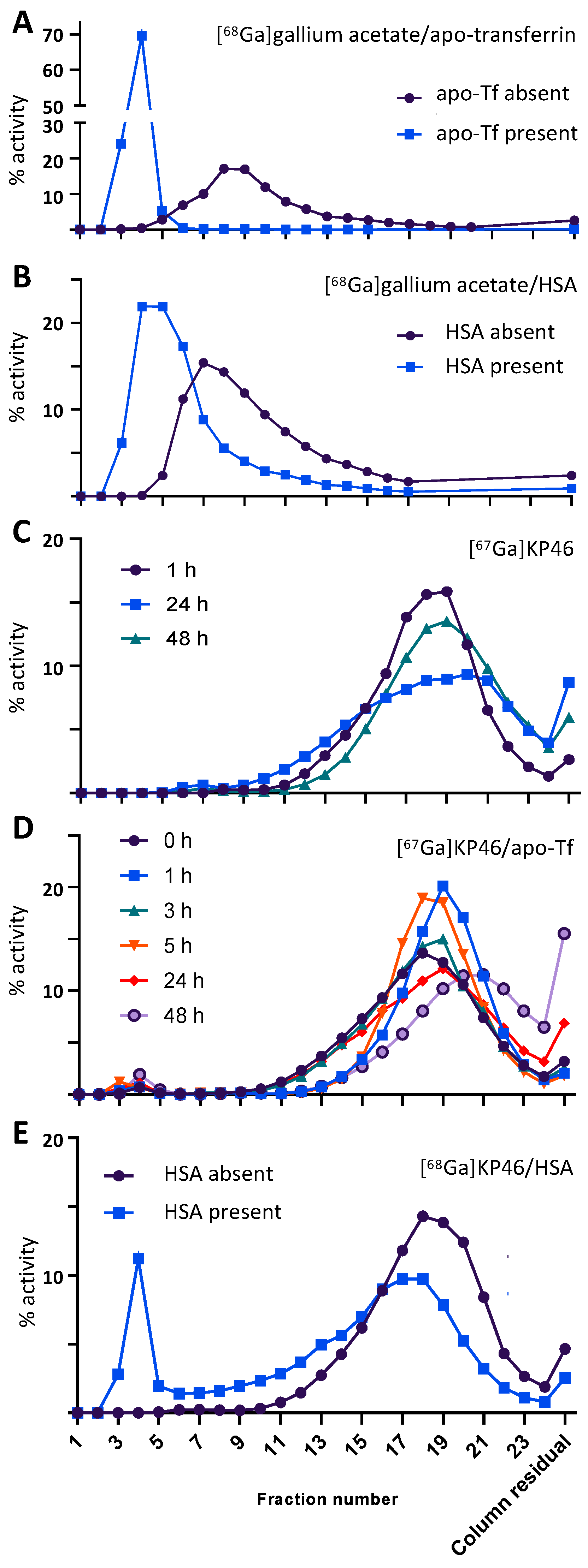
41], and HSA because of its high abundance and known possession of binding sites for metals and lipophilic molecules.
In experiments with apo-Tf, while ionic Ga
3+ (as its acetate salt) showed the expected almost complete binding to apo-Tf in the presence of bicarbonate, KP46 by contrast showed no evidence of gallium binding to apo-Tf. This shows that neither transchelation of gallium nor hydrophobic binding of the intact KP46 complex occurred under the conditions of the experiment. The elution time of the radioactivity matched that of KP46 controls (where protein was absent), suggesting that KP46 remains intact during the incubation. This result conflicts with previous studies of KP46 binding to apo-Tf in which extensive binding was observed using bulk nonradioactive KP46 [
32,
33,
42] either with or without dissociation of the KP46 complex [
31,
32]. Again, we suggest that the differing methods and conditions may account for the apparent conflict. In the “bulk drug” approaches reported in the literature, the KP46 was presented to the protein as a stoichiometric compound with a Ga:8HQ ratio of 1:3, whereas in the radiotracer experiments, gallium was present at a very low concentration with 8HQ in very large excess. In the small volume of serum used, this excess may be sufficient to overcome the greater affinity of gallium for apo-Tf than for 8HQ and shift the equilibrium away from transchelation of gallium to apo-Tf. This argument is consistent with the view that, if KP46 is absorbed intact from the digestive tract into blood, dissociative equilibria become important in determining its pharmacokinetics and biodistribution.
Upon incubation with HSA, both [
68/67Ga]KP46 and [
68/67Ga]gallium acetate showed only weak protein association, consistent with the literature [
33]. In the case of [
68/67Ga]KP46, the HSA-binding data alone provide no indication of whether the binding is due to transchelation or a hydrophobic interaction between HSA and intact KP46; however, in light of the apo-Tf binding data, under these conditions, where dissociation of KP46 is suppressed by the presence of excess 8HQ, transchelation is unlikely [
33,
42]. Indeed, studies in the literature using NMR and XANES suggest a hydrophobic binding mechanism for association of bulk KP46 with HSA in vitro [
32,
43,
44,
45].
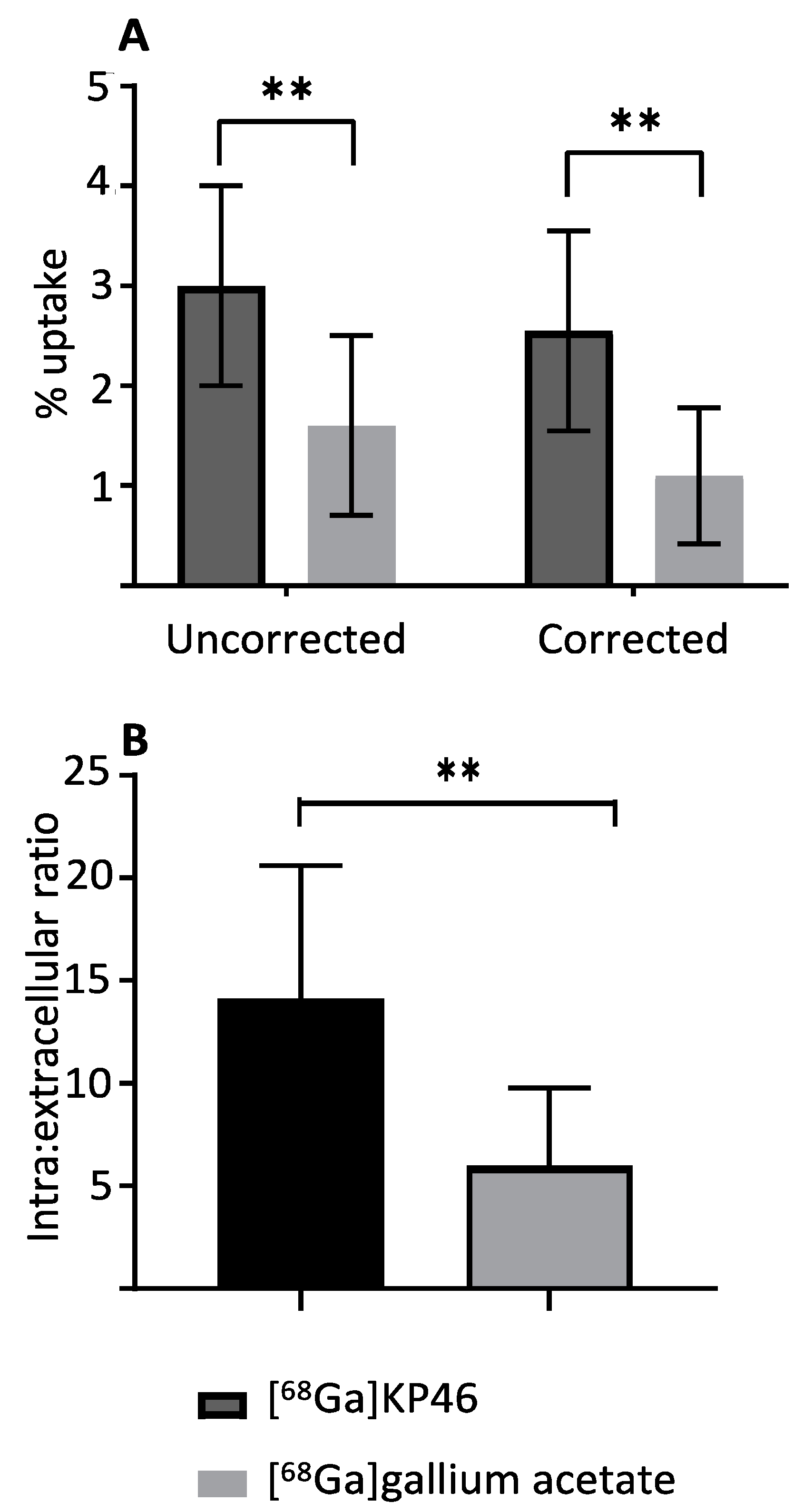
In vitro studies using the A375 cell line were conducted to compare the cellular uptake of [
68Ga]KP46 with that of ionic gallium. No-carrier-added [
68Ga]KP46 was used without addition of nonradioactive KP46. With the levels of activity used in these experiments,
68Ga concentration added to the media under these conditions was in the pM–nM range. Both [
68Ga]KP46 and unchelated
68Ga ([
68Ga]gallium acetate) were accumulated in cells, leading to higher radiogallium concentration within cells than in the medium. The fact that [
68Ga]KP46 showed a significantly higher uptake (
p < 0.01) compared to [
68Ga]Ga-acetate is attributable to the lipophilicity of [
68Ga]KP46, which allows cell entry by passive diffusion. This shows that transferrin-dependent or other specific transport mechanisms are not necessary for KP46 to deliver gallium into cells. The 8HQ complexes of
68Ga and
67Ga are known to be able to transport radiogallium nonspecifically into a variety of cells by virtue of their lipophilicity [
2,
4,
46]. Indeed, this has been used as a method of radiolabelling cells for tracking their migration in vivo [
46]. Unchelated gallium is also known to be taken up in cells via transferrin-mediated and possibly non-transferrin-mediated mechanisms [
4,
16,
17,
41,
44]. In the media used here, which contain foetal bovine serum, bovine transferrin is present; the sluggish uptake of gallium acetate in cells can be accounted for by noting that although gallium binds to bovine transferrin with similar efficiency as it does to human transferrin, human transferrin receptor has relatively poor affinity for bovine transferrin. The presence of bovine transferrin, therefore, could have an inhibitory effect on the uptake of gallium ions into human cells by diminishing the availability of gallium for transport into cells via non-transferrin-mediated routes [
47,
48,
49,
50]. This inhibition would not affect the KP46-mediated transport into cells. The experiments show that if KP46 is absorbed intact from the digestive tract into blood and survives intact in blood en route to the tumour, it could deliver gallium into tumour cells.
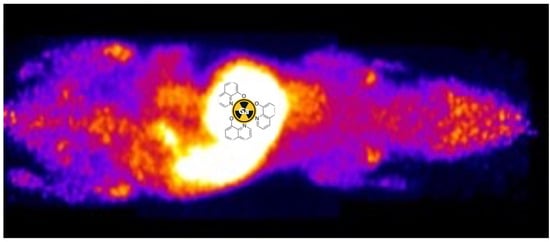
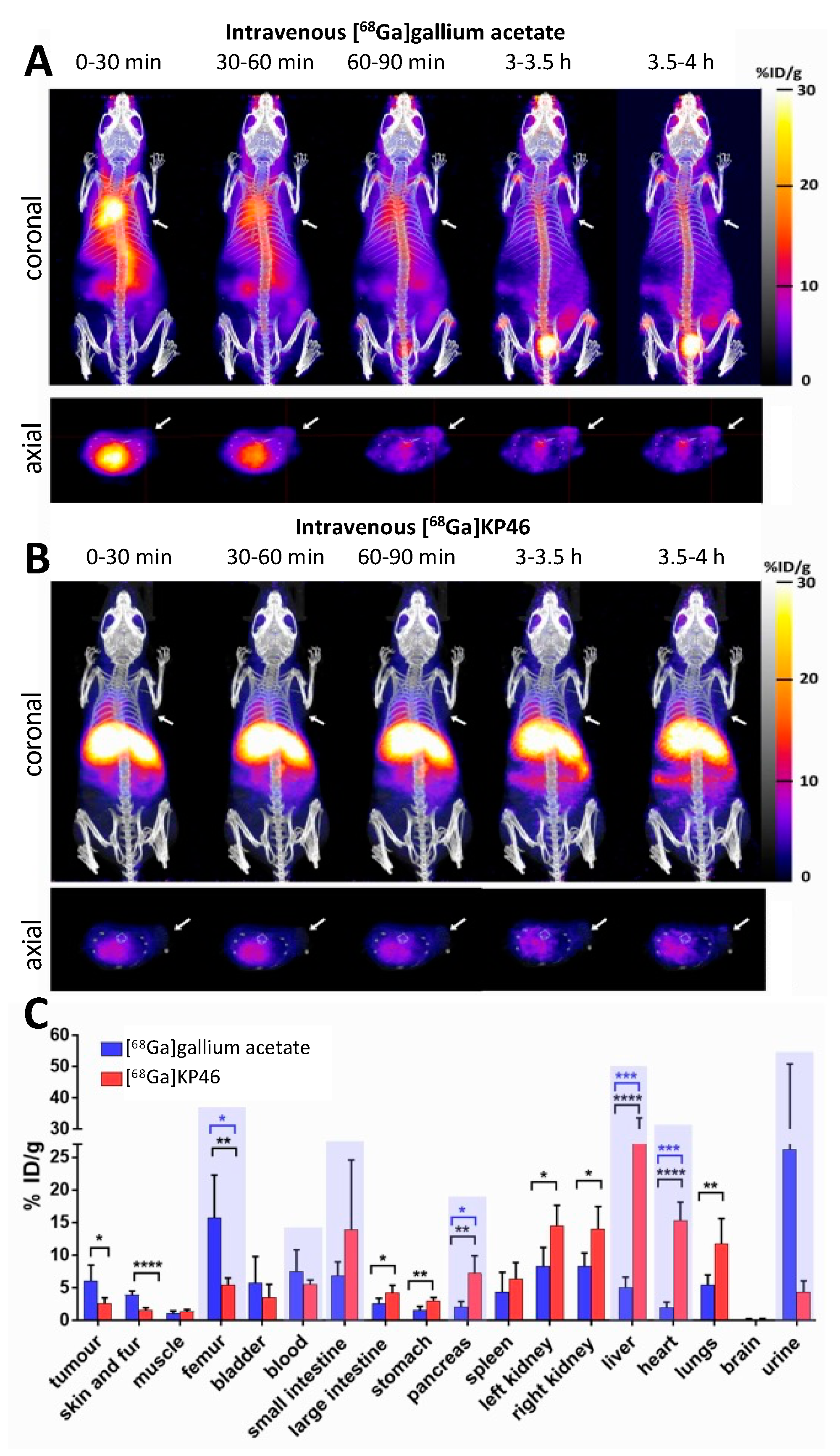
A comparison of [
68Ga]gallium acetate and [
68Ga]KP46 biodistribution after intravenous injection at the tracer level using PET imaging and ex vivo tissue counting showed clear differences between the two. In the case of [
68Ga]gallium acetate, steady clearance of
68Ga from the blood pool was observed, leading by 4 h to significant (visible above background in scans) uptake in the tumour and joints and urinary excretion. This is consistent with previous
68Ga and
67Ga imaging studies and with the accepted transferrin-dependent distribution pathways [
2,
4,
17,
38,
44]. In contrast, i.v. administration of [
68Ga]KP46 led to rapid blood clearance of
68Ga and uptake in the liver and myocardium within 30 min, with later translocation of radioactivity from the liver to intestines. This indicates that the [
68Ga]KP46 remains intact without dissociation in blood for the brief period required to prevent
68Ga from distributing via transferrin binding. This is consistent with the in vitro serum behaviour of [
68Ga]KP46 at tracer level, where the presence of excess 8HQ suppresses transchelation to transferrin. It must be noted that these conditions do not mimic those that result from the typical oral administration route of KP46 in clinical trials; instead, they indicate how KP46 might be expected to behave if absorbed intact from the digestive tract. Notably, the tumour is not among the organs receiving a significant fraction of
68Ga after intravenous administration of [
68Ga]KP46, despite the demonstrated ability of [
68Ga]KP46 to deliver
68Ga to tumour cells in vitro. We assume this difference between in vitro and in vivo behaviour is due to the rapid absorption of tracer by the liver in vivo, removing
68Ga from circulation too quickly to allow significant delivery to the tumour.
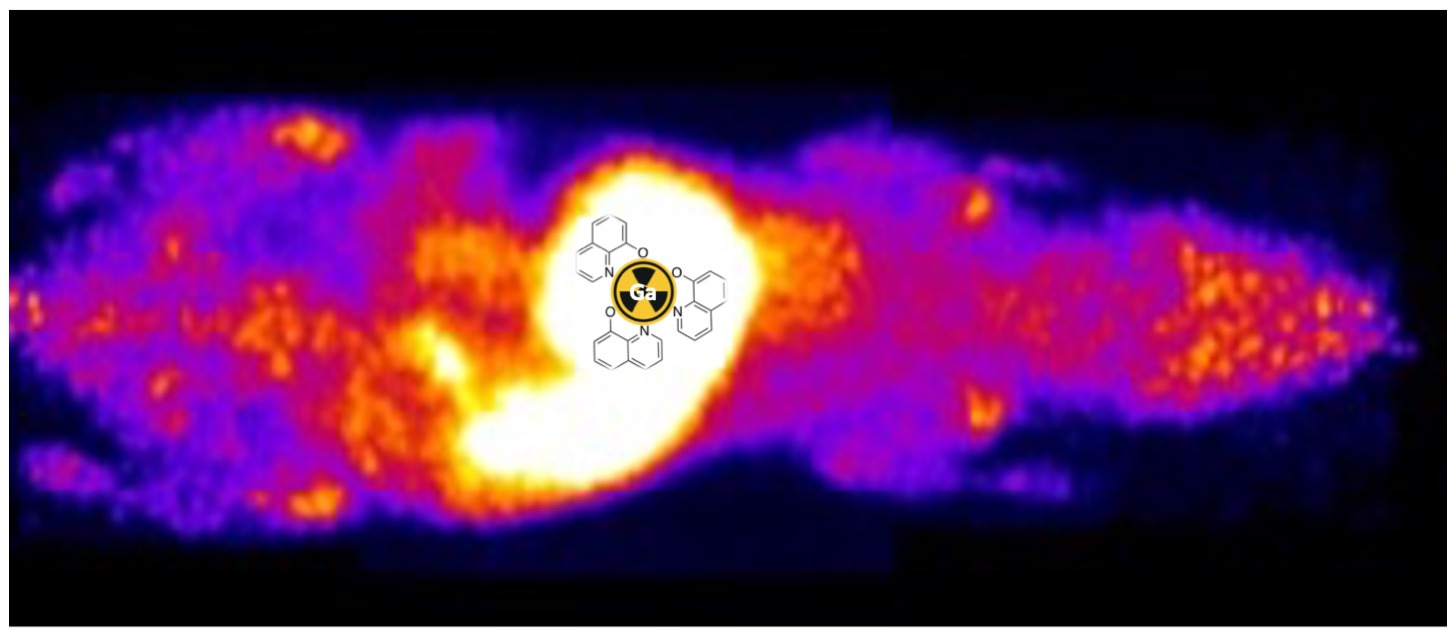
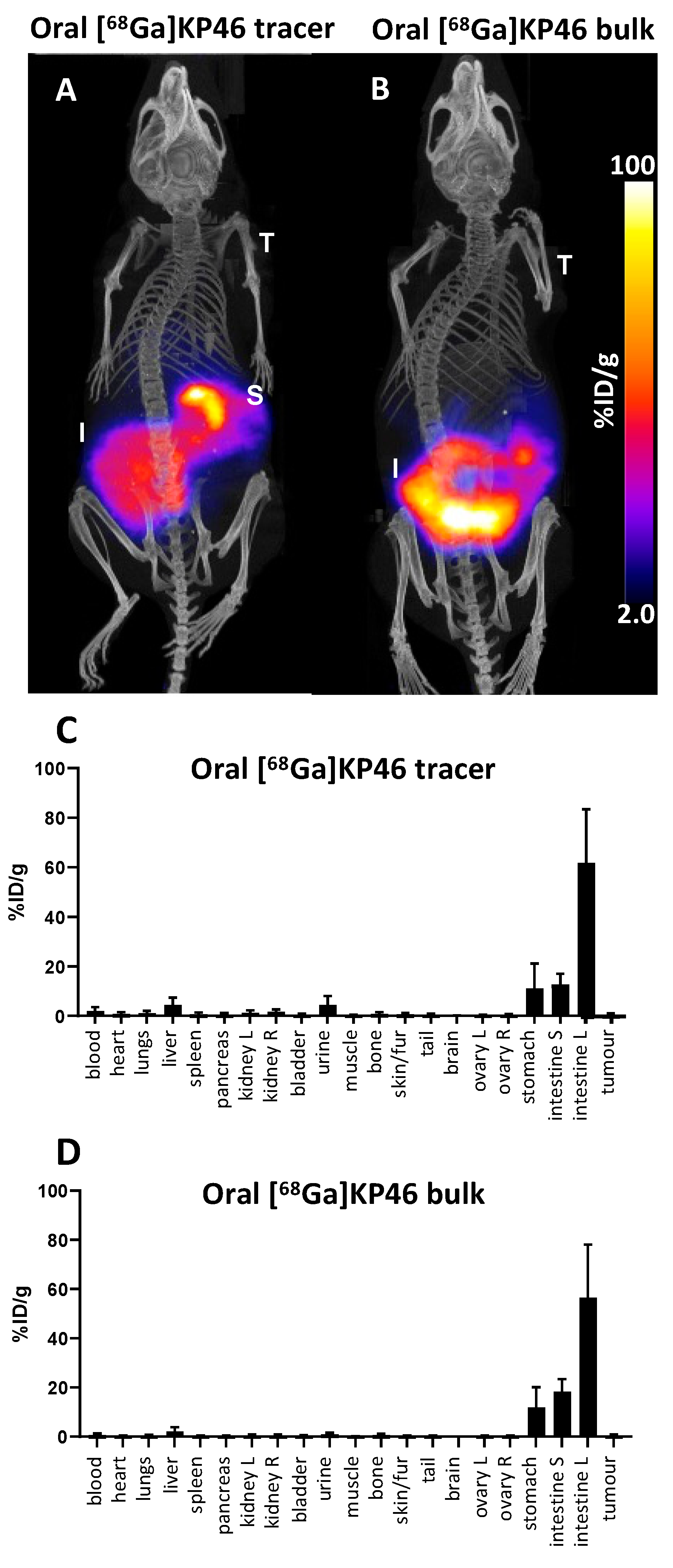
PET imaging 4 h after oral administration of [68Ga]KP46, both at tracer level and as part of a bulk dose, showed that radioactivity was largely confined to the digestive tract, with very little absorption; the little activity outside the gut was present in the liver and urine, with no significant tumour uptake. This is indicative of very poor absorption of 68Ga by 4 h.
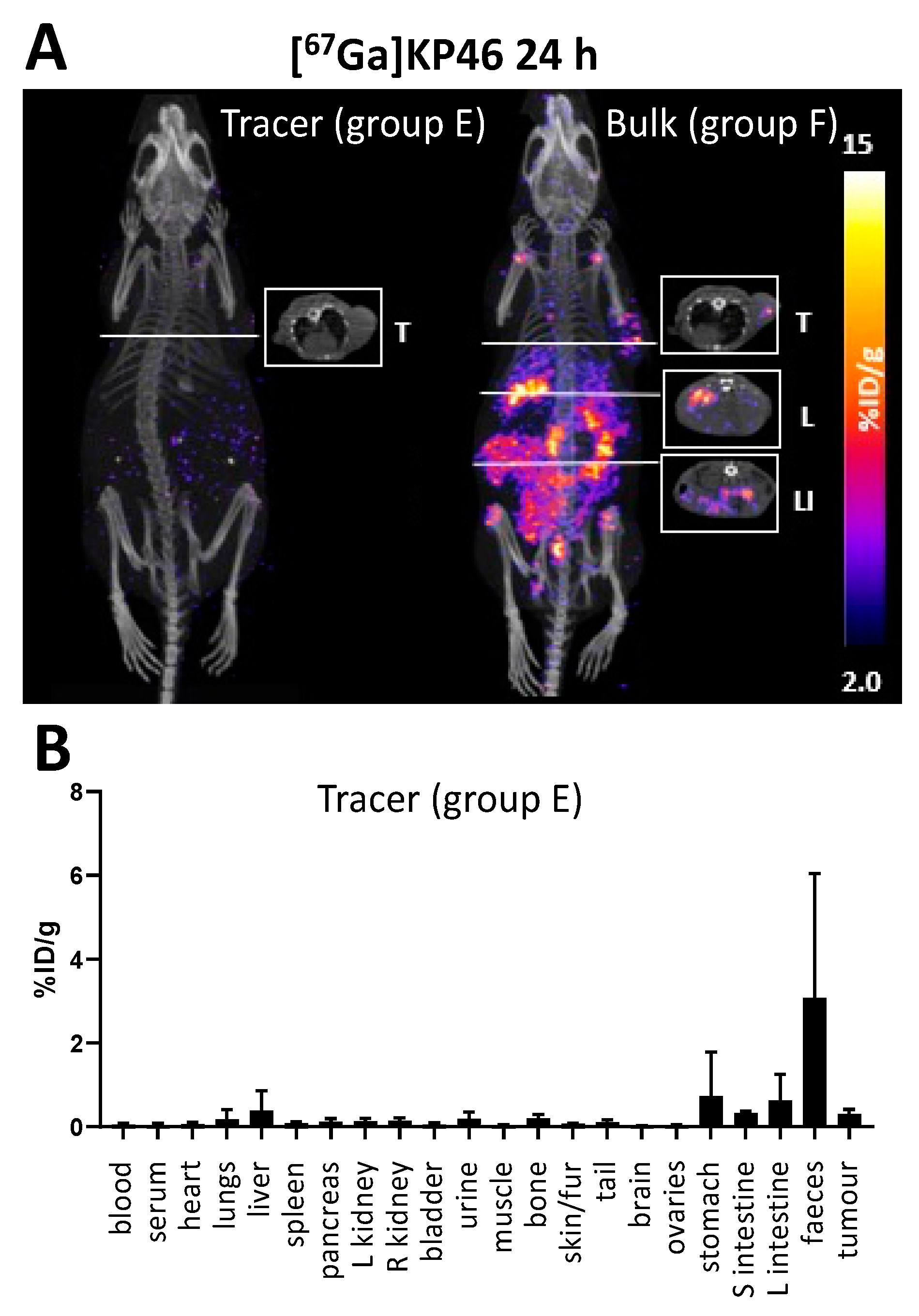
To determine whether more significant absorption occurs over a longer time period, similar studies were conducted with gallium-67. After oral administration of [
67Ga]KP46 at a tracer level or combined with bulk KP46, SPECT imaging results after 4 h were consistent with those obtained with
68Ga: most of the activity remained in the gut, with minimal absorption and trafficking to tissues (
Supplementary Materials, Figure S10). However, at the 24 h time point, the
67Ga distribution in the “tracer” and “bulk” groups diverged. [
67Ga]KP46 administered at the tracer level showed little visible uptake in any organ (including tumour) in SPECT/CT images and had been largely excreted via faeces. Ex vivo tissue counting revealed small but measurable activity in tissues outside the digestive tract; the highest activity other than the digestive tract and faeces was found in the liver and tumour, followed by the skeleton. In contrast, the presence of a therapeutic bulk dose of KP46 delayed faecal excretion, and at 24 h, while much of the radioactivity still remained in the gut, SPECT scans showed translocation of a small but measurable fraction of
67Ga to other organs, including tumour. At this point, the biodistribution of
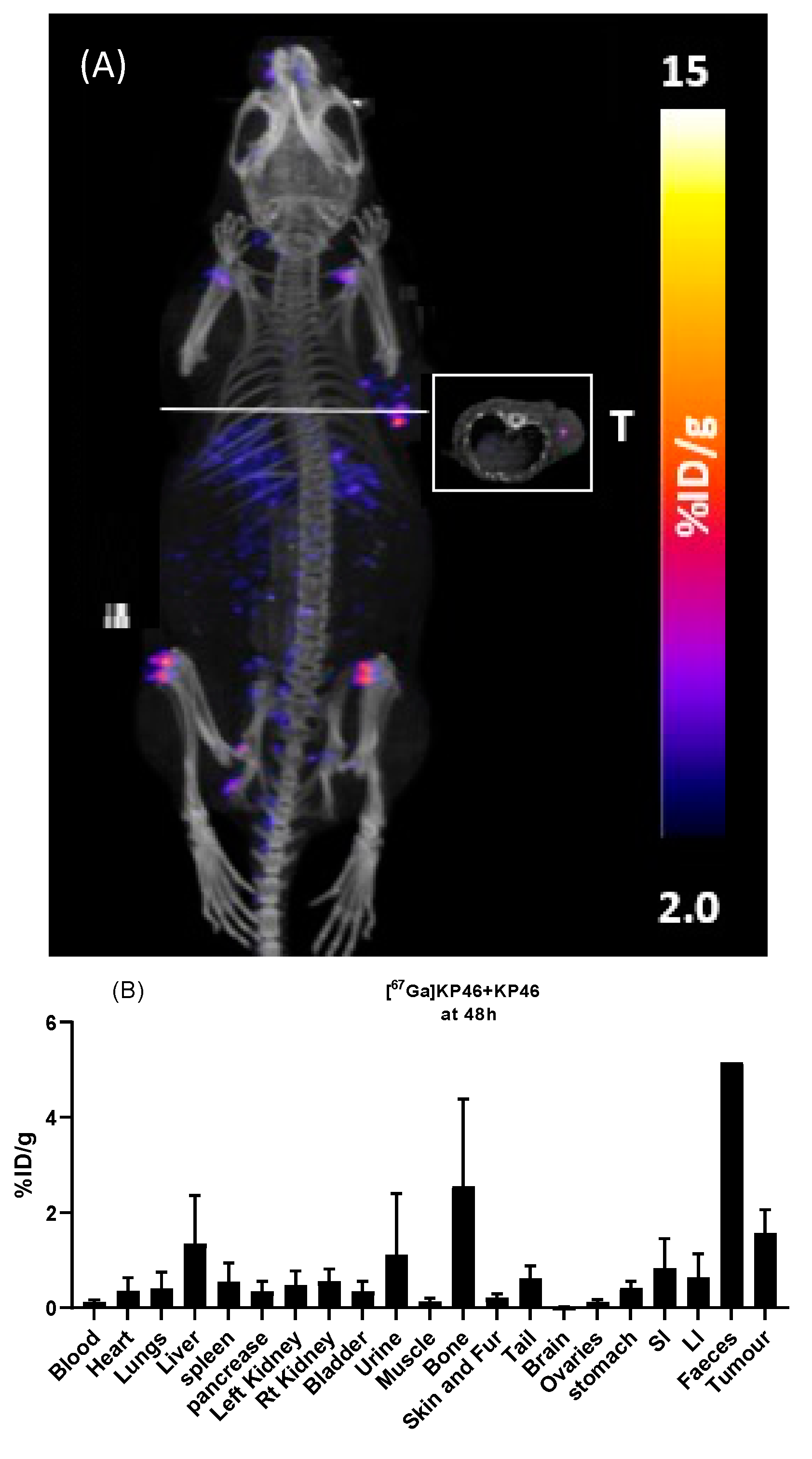
67Ga in tissues outside the digestive tract, particularly the activity in the joints, resembled that of i.v.-administered gallium acetate rather than that of i.v.-administered KP46. By 48 h, this distribution was further accentuated: most gut radioactivity had been eliminated by faecal excretion and by further absorption, leading to higher radioactivity concentration in tissues, including tumour, compared to 24 h. The highest activity was found in bone (mainly in joints as identified in SPECT scans), followed by tumour and then liver. Again, this is reminiscent of the biodistribution shown by gallium acetate rather than KP46, although the liver-to-tumour and liver-to-bone ratios were slightly higher than when
68Ga acetate was administered by i.v. injection. This difference should be viewed in light of the different delivery routes: i.v.-administered tracer reaches tissues/tumour after first passing through the pulmonary circulation, whereas orally administered tracer first passes through the portal circulation to the liver before redistribution to the pulmonary circulation and then other tissues. Hence, the liver has the greatest opportunity to capture the orally administered tracer. Attempts to determine the speciation of
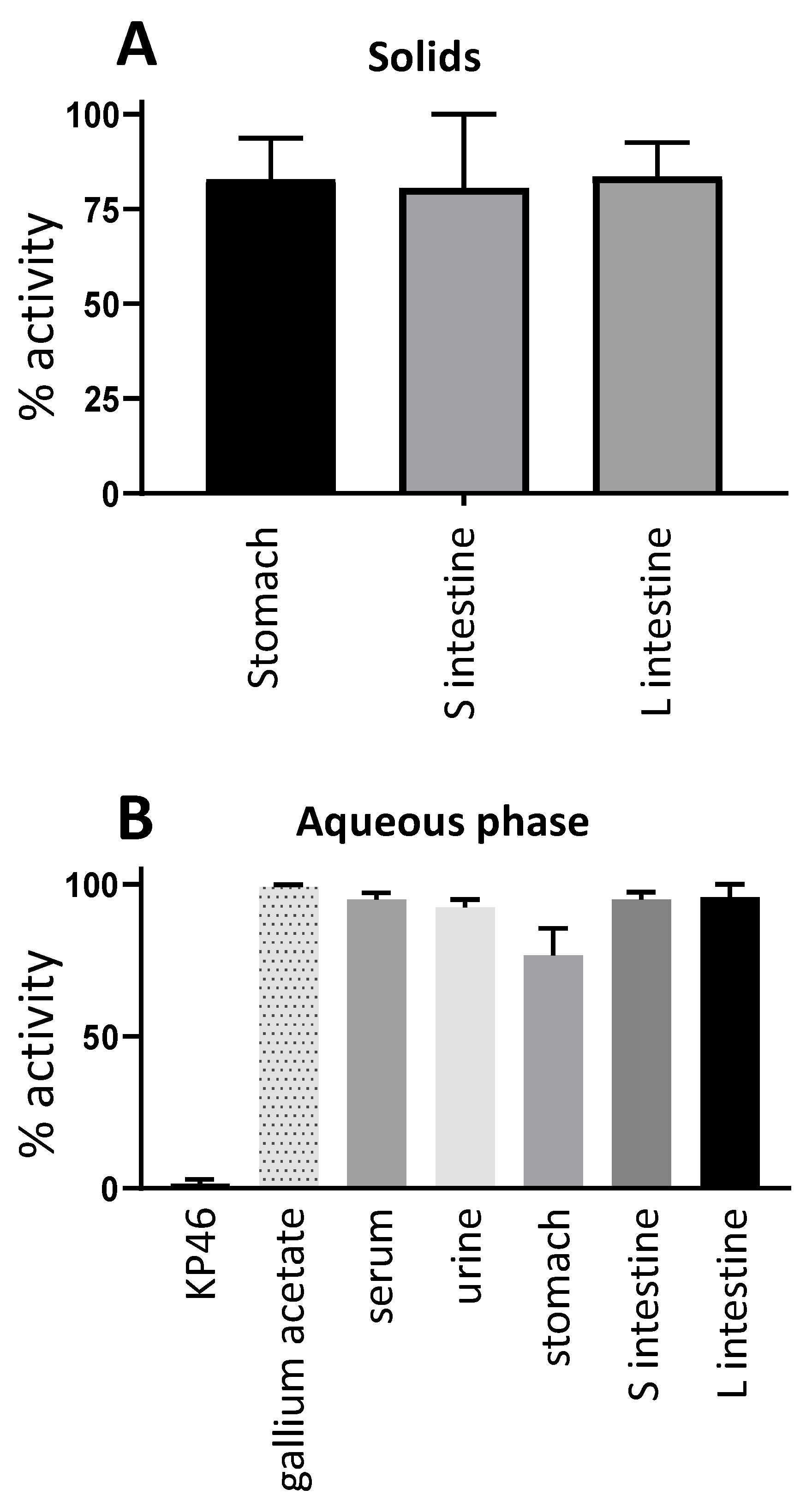
68Ga at 4 h post-oral administration by octanol extraction showed no evidence of a significant lipophilic form of
68/67Ga in any nondigestive tissue including tumour; this is consistent with the conclusion that once absorbed from the gut, the gallium is handled similarly to gallium acetate rather than intact KP46. A similar analysis of the gut contents was attempted, but the majority of radioactivity was insoluble in both the octanol and aqueous layers, and amongst the soluble fraction, less than 20% was octanol-extractable, indicating that extensive hydrolysis and dissociation of KP46 had occurred in the stomach and intestines even by 4 h. Thus, it remains unclear whether the radioactive species absorbed from the gut is KP46 or ionic gallium. However, the octanol extraction of nondigestive tract tissues and the biodistribution of absorbed radiogallium strongly suggest that, at least from the point of passing through the liver, gallium is transported as if administered intravenously in the form of gallium acetate and is, hence, delivered to tissues via transferrin-dependent pathways. The inefficient absorption from the gut ensures that the gallium concentration in the circulation remains low and is unlikely, therefore, to overload apo-transferrin capacity for gallium binding.
The radiotracer and imaging data presented here complement previous studies using non-radioactive-gallium-based drugs and provide additional insight. In this study, we did not investigate the absorption of orally administered radiogallium salts. Our conjecture is that KP46 (which itself is poorly absorbed at best) dissociates in the digestive tract, releasing ionic gallium, which is then also poorly absorbed and largely excreted faecally. This is consistent with previous studies of absorption and excretion of orally administered gallium salts in mice. In a study of orally administered [
67Ga]gallium citrate, between 87 and 100% of
67Ga was excreted in the faeces by 72 h [
19,
51]. Similarly, in mice receiving simultaneous single oral administration of bulk gallium chloride (100 nmol) and 0.037 MBq of
67Ga citrate, only 0.3 ± 0.08% of the original measured activity remained in the mice after five days [
52]. The present study was primarily concerned with the trafficking of KP46, and the results are consistent with a previous study [
22] in healthy mice given prolonged (2 weeks) daily oral doses of KP46. This study reported poor absorption, with the highest accumulation of gallium in bone (7.02 ± 3.14 µg/g, 0.16 ± 0.07%ID/g), followed by liver (3.55 ± 2.10 µg/g, 0.08 ± 0.04%ID/g) (there were no tumours in the study). This biodistribution, like that which we observed with [
67Ga]KP46, indicates that once absorbed and in circulation, the biodistribution pathway matches the known behaviour of unchelated gallium rather than intact KP46. In particular, the biodistribution is not dominated by the high liver or myocardial uptake characteristic of intravenously injected KP46. None of the data presented here suggest that KP46 offers a significant advantage over water-soluble unchelated gallium in terms of absorption and bioavailability. Data are consistent with a model in which KP46 serves as a slow-release form of gallium, releasing ionic gallium in the gut, which is then absorbed rather inefficiently (most is excreted) and which then behaves like free gallium in the blood, with much of it quickly bound to the iron-binding sites of transferrin. However, the possibility cannot be excluded that KP46 is absorbed into the blood and quickly delivered to the liver via the portal circulation, where it is metabolised and rereleased into circulation as free gallium. There is no evidence that orally administered KP46 is delivered intact to tumours in vivo.
The very low delivery of gallium to tumours, shown by the present and previous studies with orally administered KP46, can be interpreted as an indication of the potency of gallium when it does reach the tumour in very small quantities in clinical trials that show effectiveness. Alternatively, its effect on tumours may not require delivery of Ga to tumours; for example, interference with delivery of essential iron to tumours may be implicated. However, all evidence shown here points to KP46 acting as a prodrug, releasing free gallium which becomes the absorbed and active species. The corollary to this mechanism, however, is that KP46 must also release 8-hydroxyquinoline in this process. The absorption, trafficking, and cytotoxicity of 8HQ do not appear to have been considered as a potential contributor to the anticancer properties of KP46. 8HQ has shown cytotoxic effect against multiple cancer cell lines [
53,
54], and hence, the idea that KP46 could act as a prodrug not only for gallium ions but also for 8HQ should be considered [
53,
55].
5. Materials and Methods
5.1. General
Reagents and consumables were purchased from Fisher Scientific/Chemical, Crawford Scientific, or Sigma Aldrich unless specified otherwise. Athymic nude nu/nu mice and NSG mice were purchased from Charles River UK Ltd. (Margate, UK). A375 human melanoma cells were purchased from the American Type Culture Collection (ATCC) and were cultured and stocked in our laboratory. 68Ga was obtained from a 68Ge/68Ga generator (Eckert & Ziegler, Braunschweig, Germany) by eluting with 5 mL of 0.1 M ultrapure hydrochloric acid and collected in 5 × 1 mL fractions. [67Ga]Ga-citrate was purchased from Mallinckrodt, The Netherlands. iTLC was carried out using Agilent’s silica-gel-impregnated glass microfibre 10 cm strips. Developed iTLC strips were scanned with a LabLogic miniscan TLC reader with positron (β+) or gamma (γ) detectors for 68Ga and 67Ga, respectively (data analysed with Laura 6 software), or by Cyclone Plus phosphor filmless autoradiography imager (analysed with Cyclone Plus 5.0 software). HPLC was implemented using an Agilent Eclipse XDB C18 5 μm 4.6 × 150 mm reversed phase (RP) column and Agilent 1200 series HPLC with ultraviolet (UV, 220 nm) and radioactivity (Elysia raytest) detection and Gina StarTM software version 5.8. Gamma counting of tissue samples was performed using an LKB Wallac 1282 Compugamma Gamma Counter. Mass spectrometry analysis was conducted with a Thermo Exactive HR mass spectrometer with electrospray ionisation (ESI), running Xcalibur version 2.2 software. Nuclear magnetic resonance (NMR) spectra were acquired on a Bruker Avance III HD NanoBay 400 MHz NMR spectrometer (AscendTM magnet) and analysed with Bruker Topspin 4.2 software. Inductively coupled plasma-mass spectrometry (ICP-MS) determination of gallium in tissues was conducted in the London Metallomics Facility at King’s College London using a PerkinElmer NexION 350D, with Syngistix version 1.0. LC/MS was acquired through an Agilent Eclipse XDB C18 5 μm 4.6 × 150 mm RP column on an Agilent liquid chromatograph (1200 Series) with UV detection at 254 nm, connected to an Advion Expression LCMS mass spectrometer with electrospray ionisation source. Analysis was performed with Advion Mass Express software (version 6.4.16.1). Elemental microanalysis (for C, H, N) was performed by London Metropolitan University’s elemental analysis service.
5.2. Synthesis of KP46
The synthesis of KP46 was based on a method previously reported by Collery et al. [
22]. In brief, 1.175 g (8 mmol) of 8-hydroxyquinoline, dissolved in 10% acetic acid (40 mL), was added to 0.559 g (2 mmol) of gallium (III) nitrate [Ga(NO
3)
3.H
2O]. The mixture was heated under reflux to 80 °C for one hour before being filtered, and the residue was washed sequentially with hot water, cold water, and diethyl ether. The final yellow solid compound was dried overnight at 100 °C in a drying oven (yield 0.6232 g, 1.2 mmol, 60%).
5.3. Preparation of “Tracer” [68Ga]KP46, [67Ga]KP46, and [68Ga]Gallium Acetate
68Ga complex formation with 8-hydroxyquinoline was performed according to a modified method used by Yano et al. [
56], in which 200 µL of a solution of 8-hydroxyquinoline in ethanol (1 mg/mL) was mixed with 200 µL of sodium acetate (0.5 M in sterile H
2O, pH = 9) and 100 µL (8–10 MBq) of
68Ga from the most radioactive generator eluate fraction. This mixture contained 2.76 mM 8-hydroxyquinoline and approximately 0.98 nM
68Ga in a total volume of 0.5 mL at the time of preparation, with pH = 5.5–6.0. [
68Ga]Ga-acetate was prepared similarly, but we replaced the ethanolic 8-hydroxyquinoline ligand solution with ethanol only. For in vivo studies, higher activity and more concentrated samples of [
68Ga]KP46 and [
67Ga]KP46 were required, which were produced and concentrated by the following scaled-up method: for
68Ga-labelling, 1 mL of the highest-activity
68Ga generator eluate fraction (typically 240–270 MBq) was added to a mixture of 2 mL of 8-hydroxyquinoline in solution in ethanol (1 mg/mL) and 1 mL of sodium acetate (0.5 M in sterile H
2O, pH = 9). The final mixture pH was 5.5. In the case of [
67Ga]KP46 radiolabelling, after
67Ga citrate to
67Ga chloride conversion (see
Supplementary Materials), 0.5 mL of
67Ga chloride solution was added to a mixture of 1 mL of freshly dissolved 8-hydroxyquinoline in ethanol (1 mg/mL) and 0.5 mL sodium acetate (0.5 M in sterile H
2O, pH = 9). The final mixture pH was 5.5. The
68Ga and
67Ga mixtures were diluted (1:2
v/
v) with sterile water and loaded into a Sep-Pak Light C-18 cartridge that had been preconditioned with 5 mL of ethanol and 5 mL of water. The radioactivity trapped on the cartridge was eluted with 500 µL of ethanol in 5 × 100 µL fractions (recovering >90% of the radioactivity in all cases), the most radioactive of which was diluted tenfold with phosphate-buffered saline. Radiochemical purity was evaluated by iTLC (CHCl
3:CH
3OH, 95:5%,
v/
v; [
68/67Ga]KP46: Rf = 1; [
68Ga]Ga-acetate: Rf = 0).
5.4. Preparation of [68Ga]KP46 and [67Ga]KP46 for In Vivo Oral Administration Studies
To prepare radiolabelled (68Ga and 67Ga) bulk KP46, the most active ethanolic fraction (40 μL) of [68Ga]KP46 and [67Ga]KP46 eluted from the C18 column (as described above for preparation of tracer-level solutions) was combined with a 20 mM KP46 solution in dimethylsulfoxide (20 μL) and a 0.4 g/mL solution of polyethylene glycol (PEG, to maintain solubility) in water (180 µL) and was made up to 400 μL with water (160 μL). The tracer-level samples for oral administration were prepared in the same manner but with 20 μL of KP46-free DMSO in place of the KP46 solution in DMSO. The samples were analysed by HPLC and radio-iTLC prior to use in animal studies to show that radiolabelled KP46 and KP46 behaved similarly.
5.5. Determination of the Distribution/Partition Coefficients (logD and logP) of [68Ga]KP46 and [68Ga]Gallium Acetate
Tracer-level [68Ga]KP46 and [68Ga]Ga-acetate solutions (5–20 µL, 0.3–0.5 MBq) were added to a pre-equilibrated mixture (500 µL/500 µL) of octanol/water (for logP measurement) or octanol/PBS (for logD7.4 measurement). Samples were vortexed for two minutes in a Multi Vortex Mixer V-32 (Grant Bio). From each layer, a sample of 200 µL was taken and its radioactivity measured using a gamma counter.
5.6. [68/67Ga]KP46 and [68/67Ga]Gallium Acetate Binding to Serum Proteins In Vitro
Solutions of human apo-transferrin (apo-Tf, 2 mg/mL) and human serum albumin (HSA, 50 mg/mL) (both purchased from Sigma Aldrich) were dissolved in aqueous NaHCO3 (5 mM, pH = 8) and PBS (pH = 7.4), respectively. An amount of 1 MBq of tracer-level [68Ga]KP46 (13–20 µL) or [68Ga]gallium acetate (20–25 µL) was added to 1.2 mL of the apo-Tf solution and to a similar apo-Tf-free NaHCO3 solution (control). Similarly, [68Ga]KP46 and [68Ga]gallium acetate solutions were added to 1.5 mL PBS containing HSA or HSA-free PBS (control). The samples were incubated for 60 min at 37 °C (pH = 7–7.5). From each mixture, 0.5 mL was loaded into PD MidiTrap G-25 size exclusion chromatography cartridges that were preconditioned with 8 mL of NaHCO3 (5 mM) (for apo-Tf binding) or PBS (for HSA binding). Fractions (0.5 mL) were collected and counted with a Wallac gamma counter. Protein absorbance at 280 nm was measured using a Nanodrop spectrophotometer. To investigate the speciation of the Ga–KP46 complex in the presence of apo-Tf at later time points (up to 48 h), the experiment was repeated with 67Ga.
5.7. Cellular Uptake of [68Ga]KP46 In Vitro
One million A375 (human melanoma) cells were suspended in 0.5 mL of Dulbecco’s Modified Eagle’s Medium (500 mL DMEM, low-glucose medium with 50 mL 10% FBS, 5 mL of L-glutamine, and 5 mL of penicillin and streptomycin) in 1.5 mL Eppendorf tubes. An amount of 10 µL of no-carrier-added [68Ga]KP46 or [68Ga]gallium acetate was added to the cell suspensions (giving a 68Ga concentration of ca. 20 pM) and to similar tubes containing medium but no cells (to check and compensate for adhesion of radioactivity to tubes). Samples were incubated for 60 min at 37 °C under 5% CO2 and then centrifuged (Sci Spin Mini) for three minutes at 1500 RPM. The supernatants were transferred to fresh Eppendorf tubes. The pellets were washed twice with 0.5 mL PBS, following the same centrifugation protocol, and washes were added to the supernatant tubes. Supernatants and cell pellets were measured using a gamma counter to determine the average activity in cell pellets as a % of the total activity for each sample. The intracellular-to-extracellular 68Ga concentration ratio was also estimated by correcting for the different volume of supernatant and cell pellet using an estimated spherical cell diameter of 17.5 μm, which leads to an estimated volume of the cell pellet (106 cells) of 2.8 μL.
5.8. In Vivo Studies
All animal experiments were performed in accordance with the Animals (Scientific Procedures) Act, 1986, with protocols approved by the Animal Welfare and Ethical Review Body for King’s College London under project and personal licences approved by the UK Home Office. Tumour xenograft studies were performed on female NSG mice or female athymic nude mice after injecting them subcutaneously in the right shoulder with 2.5 × 10
6 A375 cells in 100 µL of PBS. All in vivo studies were conducted between 14 and 24 days after inoculation, with tumour volumes between 100 mm
3 and 580 mm
3 (see
Supplementary Materials, Figure S11).
Intravenous [68Ga]gallium acetate and [68Ga]KP46: NSG mice bearing A375 xenografts were fasted for 12–14 h before the start of the experiment. Mice were anaesthetised with isoflurane and injected with tracer level [68Ga]gallium acetate (group A, n = 5) or [68Ga]KP46 (group B, n = 5) (13–18 MBq, <200 μL) via the tail vein. Mice were kept under anaesthesia during PET/CT imaging for 4 h (see protocol below), then culled by neck dislocation. The organs were harvested, weighed, and gamma counted. Additional similar studies with athymic nu/nu mice were conducted similarly.
Oral [68/67Ga]KP46: The protocol for oral administration experiments reported here was devised on the basis of pilot experiments described in
Supplementary Materials (Figure S12). For short-term (up to 4 h) studies with
68Ga, awake (nonanaesthetised, to avoid suppressive effects of anaesthesia on digestive tract activity) mice that had been fasted for up to 14 h were given [
68Ga]KP46 (5–13 MBq) (group C, “tracer”, n = 3) or [
68Ga]KP46 and KP46 (5–8.5 MBq) (group D, “bulk”, n = 3) by oral gavage. The dose given orally to group D animals, prepared as described above, was 0.4 μmol in 400 μL, molar activity ca. 25 MBq/μmol. The mice were fasted for three hours before being anaesthetised and PET/CT scanned dynamically for one hour (see below for scanning protocol). After scanning, mice in both groups were culled by neck dislocation while still anaesthetised four hours post-oral administration for organ harvesting and gamma counting. Samples of blood, urine and contents of stomach, small intestine and large intestine (the organs/tissue with measurable activity; tumours were not included as they were insufficiently radioactive) from the mice in group C were used for octanol extraction (shake-flask method) to separate water-soluble (released) and lipid-soluble (intact KP46)
68Ga. Serum samples were prepared by placing blood samples in a serum separator tube (brand VS367954, Fisher Scientific Ltd., Loughborough, UK) and centrifuging at 3000 rpm for 10 min. Serum (200–300 µL) and urine samples (150–180 µL, taken from the excised bladders) were directly added to Eppendorf tubes containing a pre-equilibrated mixture of octanol (500 µL) and water (500 µL). The tubes were vortexed for two minutes on a Multi Vortex Mixer V-32 (Grant Bio). After separation of layers, a sample of 400 µL from each layer was transferred to fresh Eppendorf tubes and analysed with a gamma counter. Stomach content (72–77 mg), small intestine content (35–88 mg), and large intestine content (86–100 mg) samples from each mouse were analysed similarly by adding them directly to Eppendorf tubes containing a pre-equilibrated mixture of octanol (500 µL) and water (500 µL) before vortexing and centrifuging at 10,000 rpm for 10 min at 4 °C. From each layer, 430 µL was transferred to fresh Eppendorf tubes and analysed with a gamma counter; the solid residues were also counted. Tissues from mice in group D were used after radioactive decay for measurements of nonradioactive
69Ga content.
For long-term (up to 48 h) studies, a similar protocol was followed, but SPECT imaging using 67Ga was repeated at intervals up to 48 h instead of PET imaging up to only 4 h postadministration. Athymic nude nu/nu mice bearing A375 xenografts were fasted for 7–14 h and divided in two groups: E (n = 4) and F (n = 4). Awake (nonanaesthetised) mice were orally administered with [67Ga]KP46 (2–11 MBq, “tracer”, group E) or with [67Ga]KP46 and KP46 (1–11 MBq, “bulk”, group F, given 0.4 μmol KP46 with molar activity ca. 15 MBq/μmol) and fasted for three hours. Mice in both groups were then anaesthetised and SPECT/CT scanned (see scanning protocol below) for one hour. Mice were then allowed to recover and feed. At 24 h postadministration, mice were reanaesthetised and rescanned, then group E mice were culled while still anaesthetised, and tissues were harvested for ex vivo gamma counting. Group F mice, which showed sufficient remaining radioactivity for further scanning at later time points, were allowed to recover and feed after the 24 h scan, and they were reanaesthetised and rescanned 48 h post-oral administration before culling and harvesting of organs for gamma counting and 69Ga measurement by ICP-MS.
5.9. Scanning Protocols
PET/CT images were acquired with a nanoScan® PET/CT (Mediso Medical Imaging Systems, Budapest, Hungary) scanner operating in list mode using a 400–600 keV energy window and a coincidence window of 1:3. CT scans were acquired for anatomical reference and attenuation correction (55 keV X-ray, exposure time 1000 ms, and 360 projections and pitch 1). PET projection data were reconstructed using the Tera-tomo® software package provided with the scanner —a Monte Carlo-based fully 3D iterative algorithm with four iterations, six subsets, and 0.4 mm isotropic voxel size; corrections for attenuation, scatter, and dead-time were enabled. The data were then visualised and quantified using VivoQuant© v.2.50 (InviCro, Boston, MA, USA) software. SPECT/CT imaging was performed on a NanoSPECT/CT Silver Upgrade scanner (Mediso; 4 heads, 4 × 9 1.0 mm multipinhole collimators) in helical scanning mode using energy windows centred around 93.20 ± 20% keV (primary), 184.60 ± 20% keV (secondary), and 300.00 ± 20% keV (tertiary). CT images were acquired using 45 kVp tube voltage and 1000 ms exposure time in 180° projections. SPECT/CT data sets were reconstructed using the HiSPECT 1.4.2611 (SciVis, Gottingen, Germany) reconstruction software package using standard reconstruction with 35% smoothing and 9 iterations. Images were coregistered and analysed using VivoQuant v2.50 (InviCro, Needham, MA, USA).



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







