1. Introduction
Texturized vegetable proteins are healthy, sustainable [
1], and can meet the protein needs of a growing population [
2,
3]. Nutritionally, it is high in protein, low in calories, low in fat, and zero in cholesterol [
4]. As a result, texturized vegetable proteins are gaining consumer attention and providing better options for vegetarians [
5]. Texturized vegetable protein is formed by extrusion technology similar to animal meat, with abundant fiber and texture similar to animal meat [
6], which has been more studied in recent years [
7]. Some researchers have found that the main influences on textural properties of texturized vegetable protein include raw materials [
7], additives such as polysaccharides [
8,
9,
10] and enzymes [
11], and extrusion conditions [
12,
13], among which raw materials are the key influencing factors [
14].
At present, the main raw materials used in the production of texturized vegetable proteins are soybean protein [
15], peanut protein [
16], pea protein [
17], and wheat gluten [
18]. Previous studies have found that different raw materials have different effects on texturized vegetable protein. Using soybean protein as the main ingredient, a series of physical and chemical changes are produced through the extrusion process, resulting in the formation of texturized vegetable protein [
19]. Texturized vegetable proteins possess unique layered and fibrous structures, exhibiting a significant level of fibrousness and an orderly structural orientation, which affects the digestibility and absorbability of the protein and its functional properties [
20,
21]. Due to its exceptional gelling properties, superior nutritional value, and cost-effectiveness, soybean protein is widely acknowledged as an ideal option for the development of texturized vegetable protein meat substitutes [
14]. A few studies have used defatted peanut protein for texturizing under low-moisture conditions [
22,
23]. The reduced capacity of peanut proteins to form a rich fiber structure during extrusion is primarily due to their inferior gel-forming ability compared to soybean proteins [
20,
22]. The researchers discovered that the primary forces responsible for maintaining the structure of peanut proteins are non-covalent bonds, such as hydrophobic interactions and hydrogen bonds, with disulfide bonds playing a secondary role [
24]. Consequently, in contrast to soybean protein, peanut protein exhibits inadequate gelling capacity and lacks the required tensile strength and fibrous structure. As a result, it possesses a soft fibrous texture [
16,
25]. Pea proteins with a moisture content of 55% and a distinct fibrous structure can be extruded to form texturized vegetable protein [
26]. However, the susceptibility of pea proteins to damage during extreme processing, such as high temperatures and rapid heating rates during extrusion, undermines their gelling properties [
27,
28]. Therefore, when pea protein is the main raw material, attention needs to be paid to the extrusion parameter (barrel temperature) settings in order to obtain properly texturized vegetable protein [
29]. Compared to soybean protein, wheat protein lacks lysine and cannot meet the human demand for amino acids [
30]. Due to its viscoelastic properties, wheat gluten can be individually formed into a continuous network structure by high moisture extrusion [
31]. In terms of network structure formation, the processing properties of wheat gluten surpass those of other plant proteins due to its exceptional gelling properties [
32]. To improve the fiber structure, wheat gluten is usually blended with other vegetable proteins such as soybeans, peanuts, and peas [
33,
34]. Therefore, in this paper, we chose to focus on different soybean protein isolates to investigate the effect of different soybean protein isolates on texturized vegetable proteins.
Texturized vegetable proteins are affected differently by different soybean proteins. The different compositions of soybean proteins (7 s and 11 s globulins) determine their various nutritional and functional properties such as moisture, surface, and protein mechanisms [
35,
36]. It has been shown that various soybean protein fractions can alter the textural properties, WHC, and rehydration properties of texturized vegetable protein [
37]. Adding 7 s improves protein hydration properties, as well as foaming and emulsifying properties [
38]. Adding 11s increases the structural strength of proteins and improves properties such as gelling [
39]. The best textural properties of texturized vegetable protein were obtained at an 11 s/7 s ratio of 1.5 [
40]. In addition, the interactions between proteins are an important determinant of solubility, gelling, adhesion, and other properties. There are significant contributions from hydrogen bonds, disulfide bonds, and hydrophobic interactions in the processing of texturized vegetable protein, which are the primary contributors influencing its textural properties [
15,
41,
42]. Studies have found that the preservation of texturized vegetable proteins, regardless of whether soybean protein is processed at low or high moisture levels, relies on a combination of hydrophobic interactions, hydrogen bonds, disulfide bonds, and their synergistic interactions [
15]. In this process, non-covalent bonds play a more significant role than covalent bonds. Similarly, the structure of proteins has an effect on the properties of texturized vegetable proteins. Research has shown that the presence of L-cysteine can alter the interconnection of disulfide bonds between protein molecules, resulting in noticeable changes in the texture and structure of textured vegetable proteins. The addition of L-cysteine may have an effect on the binding interactions within protein molecules, thus affecting their cross-linking effect [
43]. Up to now, however, it has not been clear which properties and which structures of the raw material influence the texture of the texturized vegetable protein.
Therefore, to enhance the structural characteristics of texturized vegetable proteins, we established a specialized raw material index system. We first studied three different soybean protein isolates for functional properties (WHC, emulsifying properties, and foaming properties) and structure (amino acid composition and secondary structure). Then, we prepared three texturized vegetable proteins through low-moisture extrusion and analyzed their textural properties, degree of texturization, protein free SH and S-S content, and microstructure. Finally, we analyzed the correlation between soybean protein isolates and texturized vegetable proteins and drew relevant conclusions, which provide support for dedicated raw materials for texturized vegetable proteins.
3. Materials and Methods
3.1. Materials
We purchase three kinds of soybean protein isolates, 970 W, 971 W, 950 E, and soybean meal (Shandong Yuwang Group, Yucheng, China) and corn starch (Heilongjiang Pengcheng Biochemical Co., Ltd., Qiqihar, China). Protein content was determined by the Kjeldahl method with a conversion factor of 6.25. The protein content of soybean isolates from 971 W, 970 W, and 950 E were 82.84, 82.35, and 82.79 g/100 g.
3.2. Low-Moisture Extrusion Process
For the extrusion process, a Brabender DSE-25 pilot-scale food extruder with co-rotating (Brabender GmbH and Co., Duisburg, Germany) intermeshing twin screws was employed. The extruder has a screw diameter of 25 mm and a screw length to diameter ratio of 20:1. The ratio of raw materials used for extrusion was 6:3:1 for soybean protein isolate, soybean meal, and corn starch. The pump and feeder were calibrated and adjusted to feed moisture of 20%. On a dry basis, the yield was 1.8 kg/h. The temperatures in the extruder barrel remained constant at 60 °C, 110 °C, 130 °C, 140 °C, and 150 °C.
3.3. Functional Properties of Soybean Protein Isolates
3.3.1. Water Holding Capacity (WHC)
With slight adjustments, this method was used [
37]. Dissolve 0.1 g of protein sample (labelled M
1) in phosphate buffer solution (0.01 mol/L, 5 mL). Weigh the centrifuge tube beforehand and record its mass (labelled as M
2). Transfer to a 10 mL centrifuge tube, vortex for 3 min at room temperature, and centrifuge at 5000 rpm for 20 min. Remove the supernatant and take note of the mass (labelled as M
3). The measurements were conducted in triplicate. The WHC of the raw material is calculated using the following formula:
3.3.2. Emulsifying Properties
The Emulsification Activity Index (EAI) and the Emulsification Stability Index (ESI) are two important properties of proteins that are related to emulsification. This method was used for the determination of emulsification properties (EAI and ESI) with slight modifications [
17]. In a centrifuge tube, soybean protein isolate (0.30 g) was combined with water (30 mL) and stirred for 10 min. Following this, soybean oil (10 mL) was added and homogenized at a speed of 10,000 rpm for 1 min. At 0 min, a sample (50 μL) was collected and added to a mixed 1% sodium dodecyl sulphate (SDS) solution (5 mL). After 10 min, the sample (50 μL) was collected and added to a mixed 1% SDS solution (5 mL). Sample absorbance at 500 nm was measured by UV spectrophotometer (UV-9000 Metash Shanghai Yuan Analytical Instrument Co., Shanghai, China). The following formulas were used to calculate EAI and ESI.
The equation is as follows: T = 2.303; N represents the dilution factor; A0 denotes the initial absorbance before homogenization; C represents the concentration of soybean protein isolate (g/mL) prior to emulsion formation; ø represents the volume of the emulsion, which is equivalent to 1/4; A10 denotes the absorbance of the emulsion after 10 min of homogenization; ΔT is a time-varying value of 10 min.
3.3.3. Foaming Properties
The foam capacity (FC) and foam stability (FS) were based on this method with minor modifications [
17]. Weighing 0.30 g of soybean protein isolate, it was then combined with 30 mL of phosphate buffer solution (PBS) at pH 7.0 in a centrifuge tube. Continue to homogenize for an additional 1 min at a speed of 10,000 rpm. Next, swiftly transfer the mixture to a 50 mL graduated cylinder for quantification and reassess the foam volume after a complete duration of 25 min. The calculation of FC and FS was conducted using the following method:
where: V is the initial volume of the solution (mL); V
1 is the volume of the foam after 1 min of homogenization (mL); V
25 is the volume of the foam after 25 min of resting (mL).
3.4. Content of Amino Acids
Following this method [
47], proposed soybean protein isolate (0.1 g), 6 mol/L HCl (10 mL), and phenol (2 drops) were combined in a reaction tube. For 15 min after filling the reaction tube with nitrogen, it was subsequently sealed and incubated at a temperature of 110 °C in an oven for a period of 24 h. After the reaction tube was cooled to room temperature, the contents were passed through a cellulose filter paper for filtration. The resulting filtrate was then mixed with deionized water until a final volume of 50 mL was achieved. To supplement the filtrate, 1 mL of sodium citrate buffer with a pH of 2.2 was added. Before performing amino acid analysis on an automated amino acid analyser (Hitachi L-8900, Tokyo, Japan), the solution was further filtered using a syringe filter with a pore size of 0.22 µm.
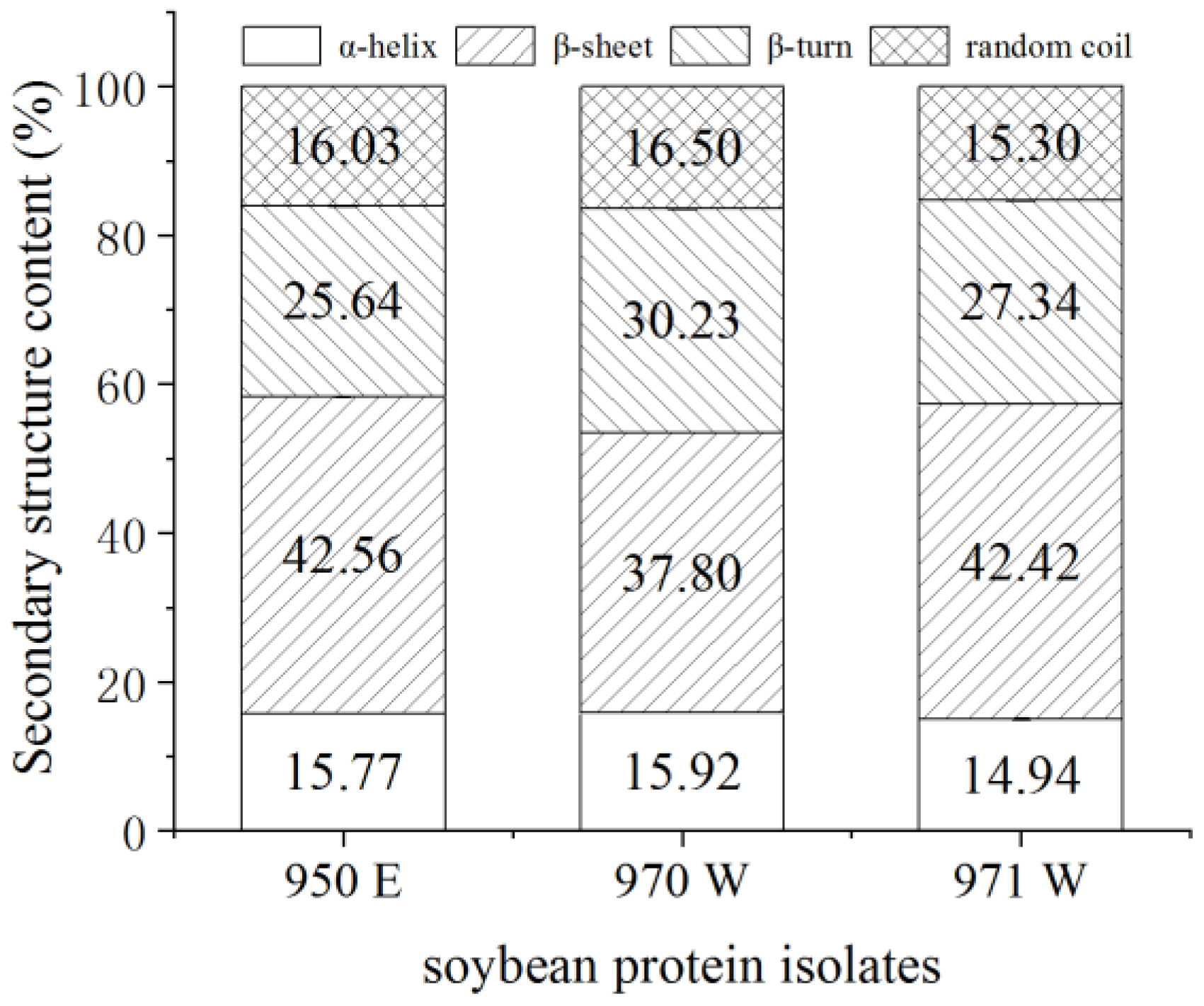
3.5. Secondary Structure of Proteins
Refer to this method [
62] with slight modifications. The first step was to collect the air background and then scan the potassium bromide background and deduct the background for the determination of the sample. Before conducting infrared spectral analysis, the sample (1 mg) and KBr (100 mg) were accurately weighed and placed into an agate mortar. Then, they were ground under an infrared lamp. After grinding, the mixture was compressed in a tablet press at a pressure of 14 kg for approximately 1 min. The resulting uniform and transparent sheets were analysed using a Bruker Equinox 55 spectrometer (Bruker Instrument Company, Karlsruhe, Germany) for infrared spectral analysis. Fourier Transform Infrared Spectrometer (FTIR) (Germany TENSOR 27 (ATR), Bruker, Karlsruhe, Germany) operating conditions: the infrared spectra were recorded in the wave number range of 400–4000 cm
−1, with a resolution of 4 cm
−1, signal scanning cumulative 64 times, each sample was measured 3 times, and the average value was obtained. The infrared curves were processed with the OMNIC 8.0 software (Thermo Fisher Scientific, Waltham, MA, USA) for the correction of the baseline, the normalization, and the Fourier deconvolution. PeakFitv4.12 software (Systat Software Inc., San Jose, CA, USA) was used to analyse and calculate the percentage of sub-peak structures in the amide I region.
3.6. Texturized Vegetable Protein
3.6.1. Textural Properties
Based on this approach [
49], the pre-treatment of the samples (three texturized vegetable proteins (971 W, 970 W, and 950 E)) involved hydrating them at room temperature for 90 min. After being drained using a sieve for 30 min, it is cut with a knife to a length of 20 mm. During the texture analysis conducted in TPA mode, a cylindrical aluminium probe (P/36R, Ø 35 mm) installed in the Texture Analyser (TA.XT2, Stable Micro Systems, Godalming, UK) was utilized to compress the sample to 50% of its initial height. The test parameters for the plus texture analyser were configured as follows: the pre-test speed was set to 2 mm/s, the test speed was set to 1 mm/s, the post-test speed was set to 2 mm/s, the trigger force was set to 5 g, and the time interval between two compressions was set to 5 s. Each sample was replicated five times.
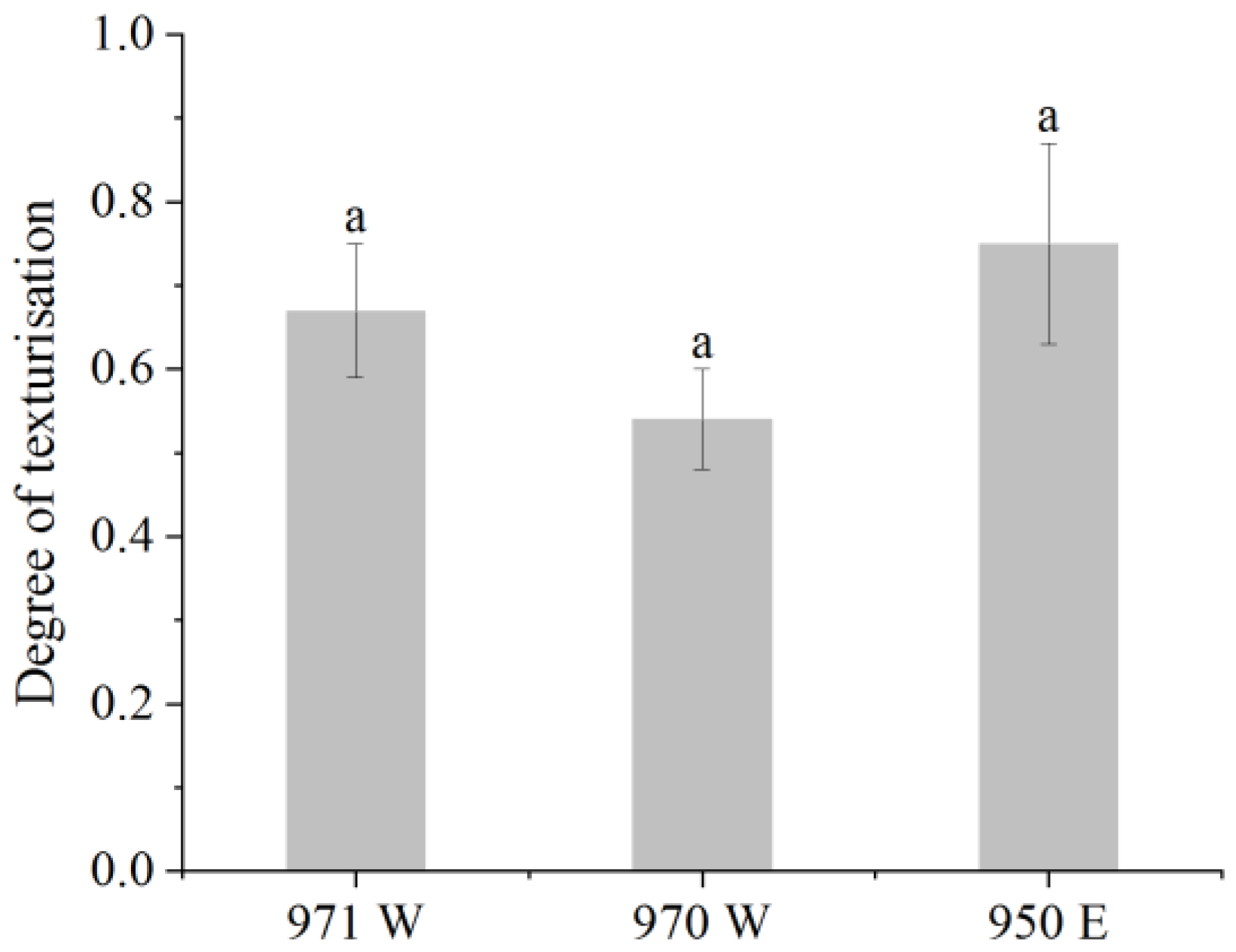
3.6.2. Degree of Texturization
These methods were referenced with slight modifications [
47]. The test method was as follows: the samples were trimmed to a length of 20 mm and placed on the sample stage for testing. Longitudinal (parallel to the direction of extrusion) and transverse (perpendicular to the direction of extrusion) shear determinations were made on the sample using the Texture Analyser’s (TA.XT2, Stable Micro Systems, Godalming, UK) A-CKB probe in shear mode. The physical property tester was operated in shear mode with the following parameters: 2 mm/s before the test, 1 mm/s during the test, and 2 mm/s after the test while maintaining 75% shear. The measurements were repeated five times for each sample, and the average value was calculated from the results. The degree of texturization was calculated using the following formula:
In the given formula, the symbol Ft represents the shear force in the transverse direction, while the symbol Fl represents the shear force in the longitudinal direction.
3.6.3. Free Sulfhydryl and Disulfide Content of Proteins (Free SH and S-S Content)
Free SH were measured using the 5,5-dithiobis (2-nitrobenzoic acid) (DTNB) method as described by [
63] and Ellman’s method [
37], in which 4 mg of DTNB was added to 1 mL of Tris-Gly in solution at a pH of 8.0. In the experiments, the sample (3 mg) was dissolved in 5 mL of Tris-Gly-EDTA buffer solution and reacted for 30 min with shaking every 10 min. After 30 min, 40 μL of DTNB was added, shaken, and mixed, and the reaction was wrapped in tinfoil and reacted at 25 °C for 30 min. The reaction was centrifuged at 25 °C (13,600
g for 10 min), and the supernatant was analysed for absorbance at 412 nm. The reagent blank was subtracted from the sample. The Tris-Gly-EDTA buffer solution consisted of Tris (0.086 mol/L), Gly (0.09 mol/L), and EDTA (0.04 mol/L), and the pH was adjusted to 8.0 with hydrochloric acid. Free SH content was calculated as follows:
In the provided formula, the value of 73.53 is derived from the ratio 106/13,600. The variable A412 represents the absorbance measured at 412 nm. The variables C and D denote the protein concentration (mg/mL) and the dilution factor, respectively.
Determination of S-S content: Sample (3 mg) was added to 5 mL Tris-Gly-EDTA, then 0.05 mL of β-mercaptoethanol (β-ME), held at 25 °C for 1 h, then added 12% trichloroacetic acid (TCA) (10 mL) and continued to be held at 25 °C for 1 h, centrifuged (4000 rpm, 10 min), and the supernatant was poured off and the precipitate was retained. Dissolve the precipitate in 5 mL of 12% TCA, wash the precipitate, continue centrifugation, pour off the supernatant, and keep the precipitate. The precipitate was dissolved in 5 mL of Tris-Gly-EDTA, 40 μL of DTNB was added, and the reaction was carried out for 30 min to measure the absorbance at 412 nm. The content of S-S was calculated according to the following formula:
where 73.53 = 106/13,600, D denotes dilution factor, and C denotes protein concentration (g/L).
3.6.4. Microstructure
We used a scanning electron microscope (SEM) (Hitachi SU8010, Tokyo, Japan) to analyse the microstructure, following the procedure outlined with minor adjustments [
47]. Prior to observation, both hydrated and non-hydrated samples were subjected to freeze-drying and further coated with a layer of gold. The observations were carried out at 15 kV accelerating voltage, using a magnification of 30× and 500× for all images.
3.7. Data Analysis
Results are expressed as the mean ± standard deviation. The statistical software SPSS 24.0 (IBM, Armonk, NY, USA) was utilized to conduct both one-way ANOVA (analysis of variance) and Pearson correlation analysis. Graphs were created using Origin 2010 software (OriginLab, Northampton, MA, USA). The mean values of the samples showed significant differences at a significance level of p < 0.05.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}