
Study Models of Drug–Drug Interactions Involving P-Glycoprotein: The Potential Benefit of P-Glycoprotein Modulation at the Kidney and Intestinal Levels

 ,
,  , ,
, ,  and
and
Abstract
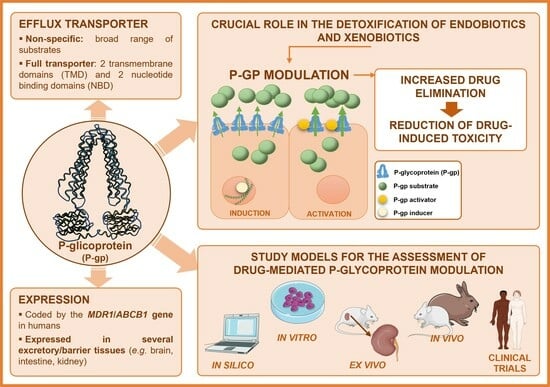
:
1. Introduction
2. Overview of P-Glycoprotein
2.1. Description
2.2. P-gp’s Structure
2.3. P-gp Efflux Mechanisms
2.4. P-gp Expression
3. Study Models of Drug–Drug Interactions Involving P-Glycoprotein Modulation
3.1. In Silico Models
3.2. In Vitro Models
3.3. Ex Vivo Models
3.4. In Vivo Models
3.5. Clinical Trials
4. Drug-Mediated P-gp Modulation
4.1. Modulation of P-gp at the Intestinal Level
4.2. P-gp Modulation at the Kidney Level

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P-gp Substrate | Experimental Conditions | Modulation Type | Alterations in Substrate Therapeutic Effect/Toxicity | Experimental Models | References |
|---|---|---|---|---|---|
| Cisplatin | In vitro: 0.02–200 nM, for 24 h; in vivo: 15 mg/kg, i.p. | P-gp induction by omeprazole (along with OCT2 inhibition) In vitro: 0.4–4000 µg/mL, for 24 h In vivo: 1.8 or 3.6 mg/kg/day, i.p., for 5 days | Decreased cisplatin-induced nephrotoxicity Enhanced cell viability; diminished intracellular ROS generation and membrane damage | In vitro, in HK-2 cells Protein expression: Western blot, immunofluorescence, and immunohistochemistry assay In vivo, in male Sprague Dawley rat kidneys | [137] |
| Cyclosporine A | 1–20 µM, for 36 h | P-gp induction by: Dexamethasone (1 µM, for 4 days) Cyclosporine A (1 µM, for 4 days) 1,25(OH)2D3 (35 nM, for 4 days) | Lower cyclosporine A-induced cytotoxicity | In vitro, in HK-2 cells Cell viability: trypan blue dye exclusion test and MTT assay | [176] |
| 0–80 µM, for 36 h | P-gp inhibition by grapefruit juice (5%, for 3 days) | Higher cyclosporine A-induced cytotoxicity | In vitro, in HK-2 cells Cell viability: trypan blue dye exclusion test and WST-1 colorimetric test | [177] | |
| 0–80 µM, for 24 h | P-gp inhibition by Zuccagnia punctata extract or 3,7-dihydroxyflavone (5 mg/mL, for 3 days) | Higher cyclosporine A-induced cytotoxicity | In vitro, in HK-2 cells Cell viability: NR assay and trypan blue exclusion test | [181] | |
| Digoxin | 0.75 mg, for 3 days, orally | P-gp inhibition by clarithromycin (2 × 250 mg/day, for 3 days, orally) | Higher rate of adverse effects Reduced non-glomerular renal clearance of digoxin | Randomized, placebo-controlled, double-blind crossover design applied to 12 healthy men Concentrations of digoxin in plasma and urine: radioimmunoassay | [135] |
| 0.1 mg/kg, i.v. | P-gp induction by 1,25(OH)2D3 (2.5 µg/kg every other day for 8 days, i.p.) | Significant increases in renal and cerebral P-gp levels (3.45-fold), and in the renal (74%) and total-body (34%) clearance of digoxin | In vivo, in fxr(−/−) mice | [183] | |
| 0.625 mg/kg, i.v. | P-gp induction by 17b-estradiol In vitro: 10 nM, for 24 h | In vitro: 17b-estradiol induced the expression of P-gp in cultured human PTECs In vivo: higher renal clearance of digoxin and mRNA expression in female mice (higher levels of 17b-estradiol) | In vitro, in renal PTECs P-gp expression: RT-PCR In vivo, in female and male ICR mice Quantification of digoxin by LC-MS-MS | [185] | |
| Vancomycin | In vivo: 2 or 4 mM for 72 h; in vivo: 400 or 600 mg/kg/day, for 7 days | P-gp induction by cilastatin In vitro: 200 µg/mL, for 0–72 h In vivo: 300 mg/kg/day, for 7 days | In vitro: Lower vancomycin-induced cytotoxicity In vivo: Significant decrease in vancomycin levels in the blood and kidneys | In vitro, in HK-2 cells Cell viability: MTT assay In vivo, in male C57BL/6J mice Quantification of vancomycin: fluorescence polarization immunoassay | [184] |
| Triptolide | 0–5000 nM, for 6, 12 and 24 h | P-gp induction by glycyrrhetinic acid (0–100 µM, for 8, 12 and 24 h) | Lower glycyrrhetinic-acid-induced cytotoxicity | In vitro, in HK-2 cells Cell viability: MTT assay Quantification of triptolide by UPLC-ESI-MS | [182] |
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Giacomini, K.; Huang, S.; Tweedie, D.; Benet, L.; Brouwer, K.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.-M. Transporters in Drug Development and Clinical Pharmacology. Clin. Pharmacol. Ther. 2013, 94, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Petersen, T.S.; Dalhoff, K.P. Clinical Implications of P-Glycoprotein Modulation in Drug–Drug Interactions. Drugs 2017, 77, 859–883. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.; Van Bambeke, F. ABC Multidrug Transporters: Target for Modulation of Drug Pharmacokinetics and Drug-Drug Interactions. Curr. Drug Targets 2011, 12, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Degorter, M.K.; Xia, C.Q.; Yang, J.J.; Kim, R.B. Drug Transporters in Drug Efficacy and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.C.; Jamshidi, N.; Chen, Y.; Eraly, S.A.; Cho, S.Y.; Bhatnagar, V.; Wu, W.; Bush, K.T.; Abagyan, R.; Palsson, B.O.; et al. An Organic Anion Transporter 1 (Oat1)-Centered Metabolic Network. J. Biol. Chem. 2016, 291, 19474–19486. [Google Scholar] [CrossRef] [PubMed]
- Döring, B.; Petzinger, E. Phase 0 and Phase III Transport in Various Organs: Combined Concept of Phases in Xenobiotic Transport and Metabolism. Drug Metab. Rev. 2014, 46, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.T.; Wu, W.; Lun, C.; Nigam, S.K. The Drug Transporter OAT3 (SLC22A8) and Endogenous Metabolite Communication via the Gut–Liver–Kidney Axis. J. Biol. Chem. 2017, 292, 15789–15803. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Liu, K. Renal Organic Anion Transporters in Drug–Drug Interactions and Diseases. Eur. J. Pharm. Sci. 2018, 112, 8–19. [Google Scholar] [CrossRef]
- Takano, M.; Yumoto, R.; Murakami, T. Expression and Function of Efflux Drug Transporters in the Intestine. Pharmacol. Ther. 2006, 109, 137–161. [Google Scholar] [CrossRef]
- Foley, S.E.; Tuohy, C.; Dunford, M.; Grey, M.J.; De Luca, H.; Cawley, C.; Szabady, R.L.; Maldonado-Contreras, A.; Houghton, J.M.; Ward, D.V.; et al. Gut Microbiota Regulation of P-Glycoprotein in the Intestinal Epithelium in Maintenance of Homeostasis. Microbiome 2021, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Khuri, N.; Giacomini, K.M.; Sali, A. Molecular Modeling and Ligand Docking for Solute Carrier (SLC) Transporters. Curr. Top. Med. Chem. 2013, 13, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharmacokinet. 2017, 56, 825–892. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; König, J.; Fromm, M.F. Clinical Aspects of Transporter-Mediated Drug–Drug Interactions. Clin. Pharmacol. Ther. 2019, 105, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical Studies on Drug–Drug Interactions Involving Metabolism and Transport: Methodology, Pitfalls, and Interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Welch, M.A.; van Vlijmen, H.; Korzekwa, K.; Swaan, P.W.; Matsson, P. Molecular Modeling of Drug-Transporter Interactions—An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 818–835. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Arya, V.; Yang, X.; Volpe, D.A.; Zhang, L. Evaluation of Transporters in Drug Development: Current Status and Contemporary Issues. Adv. Drug Deliv. Rev. 2017, 116, 100–118. [Google Scholar] [CrossRef]
- Balayssac, D.; Authier, N.; Cayre, A.; Coudore, F. Does Inhibition of P-Glycoprotein Lead to Drug-Drug Interactions? Toxicol. Lett. 2005, 156, 319–329. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A Surface Glycoprotein Modulating Drug Permeability in Chinese Hamster Ovary Cell Mutants. BBA-Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Martins, E.; Silva, V.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; Rocha-Pereira, C.; Ghanem, C.I.; Carmo, H.; Remião, F.; et al. Newly Synthesized Oxygenated Xanthones as Potential P-Glycoprotein Activators: In Vitro, Ex Vivo, and in Silico Studies. Molecules 2019, 24, 707. [Google Scholar] [CrossRef]
- Kim, R.B. Drugs as P-Glycoprotein Substrates, Inhibitors, and Inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Prescher, M.; Kroll, T.; Schmitt, L. ABCB4/MDR3 in Health and Disease—At the Crossroads of Biochemistry and Medicine. Biol. Chem. 2018, 400, 1245–1259. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; De Lourdes Bastos, M.; Remião, F. Modulation of P-Glycoprotein Efflux Pump: Induction and Activation as a Therapeutic Strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An Updated Patent Review on P-Glycoprotein Inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cascorbi, I. P-Glycoprotein: Tissue Distribution, Substrates, and Functional Consequences of Genetic Variations. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 261–283. ISBN 9783642145407. [Google Scholar]
- Konieczna, A.; Erdösová, B.; Lichnovská, R.; Jandl, M.; Čížková, K.; Ehrmann, J. Differential Expression of ABC Transporters (MDR1, MRP1, BCRP) in Developing Human Embryos. J. Mol. Histol. 2011, 42, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Pereira, C.; Silva, V.; Costa, V.M.; Silva, R.; Garcia, J.; Gonçalves-Monteiro, S.; Duarte-Araújo, M.; Santos-Silva, A.; Coimbra, S.; Dinis-Oliveira, R.J.; et al. Histological and Toxicological Evaluation, in Rat, of a P-Glycoprotein Inducer and Activator: 1-(Propan-2-Ylamino)-4-Propoxy-9H-Thioxanthen-9-One (TX5). EXCLI J. 2019, 18, 697–722. [Google Scholar] [CrossRef] [PubMed]
- Bonito, C.A.; Ferreira, R.J.; Ferreira, M.J.U.; Gillet, J.P.; Cordeiro, M.N.D.S.; dos Santos, D.J.V.A. Theoretical Insights on Helix Repacking as the Origin of P-Glycoprotein Promiscuity. Sci. Rep. 2020, 10, 9823. [Google Scholar] [CrossRef]
- Higgins, C.F.; Gottesman, M.M. Is the Multidrug Transporter a Flippase? Trends Biochem. Sci. 1992, 17, 18–21. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; Dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Altenberg, G.A.; Vanoye, C.G.; Horton, J.K.; Reuss, L. Unidirectional Fluxes of Rhodamine 123 in Multidrug-Resistant Cells: Evidence against Direct Drug Extrusion from the Plasma Membrane. Proc. Natl. Acad. Sci. USA 1994, 91, 4654–4657. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.U.; Dos Santos, D.J.V.A. Insights on P-Glycoproteins Efflux Mechanism Obtained by Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Ling, V. Positively Cooperative Sites for Drug Transport by P-Glycoprotein with Distinct Drug Specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Parveen, Z.; Stockner, T.; Bentele, C.; Pferschy, S.; Kraupp, M.; Freissmuth, M.; Ecker, G.F.; Chiba, P. Molecular Dissection of Dual Pseudosymmetric Solute Translocation Pathways in Human P-Glycoprotein. Mol. Pharmacol. 2011, 79, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and Functional Aspects of P-Glycoprotein and Its Inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, M.; Silva, R.; Rocha-Pereira, C.; Carmo, H.; Carvalho, F.; Bastos, M.D.L.; Remião, F. Cellular Models and in Vitro Assays for the Screening of Modulators of P-Gp, MRP1 and BCRP. Molecules 2017, 22, 600. [Google Scholar] [CrossRef]
- George, B.; You, D.; Joy, M.S.; Aleksunes, L.M. Xenobiotic Transporters and Kidney Injury. Adv. Drug Deliv. Rev. 2017, 116, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Toyohara, T.; Ohtsuki, S.; Moriyama, Y.; Abe, T.; Terasaki, T. Quantitative Targeted Absolute Proteomics for 28 Transporters in Brush-Border and Basolateral Membrane Fractions of Rat Kidney. J. Pharm. Sci. 2016, 105, 1011–1016. [Google Scholar] [CrossRef]
- Ranzani, G.N.; De Gregori, S.; De Gregori, M.; Regazzi, M.; Govoni, S. Interindividual Variability of Drug Transporters: Impact on Opioid Treatment in Chronic Renal Failure. Eur. J. Pain Suppl. 2009, 3, 21–28. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; Von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional Polymorphisms of the Human Multidrug-Resistance Gene: Multiple Sequence Variations and Correlation of One Allele with P-Glycoprotein Expression and Activity in Vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Rodríguez, M.; Belmonte, C.; Román, M.; Ochoa, D.; Jiang-Zheng, C.; Koller, D.; Mejía, G.; Zubiaur, P.; Wojnicz, A.; Abad-Santos, F. Effect of ABCB1 C3435T Polymorphism on Pharmacokinetics of Antipsychotics and Antidepressants. Basic Clin. Pharmacol. Toxicol. 2018, 123, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Leake, B.F.; Choo, E.F.; Dresser, G.K.; Kubba, S.V.; Schwarz, U.I.; Taylor, A.; Xie, H.G.; McKinsey, J.; Zhou, S.; et al. Identification of Functionally Variant MDR1 Alleles among European Americans and African Americans. Clin. Pharmacol. Ther. 2001, 70, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Saib, S.; Delavenne, X. Inflammation Induces Changes in the Functional Expression of P-Gp, BCRP, and MRP2: An Overview of Different Models and Consequences for Drug Disposition. Pharmaceutics 2021, 13, 1544. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on the Investigation of Drug Interactions; European Medicines Agency: London, UK, 2012; Volume 44, p. 59. [Google Scholar]
- FDA. Guidance for Industry. Drug Interaction Studies Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations; Guidance Document; Food and Drug Administration: Silver Spring, MD, USA, 2012; p. 79. [Google Scholar]
- Saeidnia, S.; Manayi, A.; Abdollahi, M. From in Vitro Experiments to in Vivo and Clinical Studies; Pros and Cons. Curr. Drug Discov. Technol. 2015, 12, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.; Yu, H.; Zhang, L.; Hou, T. Computational Models for Predicting Substrates or Inhibitors of P-Glycoprotein. Drug Discov. Today 2012, 17, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Sobarzo-Sánchez, E.; Uriarte, E. In Silico Prediction of P-Glycoprotein Binding: Insights from Molecular Docking Studies. Curr. Med. Chem. 2019, 26, 1746–1760. [Google Scholar] [CrossRef] [PubMed]
- Montanari, F.; Ecker, G.F. Prediction of Drug-ABC-Transporter Interaction—Recent Advances and Future Challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Helena Vasconcelos, M.; Pinto, M.; Fernandes, M. Structure and Ligand-Based Design of P-Glycoprotein Inhibitors: A Historical Perspective. Curr. Pharm. Des. 2012, 18, 4197–4214. [Google Scholar] [CrossRef]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New Uses for Old Drugs: Pharmacophore-Based Screening for the Discovery of P-Glycoprotein Inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef]
- Ecker, G.F.; Stockner, T.; Chiba, P. Computational Models for Prediction of Interactions with ABC-Transporters. Drug Discov. Today 2008, 13, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Ghaemian, P.; Shayanfar, A. Image-Based QSAR Model for the Prediction of P-Gp Inhibitory Activity of Epigallocatechin and Gallocatechin Derivatives. Curr. Comput. Aided. Drug Des. 2019, 15, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Parveen, Z.; Brunhofer, G.; Jabeen, I.; Erker, T.; Chiba, P.; Ecker, G.F. Synthesis, Biological Evaluation and 3D-QSAR Studies of New Chalcone Derivatives as Inhibitors of Human P-Glycoprotein. Bioorg. Med. Chem. 2014, 22, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Dehghani-Ghahnaviyeh, S.; Kapoor, K.; Tajkhorshid, E. Conformational Changes in the Nucleotide-Binding Domains of P-Glycoprotein Induced by ATP Hydrolysis. FEBS Lett. 2021, 595, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure-Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362. [Google Scholar] [CrossRef] [PubMed]
- Kopcho, N.; Chang, G.; Komives, E.A. Dynamics of ABC Transporter P-Glycoprotein in Three Conformational States. Sci. Rep. 2019, 9, 15092. [Google Scholar] [CrossRef] [PubMed]
- Vandevuer, S.; Van Bambeke, F.; Tulkens, P.M.; Prévost, M. Predicting the Three-Dimensional Structure of Human P-Glycoprotein in Absence of ATP by Computational Techniques Embodying Crosslinking Data: Insight into the Mechanism of Ligand Migration and Binding Sites. Proteins Struct. Funct. Genet. 2006, 63, 466–478. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural Insight into Substrate and Inhibitor Discrimination by Human P-Glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Domicevica, L.; Biggin, P.C. Homology Modelling of Human P-Glycoprotein. Biochem. Soc. Trans. 2015, 43, 952–958. [Google Scholar] [CrossRef]
- Didziapetris, R.; Japertas, P.; Avdeef, A.; Petrauskas, A. Classification Analysis of P-Glycoprotein Substrate Specificity. J. Drug Target. 2003, 11, 391–406. [Google Scholar] [CrossRef]
- Gombar, V.K.; Polli, J.W.; Humphreys, J.E.; Wring, S.A.; Serabjit-Singh, C.S. Predicting P-Glycoprotein Substrates by a Quantitative Structure-Activity Relationship Model. J. Pharm. Sci. 2004, 93, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, L.; Li, Y.; Tian, S.; Sun, H.; Hou, T. ADMET Evaluation in Drug Discovery. 13. Development of in Silico Prediction Models for p-Glycoprotein Substrates. Mol. Pharm. 2014, 11, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.V.; Sawada, G.A.; Watson, I.A.; Raub, T.J. Integration of in Silico and in Vitro Tools for Scaffold Optimization during Drug Discovery: Predicting P-Glycoprotein Efflux. Mol. Pharm. 2013, 10, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Swedrowska, M.; Jamshidi, S.; Kumar, A.; Kelly, C.; Rahman, K.M.; Forbes, B. In Silico and in Vitro Screening for P-Glycoprotein Interaction with Tenofovir, Darunavir, and Dapivirine: An Antiretroviral Drug Combination for Topical Prevention of Colorectal HIV Transmission. Mol. Pharm. 2017, 14, 2660–2669. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Esaki, T.; Ohashi, R.; Kuroda, M.; Kawashima, H.; Komura, H.; Natsume-Kitatani, Y.; Mizuguchi, K. Development of an in Silico Prediction Model for P-Glycoprotein Efflux Potential in Brain Capillary Endothelial Cells toward the Prediction of Brain Penetration. J. Med. Chem. 2021, 64, 2725–2738. [Google Scholar] [CrossRef]
- Guéniche, N.; Huguet, A.; Bruyere, A.; Habauzit, D.; Le Hégarat, L.; Fardel, O. Comparative in Silico Prediction of P-Glycoprotein-Mediated Transport for 2010-2020 US FDA-Approved Drugs Using Six Web-Tools. Biopharm. Drug Dispos. 2021, 42, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Li, Y.; Yang, S.L.; Yang, L. An in Silico Approach for Screening Flavonoids as P-Glycoprotein Inhibitors Based on a Bayesian-Regularized Neural Network. J. Comput. Aided. Mol. Des. 2005, 19, 137–147. [Google Scholar] [CrossRef]
- Jara, G.E.; Vera, D.M.A.; Pierini, A.B. Binding of Modulators to Mouse and Human Multidrug Resistance P-Glycoprotein. A Computational Study. J. Mol. Graph. Model. 2013, 46, 10–21. [Google Scholar] [CrossRef]
- Brewer, F.K.; Follit, C.A.; Vogel, P.D.; Wise, J.G. In Silico Screening for Inhibitors of P-Glycoprotein That Target the Nucleotide Binding Domains. Mol. Pharmacol. 2014, 86, 716–726. [Google Scholar] [CrossRef]
- Morsy, M.A.; El-Sheikh, A.A.K.; Ibrahim, A.R.N.; Khedr, M.A.; Al-Taher, A.Y. In Silico Comparisons between Natural Inhibitors of ABCB1/P-Glycoprotein to Overcome Doxorubicin-Resistance in the NCI/ADR-RES Cell Line. Eur. J. Pharm. Sci. 2018, 112, 87–94. [Google Scholar] [CrossRef]
- Ramos, P.; Schmitz, M.; Gama, S.; Portantiolo, A.; Durruthy, M.G.; de Souza Votto, A.P.; Cornetet, L.R.; dos Santos Machado, K.; Werhli, A.; Tonel, M.Z.; et al. Cytoprotection of Lipoic Acid against Toxicity Induced by Saxitoxin in Hippocampal Cell Line HT-22 through in Silico Modeling and in Vitro Assays. Toxicology 2018, 393, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, S.; Sahebkar, A.; Kalalinia, F.; Behravan, J.; Hadizadeh, F. Synthesis, in Silico and in Vitro Studies of New 1,4-Dihydropiridine Derivatives for Antitumor and P-Glycoprotein Inhibitory Activity. Bioorg. Chem. 2019, 91, 103156. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, S.; Hadizadeh, F.; Ferreira, R.J. Theoretical Studies on 1,4-Dihydropyridine Derivatives as P-Glycoprotein Allosteric Inhibitors: Insights on Symmetry and Stereochemistry. J. Biomol. Struct. Dyn. 2021, 39, 4752–4763. [Google Scholar] [CrossRef] [PubMed]
- Di Sotto, A.; Irannejad, H.; Eufemi, M.; Mancinelli, R.; Abete, L.; Mammola, C.L.; Altieri, F.; Mazzanti, G.; Di Giacomo, S. Potentiation of Low-Dose Doxorubicin Cytotoxicity by Affecting p-Glycoprotein through Caryophyllane Sesquiterpenes in Hepg2 Cells: An in Vitro and in Silico Study. Int. J. Mol. Sci. 2020, 21, 633. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Júnior, O.S.; Franco, C.D.J.P.; de Moraes, A.A.B.; Cruz, J.N.; da Costa, K.S.; do Nascimento, L.D.; de Aguiar Andrade, E.H. In Silico Analyses of Toxicity of the Major Constituents of Essential Oils from Two Ipomoea L. Species. Toxicon 2021, 195, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Durães, F.; Palmeira, A.; Cruz, B.; Freitas-Silva, J.; Szemerédi, N.; Gales, L.; da Costa, P.M.; Remião, F.; Silva, R.; Pinto, M.; et al. Antimicrobial Activity of a Library of Thioxanthones and Their Potential as Efflux Pump Inhibitors. Pharmaceuticals 2021, 14, 572. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.J.; Beh, R.C.; Prestedge, J.; Catak, S.; Hung, A.; Karagiannis, T.C. Identification of Novel Bioactive Compounds from Olea Europaea by Evaluation of Chemical Compounds in the OliveNetTM Library: In Silico Bioactivity and Molecular Modelling, and in Vitro Validation of HERG Activity. Comput. Biol. Med. 2022, 142, 105247. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; Barbosa, D.J.; Palmeira, A.; Sousa, E.; Carvalho, F.; De Lourdes Bastos, M.; Remião, F. Colchicine Effect on P-Glycoprotein Expression and Activity: In Silico and in Vitro Studies. Chem. Biol. Interact. 2014, 218, 50–62. [Google Scholar] [CrossRef]
- Wongrattanakamon, P.; Lee, V.S.; Nimmanpipug, P.; Jiranusornkul, S. 3D-QSAR Modelling Dataset of Bioflavonoids for Predicting the Potential Modulatory Effect on P-Glycoprotein Activity. Data Br. 2016, 9, 35–42. [Google Scholar] [CrossRef]
- Silva, R.; Palmeira, A.; Carmo, H.; Barbosa, D.J.; Gameiro, M.; Gomes, A.; Paiva, A.M.; Sousa, E.; Pinto, M.; Bastos, M.d.L.; et al. P-Glycoprotein Induction in Caco-2 Cells by Newly Synthetized Thioxanthones Prevents Paraquat Cytotoxicity. Arch. Toxicol. 2015, 89, 1783–1800. [Google Scholar] [CrossRef]
- Young, J.; Garikipati, N.; Durrant, J.D. BINANA 2: Characterizing Receptor/Ligand Interactions in Python and Java Script. J. Chem. Inf. Model. 2022, 62, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Overview of Transporters in Pharmacokinetics and Drug Discovery. Curr. Protoc. Pharmacol. 2018, 82, e46. [Google Scholar] [CrossRef] [PubMed]
- Saaby, L.; Brodin, B. A Critical View on In Vitro Analysis of P-Glycoprotein (P-Gp) Transport Kinetics. J. Pharm. Sci. 2017, 106, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Dunn, R.T.; Jayadev, S.; DiSorbo, O.; Pack, F.D.; Farr, S.B.; Stoll, R.E.; Blanchard, K.T. Assessment of Cisplatin-Induced Nephrotoxicity by Microarray Technology. Toxicol. Sci. 2001, 63, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, I.V.; Pande, P.; Patton, W.F. Sensitive and Specific Fluorescent Probes for Functional Analysis of the Three Major Types of Mammalian ABC Transporters. PLoS ONE 2011, 6, e22429. [Google Scholar] [CrossRef] [PubMed]
- Nieri, P.; Romiti, N.; Adinolfi, B.; Chicca, A.; Massarelli, I.; Chieli, E. Modulation of P-Glycoprotein Activity by Cannabinoid Molecules in HK-2 Renal Cells. Br. J. Pharmacol. 2006, 148, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Carmo, H.; Dinis-Oliveira, R.; Cordeiro-Da-Silva, A.; Lima, S.C.; Carvalho, F.; De Lourdes Bastos, M.; Remião, F. In Vitro Study of P-Glycoprotein Induction as an Antidotal Pathway to Prevent Cytotoxicity in Caco-2 Cells. Arch. Toxicol. 2011, 85, 315–326. [Google Scholar] [CrossRef]
- Silva, R.; Sousa, E.; Carmo, H.; Palmeira, A.; Barbosa, D.J.; Gameiro, M.; Pinto, M.; De Lourdes Bastos, M.; Remião, F. Induction and Activation of P-Glycoprotein by Dihydroxylated Xanthones Protect against the Cytotoxicity of the P-Glycoprotein Substrate Paraquat. Arch. Toxicol. 2014, 88, 937–951. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Silva, R.; Gaio, A.R.; Martins, A.M.; Lima, S.C.; Cordeiro-da-Silva, A.; de Lourdes Bastos, M.; Remião, F. P-Glycoprotein Activity in Human Caucasian Male Lymphocytes Does Not Follow Its Increased Expression during Aging. Cytom. Part A 2011, 79, 912–919. [Google Scholar] [CrossRef]
- Adan, A.; Kiraz, Y.; Baran, Y. Cell Proliferation and Cytotoxicity Assays. Curr. Pharm. Biotechnol. 2016, 17, 1213–1221. [Google Scholar] [CrossRef]
- Silva, R.; Carmo, H.; Vilas-Boas, V.; de Pinho, P.G.; Dinis-Oliveira, R.J.; Carvalho, F.; Silva, I.; Correia-de-Sá, P.; Bastos, M.d.L.; Remião, F. Doxorubicin Decreases Paraquat Accumulation and Toxicity in Caco-2 Cells. Toxicol. Lett. 2013, 217, 34–41. [Google Scholar] [CrossRef]
- Nikzad, S.; Baradaran-Ghahfarokhi, M.; Nasri, P. Dose-Response Modeling Using MTT Assay: A Short Review. Life Sci. J. 2014, 11, 7–8. [Google Scholar]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral Red Uptake Assay for the Estimation of Cell Viability/Cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Crean, D.; Bellwon, P.; Aschauer, L.; Limonciel, A.; Moenks, K.; Hewitt, P.; Schmidt, T.; Herrgen, K.; Dekant, W.; Lukas, A.; et al. Development of an in Vitro Renal Epithelial Disease State Model for Xenobiotic Toxicity Testing. Toxicol. Vitr. 2015, 30, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Valente, M.J.; Araújo, A.M.; Silva, R.; Bastos, M.d.L.; Carvalho, F.; Guedes de Pinho, P.; Carvalho, M. 3,4-Methylenedioxypyrovalerone (MDPV): In Vitro Mechanisms of Hepatotoxicity under Normothermic and Hyperthermic Conditions. Arch. Toxicol. 2016, 90, 1959–1973. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. In Vitro Blood–Brain Barrier Models: Current and Perspective Technologies. J. Pharm. Sci. 2012, 101, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.O. The HCMEC/D3 Cell Line as a Model of the Human Blood Brain Barrier. Fluids Barriers CNS 2013, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Gómez-Zepeda, D.; Auvity, S.; Menet, M.C.; Crété, D.; Labat, L.; Remião, F.; Cisternino, S.; Declèves, X. Effect of Subchronic Intravenous Morphine Infusion and Naloxone-Precipitated Morphine Withdrawal on P-Gp and Bcrp at the Rat Blood-Brain Barrier. J. Pharm. Sci. 2016, 105, 350–358. [Google Scholar] [CrossRef]
- Soldatow, V.Y.; Lecluyse, E.L.; Griffith, L.G.; Rusyn, I. In Vitro Models for Liver Toxicity Testing. Toxicol. Res. 2013, 2, 23–39. [Google Scholar] [CrossRef]
- Le Vee, M.; Jigorel, E.; Glaise, D.; Gripon, P.; Guguen-Guillouzo, C.; Fardel, O. Functional Expression of Sinusoidal and Canalicular Hepatic Drug Transporters in the Differentiated Human Hepatoma HepaRG Cell Line. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2006, 28, 109–117. [Google Scholar] [CrossRef]
- Carabias, P.; Espelt, M.V.; Bacigalupo, M.L.; Rojas, P.; Sarrias, L.; Rubin, A.; Saffioti, N.A.; Elola, M.T.; Rossi, J.P.; Wolfenstein-Todel, C.; et al. Galectin-1 Confers Resistance to Doxorubicin in Hepatocellular Carcinoma Cells through Modulation of P-Glycoprotein Expression. Cell Death Dis. 2022, 13, 79. [Google Scholar] [CrossRef]
- Romiti, N.; Tongiani, R.; Cervelli, F.; Chieli, E. Effects of Curcumin on P-Glycoprotein in Primary Cultures of Rat Hepatocytes. Life Sci. 1998, 62, 2349–2358. [Google Scholar] [CrossRef]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An Immortalized Proximal Tubule Epithelial Cell Line from Normal Adult Human Kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, T.; Kinjo, M.; Dowling, T.C. Determination of Rhodamine 123 in Cell Lysate by HPLC with Visible Wavelength Detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 814, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, L.; Fæste, C.K.; Solhaug, A. Role of P-Glycoprotein in Deoxynivalenol-Mediated in Vitro Toxicity. Toxicol. Lett. 2018, 284, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Vormann, M.K.; Gijzen, L.; Hutter, S.; Boot, L.; Nicolas, A.; van den Heuvel, A.; Vriend, J.; Ng, C.P.; Nieskens, T.T.G.; van Duinen, V.; et al. Nephrotoxicity and Kidney Transport Assessment on 3D Perfused Proximal Tubules. AAPS J. 2018, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Nieskens, T.T.G.; Vormann, M.K.; van den Berge, B.T.; van den Heuvel, A.; Russel, F.G.M.; Suter-Dick, L.; Lanz, H.L.; Vulto, P.; Masereeuw, R.; et al. Screening of Drug-Transporter Interactions in a 3D Microfluidic Renal Proximal Tubule on a Chip. AAPS J. 2018, 20, 87. [Google Scholar] [CrossRef] [PubMed]
- Zweibaum, A.; Laburthe, M.; Grasset, E.; Louvard, D. Intestinal Absorption and Secretion. In Handbook of Physiolgy; Oxford University Press: Oxford, MI, USA, 1991. [Google Scholar]
- Ezuruike, U.F.; Chieli, E.; Prieto, J.M. In Vitro Modulation of Glibenclamide Transport by P-Glycoprotein Inhibitory Antidiabetic African Plant Extracts. Planta Med. 2019, 85, 987–996. [Google Scholar] [CrossRef]
- Silva, V.; Gil-Martins, E.; Rocha-Pereira, C.; Lemos, A.; Palmeira, A.; Puthongking, P.; Sousa, E.; de Lourdes Bastos, M.; Remião, F.; Silva, R. Oxygenated Xanthones as P-Glycoprotein Modulators at the Intestinal Barrier: In Vitro and Docking Studies. Med. Chem. Res. 2020, 29, 1041–1057. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Silva, R.; Palmeira, A.; Sousa, E.; Ferreira, L.M.; Branco, P.S.; Carvalho, F.; de Bastos, M.L.; Remião, F. Development of Novel Rifampicin-Derived P-Glycoprotein Activators/Inducers. Synthesis, In Silico Analysis and Application in the RBE4 Cell Model, Using Paraquat as Substrate. PLoS ONE 2013, 8, e74425. [Google Scholar] [CrossRef]
- Saaby, L.; Helms, H.C.C.; Brodin, B. IPEC-J2 MDR1, a Novel High-Resistance Cell Line with Functional Expression of Human P-Glycoprotein (ABCB1) for Drug Screening Studies. Mol. Pharm. 2016, 13, 640–652. [Google Scholar] [CrossRef] [PubMed]
- Ozgür, B.; Saaby, L.; Langthaler, K.; Brodin, B. Characterization of the IPEC-J2 MDR1 (IP-Gp) Cell Line as a Tool for Identification of P-Gp Substrates. Eur. J. Pharm. Sci. 2018, 112, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Van de Water, F.M.; Boleij, J.M.; Peters, J.G.P.; Russel, F.G.M.; Masereeuw, R. Characterization of P-Glycoprotein and Multidrug Resistance Proteins in Rat Kidney and Intestinal Cell Lines. Eur. J. Pharm. Sci. 2007, 30, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yin, H.; Bai, P.; Miao, P.; Deng, X.; Xu, Y.; Hu, J.; Yin, J. ABC Transporters Affect the Elimination and Toxicity of CdTe Quantum Dots in Liver and Kidney Cells. Toxicol. Appl. Pharmacol. 2016, 303, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; de Graaf, I.A.M.; de Jager, M.H.; Groothuis, G.M.M. P-Gp Activity and Inhibition in the Different Regions of Human Intestine Ex Vivo. Biopharm. Drug Dispos. 2017, 38, 127–138. [Google Scholar] [CrossRef]
- Novak, A.; Carpini, G.D.; Ruiz, M.L.; Luquita, M.G.; Rubio, M.C.; Mottino, A.D.; Ghanem, C.I. Acetaminophen Inhibits Intestinal P-Glycoprotein Transport Activity. J. Pharm. Sci. 2013, 102, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Godoy, Y.C.; Martinez, S.A.; Ghanem, C.I.; Celuch, S.M. Fructose-Induced Metabolic Syndrome Decreases Protein Expression and Activity of Intestinal P-Glycoprotein. Nutrition 2015, 31, 871–876. [Google Scholar] [CrossRef]
- Rocha-Pereira, C.; Ghanem, C.I.; Silva, R.; Casanova, A.G.; Duarte-Araújo, M.; Gonçalves-Monteiro, S.; Sousa, E.; Bastos, M.D.L.; Remião, F. P-Glycoprotein Activation by 1-(Propan-2-Ylamino)-4-Propoxy-9H-Thioxanthen-9-One (TX5) in Rat Distal Ileum: Ex Vivo and in Vivo Studies. Toxicol. Appl. Pharmacol. 2020, 386, 114832. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Animal Research: Reporting in Vivo Experiments: The ARRIVE Guidelines. Br. J. Pharmacol. 2010, 160, 1577–1579. [Google Scholar] [CrossRef]
- Hernández-Lozano, I.; Mairinger, S.; Filip, T.; Sauberer, M.; Wanek, T.; Stanek, J.; Sake, J.A.; Pekar, T.; Ehrhardt, C.; Langer, O. PET Imaging to Assess the Impact of P-Glycoprotein on Pulmonary Drug Delivery in Rats. J. Control. Release 2022, 342, 44–52. [Google Scholar] [CrossRef]
- De Bruyne, S.; Wyffels, L.; Boos, T.L.; Staelens, S.; Deleye, S.; Rice, K.C.; De Vos, F. In Vivo Evaluation of [123I]-4-(2-(Bis(4-Fluorophenyl)Methoxy)Ethyl)-1-(4-Iodobenzyl)Piperidine, an Iodinated SPECT Tracer for Imaging the P-Gp Transporter. Nucl. Med. Biol. 2010, 37, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Ceré, L.I.; Sedlmeier, M.G.; Semeniuk, M.; Luquita, M.G.; Francés, D.; Ronco, M.T.; Rigalli, J.P.; Ruiz, M.L.; Catania, V.A. Induction of P-Glycoprotein Expression and Activity by Prolactin in Female Rat Liver. Life Sci. 2021, 287, 119936. [Google Scholar] [CrossRef] [PubMed]
- Takeda, F.; Oda, M.; Terasaki, M.; Ichimura, Y.; Kojima, H.; Saitoh, H. Downregulated Expression of Intestinal P-Glycoprotein in Rats with Cisplatin-Induced Acute Kidney Injury Causes Amplification of Its Transport Capacity to Maintain “Gatekeeper” Function. Toxicol. Appl. Pharmacol. 2021, 423, 115570. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.S.; Hassan, M.A.; Kondo, T. Effect of Lyophilized Grapefruit Juice on P-Glycoprotein-Mediated Drug Transport in-Vitro and in-Vivo. Drug Dev. Ind. Pharm. 2015, 41, 375–381. [Google Scholar] [CrossRef]
- Weinheimer, M.; Fricker, G.; Burhenne, J.; Mylius, P.; Schubert, R. The Application of P-Gp Inhibiting Phospholipids as Novel Oral Bioavailability Enhancers—An in Vitro and in Vivo Comparison. Eur. J. Pharm. Sci. 2017, 108, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Ballent, M.; Lifschitz, A.; Virkel, G.; Sallovitz, J.; Lanusse, C. Modulation of the P-glycoprotein-mediated intestinal secretion of ivermectin: In vitro and in vivo assessments. Drug Metab. Dispos. 2006, 34, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Srirangam, P.; Vidya Sagar, J. Modulation of the P-Glycoproein-Mediated Intestinal Secretion of Glibenclamide: In Vitro and in Vivo Assessments. J. Young Pharm. 2010, 2, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.P.; Huang, C.Y.; Lin, S.P.; Hou, Y.C. Activation of P-Glycoprotein and CYP 3A by Coptidis Rhizoma in Vivo: Using Cyclosporine as a Probe Substrate in Rats. J. Food Drug Anal. 2018, 26, S125–S132. [Google Scholar] [CrossRef]
- Bedada, S.K.; Appani, R.; Boga, P.K. Capsaicin Pretreatment Enhanced the Bioavailability of Fexofenadine in Rats by P-Glycoprotein Modulation: In Vitro, in Situ and in Vivo Evaluation. Drug Dev. Ind. Pharm. 2017, 43, 932–938. [Google Scholar] [CrossRef]
- Kukreja, J.B.; Thompson, I.M.J.; Chapin, B.F. Organizing a Clinical Trial for the New Investigator. Urol. Oncol. 2019, 37, 336–339. [Google Scholar] [CrossRef]
- Rengelshausen, J.; Göggelmann, C.; Burhenne, J.; Riedel, K.-D.; Ludwig, J.; Weiss, J.; Mikus, G.; Walter-Sack, I.; Haefeli, W.E. Contribution of Increased Oral Bioavailability and Reduced Nonglomerular Renal Clearance of Digoxin to the Digoxin-Clarithromycin Interaction. Br. J. Clin. Pharmacol. 2003, 56, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Chieli, E.; Romiti, N.; Rodeiro, I.; Garrido, G. In Vitro Effects of Mangifera Indica and Polyphenols Derived on ABCB1/P-Glycoprotein Activity. Food Chem. Toxicol. 2009, 47, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Zhang, S.; Hu, T.; Qu, X.; Zhai, J.; Zhang, Y.; Tao, L.; Yin, J.; Song, Y. Omeprazole Protects against Cisplatin-Induced Nephrotoxicity by Alleviating Oxidative Stress, Inflammation, and Transporter-Mediated Cisplatin Accumulation in Rats and HK-2 cells. Chem. Biol. Interact. 2019, 297, 130–140. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Müller, F.; Fromm, M.F. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [PubMed]
- Momper, J.D.; Yang, J.; Gockenbach, M.; Vaida, F.; Nigam, S.K. Dynamics of Organic Anion Transporter-Mediated Tubular Secretion during Postnatal Human Kidney Development and Maturation. Clin. J. Am. Soc. Nephrol. 2019, 14, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, N.; Li, F.; Zhang, G.; He, S.; Zhu, Y.; Ou, R.; Li, N.; Liu, S.; Feng, L.; et al. Induction of P-Glycoprotein Expression and Activity by Aconitum Alkaloids: Implication for Clinical Drug-Drug Interactions. Sci. Rep. 2016, 6, 25343. [Google Scholar] [CrossRef] [PubMed]
- Masereeuw, R.; Russel, F.G.M. Regulatory Pathways for ATP-Binding Cassette Transport Proteins in Kidney Proximal Tubules. AAPS J. 2012, 14, 883–894. [Google Scholar] [CrossRef]
- Lopes, A.; Martins, E.; Silva, R.; Pinto, M.M.M.; Remião, F.; Sousa, E.; Fernandes, C. Chiral Thioxanthones as Modulators of P-Glycoprotein: Synthesis and Enantioselectivity Studies. Molecules 2018, 23, 626. [Google Scholar] [CrossRef]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-Glycoprotein Transport System and Cardiovascular Drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual Inhibitors of P-Glycoprotein and Tumor Cell Growth: (Re)Discovering Thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Ambudkar, S. V Drug-Protein Hydrogen Bonds Govern the Inhibition of the ATP Hydrolysis of the Multidrug Transporter P-Glycoprotein. Biochem. Pharmacol. 2016, 101, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Küng, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a Zosuquidar and UIC2-Bound Human-Mouse Chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Matos, J.; Remião, F.; Morales, A.I. Pharmacokinetics and Toxicokinetics Roles of Membrane Transporters at Kidney Level. J. Pharm. Pharm. Sci. 2020, 23, 333–356. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Láng, I.; Smolyarchuk, E.A.; Chung, C.H.; Plotka, A.; Shi, H.; Wang, D. Evaluation of the Effect of P-Glycoprotein Inhibition and Induction on Talazoparib Disposition in Patients with Advanced Solid Tumours. Br. J. Clin. Pharmacol. 2020, 86, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Zakeri-milani, P.; Valizadeh, H. Intestinal Transporters: Absorption through p-Gp Interactions. Expert Opin. Drug Metab. Toxicol. 2014, 10, 859–871. [Google Scholar] [CrossRef]
- Taubert, D.; von Beckerath, N.; Grimberg, G.; Lazar, A.; Jung, N.; Goeser, T.; Kastrati, A.; Schömig, A.; Schömig, E. Impact of P-Glycoprotein on Clopidogrel Absorption. Clin. Pharmacol. Ther. 2006, 80, 486–501. [Google Scholar] [CrossRef]
- Shen, Q.; Lin, Y.; Handa, T.; Doi, M.; Sugie, M.; Wakayama, K.; Okada, N.; Fujita, T.; Yamamoto, A. Modulation of Intestinal P-Glycoprotein Function by Polyethylene Glycols and Their Derivatives by in Vitro Transport and in Situ Absorption Studies. Int. J. Pharm. 2006, 313, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Ampasavate, C.; Sotanaphun, U.; Phattanawasin, P.; Piyapolrungroj, N. Effects of Curcuma Spp. on P-Glycoprotein Function. Phytomedicine 2010, 17, 506–512. [Google Scholar] [CrossRef]
- Costa, J.; Almonti, V.; Cacopardo, L.; Poli, D.; Rapposelli, S.; Ahluwalia, A. Investigating Curcumin/Intestinal Epithelium Interaction in a Millifluidic Bioreactor. Bioengineering 2020, 7, 100. [Google Scholar] [CrossRef]
- Mahmoud, N.; Hegazy, M.E.F.; Wadie, W.; Elbadawi, M.; Fleischer, E.; Klinger, A.; Bringmann, G.; Khayyal, M.T.; Efferth, T. Naphthoquinone Derivatives as P-Glycoprotein Inducers in Inflammatory Bowel Disease: 2D Monolayers, 3D Spheroids, and in Vivo Models. Pharmacol. Res. 2022, 179, 106233. [Google Scholar] [CrossRef]
- Silva, V.; Gil-Martins, E.; Silva, B.; Rocha-Pereira, C.; Sousa, M.E.; Remião, F.; Silva, R. Xanthones as P-Glycoprotein Modulators and Their Impact on Drug Bioavailability. Expert Opin. Drug Metab. Toxicol. 2021, 17, 441–482. [Google Scholar] [CrossRef] [PubMed]
- Barone, G.W.; Gurley, B.J.; Ketel, B.L.; Lightfoot, M.L.; Abul-Ezz, S.R. Drug Interaction between St. John’s Wort and Cyclosporine. Ann. Pharmacother. 2000, 34, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Contino, M.; Guglielmo, S.; Riganti, C.; Antonello, G.; Perrone, M.G.; Giampietro, R.; Rolando, B.; Fruttero, R.; Colabufo, N.A. One Molecule Two Goals: A Selective P-Glycoprotein Modulator Increases Drug Transport across Gastro-Intestinal Barrier and Recovers Doxorubicin Toxicity in Multidrug Resistant Cancer Cells. Eur. J. Med. Chem. 2020, 208, 112843. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Amidon, G.L. Grapefruit Juice and Its Constituents Augment Colchicine Intestinal Absorption: Potential Hazardous Interaction and the Role of P-Glycoprotein. Pharm. Res. 2009, 26, 883–892. [Google Scholar] [CrossRef]
- Wang, Y.H.; Chao, P.D.L.; Hsiu, S.L.; Wen, K.C.; Hou, Y.C. Lethal Quercetin-Digoxin Interaction in Pigs. Life Sci. 2004, 74, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.B.; Kahnt, A.; Dillen, L.; Wuyts, K.; Snoeys, J.; Nielsen, U.G.; Holm, R.; Nielsen, C.U. Montmorillonite-Surfactant Hybrid Particles for Modulating Intestinal P-Glycoprotein-Mediated Transport. Int. J. Pharm. 2019, 571, 118696. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Lim, D.Y.; Lee, C.H.; Jeon, J.H.; Choi, M.K.; Song, I.S. Enhanced Intestinal Absorption and Pharmacokinetic Modulation of Berberine and Its Metabolites through the Inhibition of P-Glycoprotein and Intestinal Metabolism in Rats Using a Berberine Mixed Micelle Formulation. Pharmaceutics 2020, 12, 882. [Google Scholar] [CrossRef]
- Alshogran, O.Y.; Al Ghraiybah, N.F.; Al-Azzam, S.I. Evaluation of the Effect of Isobutyl Paraben and 2-Ethyl Hexyl Paraben on P-Glycoprotein Functional Expression in Rats: A Pharmacokinetic Study. Curr. Mol. Pharmacol. 2022, 15, 987–995. [Google Scholar] [CrossRef]
- Bhutto, Z.A.; He, F.; Zloh, M.; Yang, J.; Huang, J.; Guo, T.; Wang, L. Use of Quercetin in Animal Feed: Effects on the P-Gp Expression and Pharmacokinetics of Orally Administrated Enrofloxacin in Chicken. Sci. Rep. 2018, 8, 4400. [Google Scholar] [CrossRef]
- Do Nascimento, S.B.; de Lima Nascimento, M.; de Araújo, L.L.; de Oliveira, F.M.; do Carmo Vieira, M.; Duarte-Almeida, J.M.; Siqueira, J.M.; da Costa César, I.; Derendorf, H.; de Castro, W.V. Evaluation of the Effects of Maytenus Ilicifolia on the Activities of Cytochrome P450 3A and P-Glycoprotein. Curr. Drug Metab. 2020, 21, 281–290. [Google Scholar] [CrossRef]
- Houshaymi, B.; Nasreddine, N.; Kedees, M.; Soayfane, Z. Oleic Acid Increases Uptake and Decreases the P-Gp-Mediated Efflux of the Veterinary Anthelmintic Ivermectin. Drug Res. 2019, 69, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kan, J.W.Y.; Yan, C.S.W.; Wong, I.L.K.; Su, X.; Liu, Z.; Chan, T.H.; Chow, L.M.C. Discovery of a Flavonoid FM04 as a Potent Inhibitor to Reverse P-Glycoprotein-Mediated Drug Resistance in Xenografts and Improve Oral Bioavailability of Paclitaxel. Int. J. Mol. Sci. 2022, 23, 15299. [Google Scholar] [CrossRef] [PubMed]
- Pala Kara, Z.; Ozturk Civelek, D.; Ozturk, N.; Okyar, A. The Effects of P-Glycoprotein Inhibitor Zosuquidar on the Sex and Time-Dependent Pharmacokinetics of Parenterally Administered Talinolol in Mice. Eur. J. Pharm. Sci. 2021, 156, 105589. [Google Scholar] [CrossRef] [PubMed]
- Radi, Z.A. Kidney Pathophysiology, Toxicology, and Drug-Induced Injury in Drug Development. Int. J. Toxicol. 2019, 38, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.E.; Rankin, G.O. Human Renal Organic Anion Transporters: Characteristics and Contributions to Drug and Drug Metabolite Excretion. Pharmacol. Ther. 2006, 109, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Augustine, L.M.; Scheffer, G.L.; Cherrington, N.J.; Manautou, J.E. Renal Xenobiotic Transporters Are Differentially Expressed in Mice Following Cisplatin Treatment. Toxicology 2008, 250, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.D.A.; Sayer, R.; Windass, A.S.; Haslam, I.S.; De Broe, M.E.; D’Haese, P.C.; Verhulst, A. Characterisation of Human Tubular Cell Monolayers as a Model of Proximal Tubular Xenobiotic Handling. Toxicol. Appl. Pharmacol. 2008, 233, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Ramalakshmi, S.; Bastacky, S.; Johnson, J.P. Levofloxacin-Induced Granulomatous Interstitial Nephritis. Am. J. Kidney Dis. 2003, 41, e7.1–e7.5. [Google Scholar] [CrossRef]
- Kotowski, M.J.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Tejchman, K.; Dziewanowski, K.; Ostrowski, M.; Czerny, B.; Grzeskowiak, E.; Machalinski, B.; Sienko, J. Effect of Multidrug-Resistant 1 (MDR1) and CYP3A4*1B Polymorphisms on Cyclosporine-Based Immunosuppressive Therapy in Renal Transplant Patients. Ann. Transplant. 2019, 24, 108–114. [Google Scholar] [CrossRef]
- Orr, S.E.; Bridges, C.C. Chronic Kidney Disease and Exposure to Nephrotoxic Metals. Int. J. Mol. Sci. 2017, 18, 1039. [Google Scholar] [CrossRef]
- Tramonti, G.; Romiti, N.; Norpoth, M.; Chieli, E. P-Glycoprotein in HK-2 Proximal Tubule Cell Line. Ren. Fail. 2001, 23, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Romiti, N.; Tramonti, G.; Chieli, E. Influence of Different Chemicals on MDR-1 P-Glycoprotein Expression and Activity in the HK-2 Proximal Tubular Cell Line. Toxicol. Appl. Pharmacol. 2002, 183, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Romiti, N.; Tramonti, G.; Donati, A.; Chieli, E. Effects of Grapefruit Juice on the Multidrug Transporter P-Glycoprotein in the Human Proximal Tubular Cell Line HK-2. Life Sci. 2004, 76, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Romiti, N.; Pellati, F.; Nieri, P.; Benvenuti, S.; Adinolfi, B.; Chieli, E. P-Glycoprotein Inhibitory Activity of Lipophilic Constituents of Echinacea Pallida Roots in a Human Proximal Tubular Cell Line. Planta Med. 2008, 74, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Romiti, N.; Tramonti, G.; Corti, A.; Chieli, E. Effects of Devil’s Claw (Harpagophytum Procumbens) on the Multidrug Transporter ABCB1/P-Glycoprotein. Phytomedicine 2009, 16, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Chieli, E.; Romiti, N.; Rodeiro, I.; Garrido, G. In Vitro Modulation of ABCB1/P-Glycoprotein Expression by Polyphenols from Mangifera Indica. Chem. Biol. Interact. 2010, 186, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Chieli, E.; Romiti, N.; Catiana Zampini, I.; Garrido, G.; Inés Isla, M. Effects of Zuccagnia Punctata Extracts and Their Flavonoids on the Function and Expression of ABCB1/P-Glycoprotein Multidrug Transporter. J. Ethnopharmacol. 2012, 144, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yan, M.; Cao, L.; Fang, P.; Guo, Z.; Hou, Z.; Zhang, B. Glycyrrhetinic Acid Accelerates the Clearance of Triptolide through P-Gp In Vitro. Phyther. Res. 2017, 31, 1090–1096. [Google Scholar] [CrossRef]
- Chow, E.C.Y.; Durk, M.R.; Cummins, C.L.; Pang, K.S. 1α,25-Dihydroxyvitamin D3 up-Regulates P-Glycoprotein via the Vitamin D Receptor and Not Farnesoid X Receptor in Both Fxr(-/-) and Fxr(+/+) Mice and Increased Renal and Brain Efflux of Digoxin in Mice in Vivo. J. Pharmacol. Exp. Ther. 2011, 337, 846–859. [Google Scholar] [CrossRef]
- Im, D.S.; Shin, H.J.; Yang, K.J.; Jung, S.Y.; Song, H.Y.; Hwang, H.S.; Gil, H.-W. Cilastatin Attenuates Vancomycin-Induced Nephrotoxicity via P-Glycoprotein. Toxicol. Lett. 2017, 277, 9–17. [Google Scholar] [CrossRef]
- Kanado, Y.; Tsurudome, Y.; Omata, Y.; Yasukochi, S.; Kusunose, N.; Akamine, T.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. Estradiol Regulation of P-Glycoprotein Expression in Mouse Kidney and Human Tubular Epithelial Cells, Implication for Renal Clearance of Drugs. Biochem. Biophys. Res. Commun. 2019, 519, 613–619. [Google Scholar] [CrossRef]
- Semeniuk, M.; Ceré, L.I.; Ciriaci, N.; Bucci-Muñoz, M.; Villanueva, S.S.M.; Mottino, A.D.; Catania, V.A.; Rigalli, J.P.; Ruiz, M.L. Regulation of Hepatic P-Gp Expression and Activity by Genistein in Rats. Arch. Toxicol. 2020, 94, 1625–1635. [Google Scholar] [CrossRef]






| Research Models | Advantages | Limitations | References |
|---|---|---|---|
| In silico | Allows for the simulation of biological systems; represents a reliable alternative to excessive animal use and other experimental techniques; enables larger and faster screenings to identify P-gp modulators/substrates; is less expensive, laborious, and time-consuming | Complexity of the methods; method selection can be difficult; difficult extrapolation to the in vitro, ex vivo, and in vivo experimental approaches; need for the acquisition of specific and sometimes expensive software | [33,49] |
| In vitro | Possibility of screening for a large number of substrates and/or modulators; possibility of testing several experimental conditions at the same time; allows for the assessment of the IC50 (for inhibitors), kinetic studies, etc.; allows for a high-throughput mode | Difficult clarification of the precise targets of xenobiotics, given the expression of multiple transporters in a particular cell line; difficult extrapolation of the results to the human body; laborious, time-consuming, and expensive; the cells need to be maintained under aseptic cell culture conditions prior to use | [38,48] |
| Ex vivo | Higher accuracy in the determination of the transporter’s function in absorption and/or elimination processes; allows for a simple understanding of the transporter’s role in one specific organ; allows for the evaluation of the interactions between transporters and metabolic enzymes | Difficult clarification of the modulation of one specific efflux transporter; requirement of ethics committees’ approval; requirement of surgical skills and equipment; expensive, laborious, and time-consuming; difficult data extrapolation to humans | [38] |
| In vivo | Better perspective on the (pharmaco)toxicokinetic characteristics; easier extrapolation to the human organism | Requirement of ethics committees’ approval; does not permit screenings of a large number of compounds and/or distinct experimental conditions | [48] |
| Substrates (actively transported by P-gp) | Analgesics: morphine; anti-arrhythmics: amiodarone, propafenone, quinidine; anti-alcoholism drugs: disulfiram; anticancer drugs: bisantrene, catharanthine, cisplatin, daunorubicin, docetaxel, doxorubicin, etoposide, irinotecan, mitoxantrone, paclitaxel, teniposide, topotecan, vinblastine, vincristine; anticonvulsants: phenobarbital, phenytoin; antidepressants: amitriptyline, nortriptyline, doxepin, venlafaxine, paroxetine; antidiarrheal agents: loperamide; anti-inflammatory agents: flunisolide; antiemetics: ondansetron, domperidone; antiepileptics: phenytoin, felbamate, topiramate, carbamazepine, lamotrigine, phenobarbital, gabapentin, topiramate; anti-gout agents: colchicine; antihistamines: terfenadine, fexofenadine; anti-hypertensives: debrisoquine, reserpine, propranolol, celiprolol, diltiazem, losartan, talinolol, prazosin; antimicrobial agents: actinomycin D, amoxicillin, clarithromycin, doxycycline, erythromycin, fluoroquinolones, gramicidin, grepafloxacin A, itraconazole, ketoconazole, levofloxacin, rifampin, sparfloxacin, valinomycin, tetracyclines, tetracycline; anti-tuberculous agents: erythromycin, rifampin; anthelmintics: abamectin, ivermectin; calcium channel blockers: verapamil, nifedipine, azidopine, diltiazem, nicardipin; cardiac glycosides: digitoxin, digoxin, quinidine; calmodulin antagonists: trifluoperazine, chlorpromazine, trans-flupentixol; hepatitis C treatments: ledipasvir/sofosbuvir; histamine H2 receptor antagonists: cimetidine; HIV protease inhibitors: ritonavir, saquinavir, nelfinavir, amprenavir, indinavir, maraviroc, darunavir; HMG-CoA reductase inhibitors: lovastatin, simvastatin; immunosuppressive agents: sirolimus, valspodar, cyclosporine A, tacrolimus (FK506); fluorescent compounds: berberine, calcein-AM, Hoechst 33342, rhodamine 123; natural products: flavonoids, curcuminoids, rhizome extract; opioids: loperamide, morphine, pentazocine; pesticides: methyl parathion, endosulfan, paraquat, cypermethrin, fenvalerate; steroids: budesonide, dexamethasone, hydrocortisone, corticosterone, cortisol, aldosterone, methylprednisolone, triamcinolone acetonide; tyrosine kinase inhibitors: imatinib mesylate, gefitinib, nilotinib, tandutinib; thrombin inhibitors: dabigatran |
| Inhibitors (block P-gp-mediated transport by different mechanisms) | First-generation: verapamil, cyclosporine A, nifedipine, quinidine, quinine, amiodarone, tamoxifen detergents; second-generation: R-verapamil, PSC833, dexniguldipine, valspodar, elacridar, biricodar, dexverapamil, dofequine fumarate; third-generation: ontogen (OC 144-093), zosuquidar, tariquidar, elacridar, laniquidar, biricodar; fourth-generation: anti-arrhythmics: propafenone; hepatitis C treatments: dasabuvir, ledipasvir, paritaprevir, ritonavir; natural products: flavonoids like tangeretin, sinensetin, baicalein, quercetin, ellipticine, cnidiadin, praeruptorin, capsaicin; surfactants: sodium dodecyl sulfate, Tween-20 and Span-80, and lipids; others: clarithromycin, itraconazole |
| Inducers (increase P-gp protein expression by prompting the transcription of the MDR1/ABCB1 gene) | Abacavir, actinomycin D, aflatoxin B1, aldosterone, ambrisentan, amiodarone, amprenavir, m-amsacrine, apigenin, artemisinin, asiatic acid, atazanavir, atorvastatin, avermectin, beclomethasone, benzo(a)pyrene, benzo(e)pyrene, berberine, betamethasone, bilirubin, bosentan, bromocriptine, budesonide, caffeine, cadmium chloride, capsaicin, carbamazepine, catechin, celiprolol, cembratriene, R-cetirizine, CITCO, chlorambucil, cholate, chrysin, ciclesonide, cisplatin, clotrimazole, colchicine, corticosterone, curcuma family extracts, curcumin, cyanidin, cyclophosphamide, cyclosporine A, cytarabine, daidzein daunorubicin, daurunavir, depsipeptide (FR901228, FK228, NSC630176), desvenlafaxine, dexamethasone, diclofenac, digoxin, dihydroxylated xanthones, 1α,25-dihydroxyvitamin D3, diltiazem, dimethylformamide, 6,16α-dimethylpregnenolone, dimethyl sulfoxide, docetaxel, doxorubicin, doxycycline, efavirenz, emetined, epigallocatechin-3-gallate, epirubicin, eriodictyol, erythromycin, β-estradiol, ethinylestradiol, etoposide, fenbufen, flavone, 5-fluorouracil, flutamide, fluticasone, genistein, ginkgolides a and b, hydroxyurea, hyperforin, hypericin, Hypericum perforatum extracts (Saint John’s wort), idarubicin, ifosfamide, indinavir, indomethacin, insulin, isosafrole, isoxanthohumol, ivermectin, lopinavir, LY191401, mangiferin, meloxicam, mepirizole, methotrexate, methylprednisolone, midazolam, mifepristone, mitoxantrone, morphine, mx2, myricetin, naringenin, nefazodone, nelfinavir, nevirapine, nicardipine, nifedipine, nimesulide, norathyriol, oleocanthal, ouabain, oxycodone, paclitaxel, parthenolide, pentylenetetrazole, phenobarbital, phenothiazine, phenytoin, phorbol 12-myristate 13-acetate, piperine, platelet-activating factor, prednisolone, 5β-pregnane-3,20-dione, pregnenolone-16α-carbonitrile, probenecid propranolol, quercetin, quinidine, rapamycin or sirolimus, rescinnamine, reserpine, retinoic acid,, rhinacanthin-C, rifabutin, rifampicin and derivatives, rilpivirinem, ritonavir, saquinavir, small-molecule tyrosine kinase inhibitors (e.g., erlotinib, gefitinib, nilotinib, sorafenib, vandetanib), sildenafil, sodium arsenite, sodium butyrate, spironolactone, SR12813, sulindac, tacrolimus, tadalafil, tamoxifen, tangeretin, talazoparib, taurocholate, taxifolin, TCDD, thioxanthonic derivatives (e.g., 1-(propan-2-ylamino)-4-propoxy-9H-thioxanthen-9-one (TX5)), γ-tocotrienol, topotecan, trazodone, triactyloleandomycin, trichostatin A, trimethoxybenzoylyohimbine, venlafaxine, verapamil, vinblastine, vincristine [3], tetrahydrocurcumin |
| Activators (promote an immediate increase in P-gp transport activity without interfering with P-gp protein expression) | Dihydroxylated xanthones (X1–X5): 3,4-dihydroxy-9H-xanthen-9-one, 1,2-dihydroxy-9H-xanthen-9-one, 1,3-dihydroxy-9H-xanthen-9-one, 2,3-dihydroxy-9H-xanthen-9-one, 3,6-dihydroxy-9H-xanthen-9-one; thioxanthonic derivatives (TX1-TX5): 1-[(3-hydroxypropyl)amino]-4-propoxy-9H-thioxanthen-9-one, 1-chloro-4-hydroxy-9H-thioxanthen-9-one, 1-{[2-(1,3-benzodioxol-5-yl) ethyl]amino}-4-propoxy-9H-thioxanthen-9-one, 1-[(2-methylpropyl) amino]-4-propoxy-9H-thioxanthen-9-one, 1-(propan-2-ylamino)-4-propoxy-9H-thioxanthen-9-one; aminated thioxanthones (ATX1-ATX8): (S)-1-((1-hydroxypropan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one, (R)-1-((1-hydroxypropan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one, (S)-1-((2-hydroxypropyl)amino)-4-propoxy-9H-thioxanthen-9-one, (R)-1-((2-hydroxypropyl)amino)-4-propoxy-9H-thioxanthen-9-one, (S)-1-((1-hydroxy-4-methylpentan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one, (R)-1-((1-hydroxy-4-methylpentan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one, (S)-1-((1-hydroxy-3-methylbutan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one, (R)-1-((1-hydroxy-3-methylbutan-2-yl)amino)-4-propoxy-9H-thioxanthen-9-one; oxygenated xanthones (OX1-OX2, OX4-OX6): 3,4-dimethoxy-1-methyl-9H-xanthen-9-one, 1-(dibromomethyl)-3,4-dimethoxy-9H-xanthen-9-one, 4-hydroxy-3-methoxy-9-oxo-9H-xanthene-1-carbaldehyde, 3,4-dimethoxy-9-oxo-9H-xanthene-1-carbaldehyde, 1-(hydroxymethyl)-3,4-dimethoxy-9H-xanthen-9-one |
| P-gp Substrate | Experimental Conditions | Modulation Type | Alterations in Substrate Therapeutic Effect/Toxicity | Experimental Models | References |
|---|---|---|---|---|---|
| [3H]-carbamazepine [3H]-dexamethasone [3H]-imipramine [3H]-lansoprazole [3H]-prazosin [3H]-quinidine [3H]-rifampicin [3H]-ritonavir [3H]-tamoxifen | 1 mCi, for 30 min | P-gp induction by tetrahydroisoquinoline derivative 8b (N-{4′-[(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl) methyl]-[1,1′-biphenyl]-4-yl}-4-methoxy-N-(4-methoxy- benzenesulfonyl)benzene-1-sulfonamide, 100 nM, for 2 h) | Higher efflux transport of P-gp substrates | In vitro, in Caco-2 cells Efflux/accumulation assay Substrates’ radioactivity measured by liquid scintillation | [157] |
| Colchicine | 0.1 mM, for 0–120 min | P-gp inhibition: In vitro, grapefruit juice (1–10%), 6′,7′-dihydroxybergamottin (1–500 µM), naringin (100–2000 µM), naringenin (10–500 µM), verapamil and quinidine (0.01, 0.05 and 0.1 mM), for 30 min; In situ, grapefruit juice (10%, for 1 h) | Inhibition of efflux transport of colchicine, proven by the increase in AP–BL permeability and decrease in BL–AP Higher oral bioavailability of colchicine | In vitro, in Caco-2 cell monolayers AP–BL and BL–AP directions in the absence/presence of known P-gp inhibitors (verapamil and quinidine) In situ, in male albino Wistar rats Single-pass intestinal perfusion studies, in both the jejunum and ileum | [158] |
| Clopidogrel | 10 µM, for 20–120 min | P-gp inhibition by omeprazole (6–60 µM), cyclosporin (2–20 µM), verapamil (25–250 µM), quinidine (20–200 µM), and elacridar (0.12–1.2 µM), for 20–120 min | Higher intracellular accumulation of clopidogrel Higher bioavailability of clopidogrel | In vitro, in Caco-2 cells Quantification of clopidogrel in LC-MS-MS | [150] |
| Cyclosporine A | 100 mg, twice daily | P-gp induction by St. Johns’ wort (300–600 mg/day) | Lower absorption of cyclosporine A Lower serum levels of cyclosporine | Case report, in a 29-year-old white woman Quantification of serum cyclosporine A levels | [156] |
| Digoxin | 0.02 mg/kg, orally | P-gp inhibition by quercetin (40 or 50 mg/kg, orally) | Higher Cmax and AUC of digoxin | In vivo, in male Yorkshire pigs Quantification of serum digoxin levels through fluorescence polarization immunoassays | [159] |
| 0.04 mg/mL, orally | P-gp inhibition by montmorillonite–surfactant hybrid particles (5.0 mL/kg, orally) | Higher Cmax and AUC of digoxin | In vivo, in male Sprague Dawley rats LC-MS-MS: blood levels of digoxin | [160] | |
| 0.1 µM of [3H]-digoxin, for 30 min | P-gp inhibition: Berberine formulation with P85 (0.1% and 1%, for 30 min) and Tween 80 (0.1% and 1%, for 30 min) | Lower P-gp-mediated efflux of digoxin Higher intestinal accumulation of digoxin | In vitro, in LLC-PK1-P-gp cells [3H]-digoxin radioactivity measured by a liquid scintillation counter | [161] | |
| 0.25 mg/kg, orally | P-gp activation by thioxanthone 5 (30 mg/kg, orally) | Lower AUC of digoxin | In vivo, in adult male Wistar Han rats Digoxin plasma levels measured on an AutoAnalyzer | [122] | |
| 0.2 mg/kg, orally | P-gp inhibition by verapamil (25 mg/kg, orally) | Higher Cmax and AUC of digoxin | In vivo, in male Sprague Dawley rats LC-MS-MS: blood levels of digoxin | [162] | |
| Doxorubicin | 1.0 µM, every two weeks in the cell culture medium | P-gp inhibition by aminated thioxanthones (1 or 10 µM, for 48 h) | Higher cell-growth-inhibitory effect | In vitro, in K562 and K562Dox cells Sulforhodamine-B assay: measurement of the cell-growth-inhibitory effect | [144] |
| Enrofloxacin | 10 mg/kg, orally | P-gp induction by quercetin (15 and 60 mg/kg, for 5 days, orally) | Higher P-gp-mediated function Lower AUC, peak concentration, lower intestinal absorption; higher clearance | In vivo, in Arbor Acres chickens Quantification of enrofloxacin plasma levels through HPLC | [163] |
| Fexofenadine | 50 µM, for 15 min | P-gp inhibition by Maytenus ilicifolia (obtained by infusion or turbo-extraction using hydroacetonic solvent, 200 µg/mL, for 30 min) | Lower intestinal metabolism Lower transport of drugs | In vitro, in Caco-2 cells Quantification of fexofenadine through HPLC | [164] |
| Glibenclamide | Ex vivo: 1 mg/mL, for 2 h; in vivo: 3.6 mg/kg, orally, for 8 h | P-gp inhibition by carbamazepine (ex vivo: 25 mM, for 2 h; in vivo: 90 mg/kg, orally, for 1 + 8 h) | Ex vivo: lower accumulation of glibenclamide In vivo: higher absorption of glibenclamide | Ex vivo, in ileal sacs of albino rats In vivo, in albino rats Quantification of glibenclamide through HPLC | [131] |
| Ivermectin | Ex vivo: 3 µM; in vivo: 200 µg/kg, subcutaneously | P-gp inhibition by itraconazole (ex vivo: 10 µM; in vivo: 5 mg, two doses, subcutaneously) | Ex vivo: higher levels of ivermectin In vivo: higher concentrations of ivermectin in plasma and gastrointestinal tissues; enhanced ivermectin absorption | Ex vivo, in everted ileal sacs In vivo, in male Wistar rats Quantification of ivermectin levels using HPLC | [130] |
| In vitro: 10 µM, for 2–24 h; in vivo: 0.2 mg/kg, orally | P-gp inhibition by oleic acid (In vitro: 500 µM, for 2–24 h; in vivo: 1 g/kg, orally) | Ex vivo: higher ivermectin levels In vivo: higher concentrations of ivermectin in plasma and intestinal mucosa | In vitro, in Caco-2 cells In vivo, in wild-type mice Quantification of ivermectin levels using HPLC | [165] | |
| Paraquat | 0–5000 µM, for 24 h | P-gp induction by doxorubicin (0–100 µM, for 24 h) | Lower paraquat-induced cytotoxicity | In vitro, in Caco-2 cells Cellular viability: MTT assays | [90] |
| 100–5000 µM, for 24 h | P-gp induction by doxorubicin (10, 50 and 100 µM, for 18 h) | Lower paraquat-induced cytotoxicity Lower intracellular accumulation of paraquat | In vitro, in Caco-2 cells Cellular viability: MTT assays Quantification of intracellular accumulation of paraquat: GC–IT-MS analyses | [94] | |
| 0.5–50 mM, for 48 h | P-gp induction by reduced rifampicin (10 µM, for 24, 48 and 72 h) | Lower paraquat-induced cytotoxicity | In vitro, in RBE4 cells Cellular viability: NR uptake assay | [114] | |
| 0–7500 µM, for 24 h | P-gp induction by dihydroxylated xanthones 1–5 (20 µM, for 24 h) | Lower paraquat-induced cytotoxicity | In vitro, in Caco-2 cells Cellular viability: NR uptake assay | [91] | |
| 0–7500 µM, for 24 h | P-gp induction by thioxanthones 1–5 (20 µM, for 24 h) | Lower paraquat-induced cytotoxicity | In vitro, in Caco-2 cells Cellular viability: NR uptake assay | [83] | |
| 250–2500 µM, for 24 h | P-gp induction by oxygenated xanthones 1–6 (20 µM, for 24 h) | Lower paraquat-induced cytotoxicity | In vitro, in Caco-2 cells Cellular viability: NR uptake assay | [21] | |
| Paclitaxel | 40–70 mg/kg, orally | P-gp inhibition by flavonoid FM04 (45 mg/kg, orally) | Higher intestinal absorption of paclitaxel; higher AUC | In vivo, in male Sprague Dawley rats Quantification of paclitaxel using UPLC-MSMS | [166] |
| Mitoxantrone | 0–150 µM, for 24 h | P-gp induction by oxygenated xanthones 1–6 (20 µM, for 24 h) | Lower mitoxantrone-induced cytotoxicity | In vitro, in SW480 cells and Caco-2 cells Cellular viability: NR uptake assay | [113] |
| Talinolol | 20 mg/kg, i.p. | P-gp inhibition by zosuquidar (30 mg/kg, i.p.) | Higher AUC0–5 h and AUCtotal | In vivo, in C57BL/6 J mice Expression: PCR and Western blot techniques Quantification of talinolol through HPLC-UV | [167] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veiga-Matos, J.; Morales, A.I.; Prieto, M.; Remião, F.; Silva, R. Study Models of Drug–Drug Interactions Involving P-Glycoprotein: The Potential Benefit of P-Glycoprotein Modulation at the Kidney and Intestinal Levels. Molecules 2023, 28, 7532. https://doi.org/10.3390/molecules28227532
Veiga-Matos J, Morales AI, Prieto M, Remião F, Silva R. Study Models of Drug–Drug Interactions Involving P-Glycoprotein: The Potential Benefit of P-Glycoprotein Modulation at the Kidney and Intestinal Levels. Molecules. 2023; 28(22):7532. https://doi.org/10.3390/molecules28227532
Chicago/Turabian StyleVeiga-Matos, Jéssica, Ana I. Morales, Marta Prieto, Fernando Remião, and Renata Silva. 2023. "Study Models of Drug–Drug Interactions Involving P-Glycoprotein: The Potential Benefit of P-Glycoprotein Modulation at the Kidney and Intestinal Levels" Molecules 28, no. 22: 7532. https://doi.org/10.3390/molecules28227532
APA StyleVeiga-Matos, J., Morales, A. I., Prieto, M., Remião, F., & Silva, R. (2023). Study Models of Drug–Drug Interactions Involving P-Glycoprotein: The Potential Benefit of P-Glycoprotein Modulation at the Kidney and Intestinal Levels. Molecules, 28(22), 7532. https://doi.org/10.3390/molecules28227532