1. Introduction
The Betti reaction, alternatively referred to as the Aza-Friedel–Crafts reaction, represents an important variant of the Mannich reaction and is one of the most important C-C bond-forming reactions in organic chemistry. The Betti reaction is a simple multicomponent condensation between 2-naphthol, aryl aldehydes, and amines that leads to aminobenzylnaphthols, the so-called Betti bases [
1,
2,
3]. Over the past decade, many research groups have reported different variants of this reaction, generally using a pre-formed imine that is often protected as a sulfonamide [
4,
5,
6]. Aminoarylnaphthols are a class of molecules found in natural and synthetic compounds with a wide range of interesting activities and applications. Chiral Betti bases have been widely used in optoelectronics, but their biological properties, such as anticancer, anti-bacterial, antioxidant, anti-inflammatory, antipain, antihypertensive, anti-Alzheimer, and bradycardia activities, deserve special attention [
7]. In addition, chiral Betti bases have been proven to be useful ligands and auxiliaries in asymmetric synthesis [
7,
8,
9,
10]. Their widespread applications have generated considerable interest in developing asymmetric methods for their preparation. In 2010, the Hui group reported an enantioselective Betti reaction of 2-naphthol with tosylimines catalyzed by a dinuclear zinc complex, to prepare chiral Betti base derivatives with high yields and excellent enantioselectivity of up to 96% [
11]. Subsequent studies were carried out using quinine-squaramide organocatalysts [
12]. The results obtained by Chimni and Wang’s group demonstrated that α- and β-naphthol derivatives react readily with various aromatic and heteroaromatic
N-sulfonylimines in the presence of
Cinchona alkaloid derived bifunctional organocatalyst [
13,
14]. The reaction of naphthols with isatin-derived ketimines turned out to be more demanding. In 2015, Pedro and Khan’s groups, respectively, reported an enantioselective version of this reaction organocatalyzed by the same quinine-derived thiourea to yield chiral 3-amino-2-oxindoles in high yields with excellent enantioselectivity [
15,
16,
17]. Two years later, Tanyeli used bulky cinchona-derived squaramides in the same transformation on naphthols [
18]. In 2020, Pedrosa and coworkers reported that aminoethyl polystyrene-supported cinchona-thiourea derivatives could be recovered and reused in a highly enantioselective aza-Friedel–Crafts reaction of different naphthols with a variety of
N-Boc ketimines derived from isatin [
19]. Recently, Wang and Jin reported simple Takemoto-type thiourea-catalyzed reactions of β-naphthols with isatines leading to 3-(naphthalen-1-yl)-3-amino-2-oxindoles with excellent 89–95% yield and high enantioselectivity (90–97% ee) [
20]. We would also like to mention the enantio- and diastereoselective cascade Betti/aza-Michael reaction of phenols and
N-tosylimin containing Michael acceptor catalyzed by bifunctional thiourea organocatalysts leading to 1,3-disubstituted isoindolines [
21]. To our knowledge, these are the most important, if not the only, literature reports on the asymmetric Betti reaction. Due to the high demand for the development of this reaction, we decided to test thiourea and, for the first time, thiosquaramide catalysts, compare the obtained results with those already published, and then draw general conclusions regarding the use of organocatalysts of this type in the reactions of α- and β-naphthols and 6-hydroxyquinoline with tosylimine and ketimine. Catalysts of this type have a number of advantages: their synthesis is uncomplicated, and the reactions involving them proceed under mild conditions, without metal cation. Moreover, the presence of a thiourea/thiosquaramide bridge and strongly acidic protons leads to the formation of strong hydrogen bonds and activation of the substrate. While bifunctional chiral thioureas have been widely used in asymmetric synthesis [
22,
23,
24], including the Betti reaction, bifunctional chiral thiosquaramides were used only in Michael addition [
25,
26,
27,
28,
29], asymmetric Michael–Henry tandem reaction [
30], and the addition of lawsone to β,γ-unsaturated α-keto ester [
31].
2. Results and Discussion
Bifunctional thioureas were prepared in one step, as previously described [
32,
33,
34]. The reaction of appropriate, commercially available isothiocyanates with amines leads to thioureas in near quantitative yields. Two of the synthesized thioureas,
2 and
5, are new compounds, while the bifunctional Takemoto catalyst
3 is commercially available (
Figure 1). Thiosquaramides
8–
11 were prepared in four steps from 3,4-dihydroxycyclobut-3-en-1,2-dione (squaric acid). In the first step, esterification of the square acid with cyclopentanol gave dicyclopentyl squarate [
25,
31]. Subsequently, dithionation with Lawesson’s reagent led to dicyclopentyl dithiosquarate, which was found to be an excellent platform for the synthesis of
8–
11 (
Figure 1). Disubstituted thiosquaramide derivatives reacted vigorously with primary amines—first with 3,4-bis(trifluoromethyl)aniline and then with the appropriate amine having a chiral center and a tertiary amine. The last step was carried out using the traditional solvent method, but also using mechanochemistry. It should be emphasized that the use of a ball mill resulted in a significant increase in chemical efficiency. Due to the possibility of creating rotamers and zwitterionic species by bifunctional thiosquaramides, especially those possessing an aryl substituent, we transformed them into appropriate hydrochlorides, which resulted in simplifying their spectra and sharpening many signals [
31].
To study the feasibility and enantioselectivity of the Betti reaction, a bifunctional catalyst
1 which consists of H-bond donor and amine moieties was screened for a model reaction of 1-naphthol (
12) with
N-tosylimine (
13) in toluene at room temperature (
Scheme 1 and
Table 1). The desired product
14 was isolated in good yield (98%) and with 75% ee after 20 h (
Table 1, entry 2) when a threefold excess of 1-naphthol relative to imine was used.
To further improve the enantioselectivity of the transformation, we investigated a variety of different reaction conditions, including the solvent, temperature, and catalyst loading (
Table 2).
The obtained results revealed that the solvent effect had a significant impact on the efficiency of the reaction. Toluene was found to be optimal (
Table 1, entry 2), while reactions carried out in THF, ACN,
o-xylene, and DCM resulted in a decrease in yield and enantioselectivity (
Table 1, entries 9–12). The screening of catalyst loading exhibited that 10 mol% equivalent of
1 was optimal, while 5 mol% and 30 mol% offered no improvement in the asymmetric induction (
Table 1, entries 7–8). It is noteworthy that the temperature also influenced the reaction—after lowering it from rt to 0 °C, a slight increase in enantioselectivity was observed (
Table 1, entry 6). Based on these experiments, the optimized conditions were determined to be toluene as the solvent with a 10 mol% loading of catalyst
1 at 0 °C (
Table 1, entry 6). The absolute configuration of the product was assigned as (
S) based on a comparison of the optical rotation of the product material with a value from the literature [
14,
32].
With the optimized conditions in hand, we evaluated the general applicability of this asymmetric Betti reaction using bifunctional thioureas
1–
7 (
Table 2 entries 1–7). All tested organocatalysts gave the expected product in high yields (80–98%) with moderate to high enantioselectivities (58–80% ee). (
R,
R)-piperidine-based thiourea
1 and Takemoto catalyst
3 with the 3,5-bis(trifluoromethyl)phenyl moiety provided the best yield and enantioselectivity (
Table 2, entries 1 and 3). The obtained results prove the higher efficiency of thiourea catalysts having at one of the nitrogen atoms an aryl group with strongly electron-acceptor substituents (CF
3), which by increasing the acidity of the N-H bond facilitates the formation of a hydrogen bond with the substrate. In contrast, bifunctional organocatalysts
6–
7 without the aminocyclohexyl moiety showed lower asymmetric induction (
Table 2, entries 6–7).
Thiosquaramide organocatalysts
8–
11, used for the first time in the Betti reaction, promoted the reaction but gave lower yields and ee values (
Table 2, entries 8–15). Organocatalysts
8–
9 containing ((1
R,2
R)-2-(dimethylamino)cyclohexyl)amine or ((1
R,2
R)-2-(piperidin-1-yl)cyclohexyl)amine moiety gave predominantly (
S)-product, while those not containing these moieties had the opposite enantiomer of
14. A similar relationship was observed for thioureas
1–
7. The higher activity of thiosquaramides
8–
11 (
Table 2, entries 8, 10, 12, and 14) should also be noted compared to their appropriate hydrochlorides (
Table 2, entries 9, 11, 13, and 15).
To rationalize the stereochemical outcome, a dual activation for the thiourea and thiosquaramide-catalyzed asymmetric Betti reaction of 1-naphthol (
12) with
N-tosylimine
13 is proposed in
Scheme 2. Based on previous literature reports [
13,
22,
23,
24] and the observed stereochemistry, a transition state involving a ternary complex between the catalyst and the substrates can be proposed. Both thiourea and thiosquaramide catalysts promote the reaction in a dual manner: activating
N-tosylimine through forming H-bond with the thiourea/thiosquaramide motif and enhancing the nucleophilicity of 1-naphthol by the tertiary amine moiety of catalyst. As shown in
Scheme 2, a configuration of the product was determined by the absolute configuration of the carbon atom at the thioamide nitrogen atom of the thiourea/thiosquaramide moiety. For organocatalysts
1–
5 and
8–
9, the activated 1-naphthol is capable of nucleophilic attack from C2 on the
Re face of the imine providing the
S enantiomer of the product. On the other hand, for organocatalysts
6–
7 and
10–
11 with the opposite absolute configuration (the compatibility of the relative configuration results from the different priority of the substituents), the attack of the nucleophile on the
Si face of the imine is favored which leads to (
R)-
16.
To demonstrate the generality of the
1–
11-promoted asymmetric Betti reaction, other imines and naphthols were explored (
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7). The optimization of the reaction conditions of 2-naphthol (
15) with
N-tosylimine
13 (
Scheme 3) catalyzed by thiourea
1 indicated that the use of substrates in the molar ratio naphthol:imine = 3:1, 10 mol% of
1 in toluene at 0 °C was optimal for this transformation and led to chiral product
16 in 98% yield and 54% ee value (
Table 3, entry 2).
In the next step, we decided to react 2-naphthol (
15) with imine
13 in the presence of other organocatalysts
2–
11 (
Table 4).
All tested bifunctional thioureas
1–
7 gave product
16 in high yields (65–98%) but with low enantiomeric excesses (6–61% ee,
Table 4, entries 1–7). The obtained results showed that Takemoto-type organocatalysts
1–
5 were less effective in this transformation. In contrast, all bifunctional thiosquaramides
8–
11 and their hydrochlorides slightly improved the asymmetric induction (
Table 4, entries 8–15). Although the yields of reactions catalyzed by thiosquaramides were low compared to thioureas, the ee values were higher (46–71% ee). Nevertheless, this is the first example of the use of thiosquaramide organocatalysts in the reaction of 2-naphthol with imine
13. Their higher catalytic activity in the reaction of 2-naphthol compared to 1-naphthol should also be noted (
Table 2 vs.
Table 4).
In the next stage of our research, we decided to test the catalytic activity of thiourea 1 in the reaction of 6-hydroxyquinoline as a nucleophile (17) with N-tosylimine 13. Unfortunately, despite many attempts, we were unable to obtain the desired product.
Based on literature reports [
17], we decided to react 6-hydroxyquinoline (
17) with isatin-derived
N-Boc ketimine
18 (
Scheme 4,
Table 5). We carried out the first test under the conditions developed by Pedro for this transformation [
17]. Thiourea
1 (5 mol%) catalyzed the reaction in toluene to afford regioselectively alkylated at C-5 carbon atom chiral product
19 with 68% yield after 48 h (
Table 5, entry 1) with a good enantiomeric excess (76% ee). When the reaction temperature was lowered to 0 °C, the ee value could be further enhanced to 87% (
Table 5, entry 2). Changing the solvent to DCM resulted in a drastic decrease in the yield and selectivity of the reaction (
Table 5, entry 3). The screening of catalyst loading exhibited that 5 mol% equivalent of
1 was optimal, while 10 mol% and 30 mol% offered no improvement in the asymmetric induction (
Table 5, entries 4–5).
Next, we investigated the Betti reaction under optimal conditions with the other catalysts (
Table 6). Thiourea
3 proved to be the most effective in terms of enantioselectivity of all Takemoto’s type organocatalysts (92% ee), although with unsatisfactory yield—only 65% (
Table 6, entries 1–6). Thioureas
6 and
7, which lacked the cyclohexylamine grouping, performed better under these conditions (
Table 6, entries 7–9) (
Table 6, entries 7–9). The chiral product
19 was obtained with 75% yield and 98% ee at 0 °C. Thiosquaramides
8–
11, as well as their hydrochlorides, showed very low catalytic activity (
Table 6, entries 10–17), although quinine-derived squaramides gave excellent enantioselectivity under these conditions, as reported by Pedro [
17] and Tanyeli [
18].
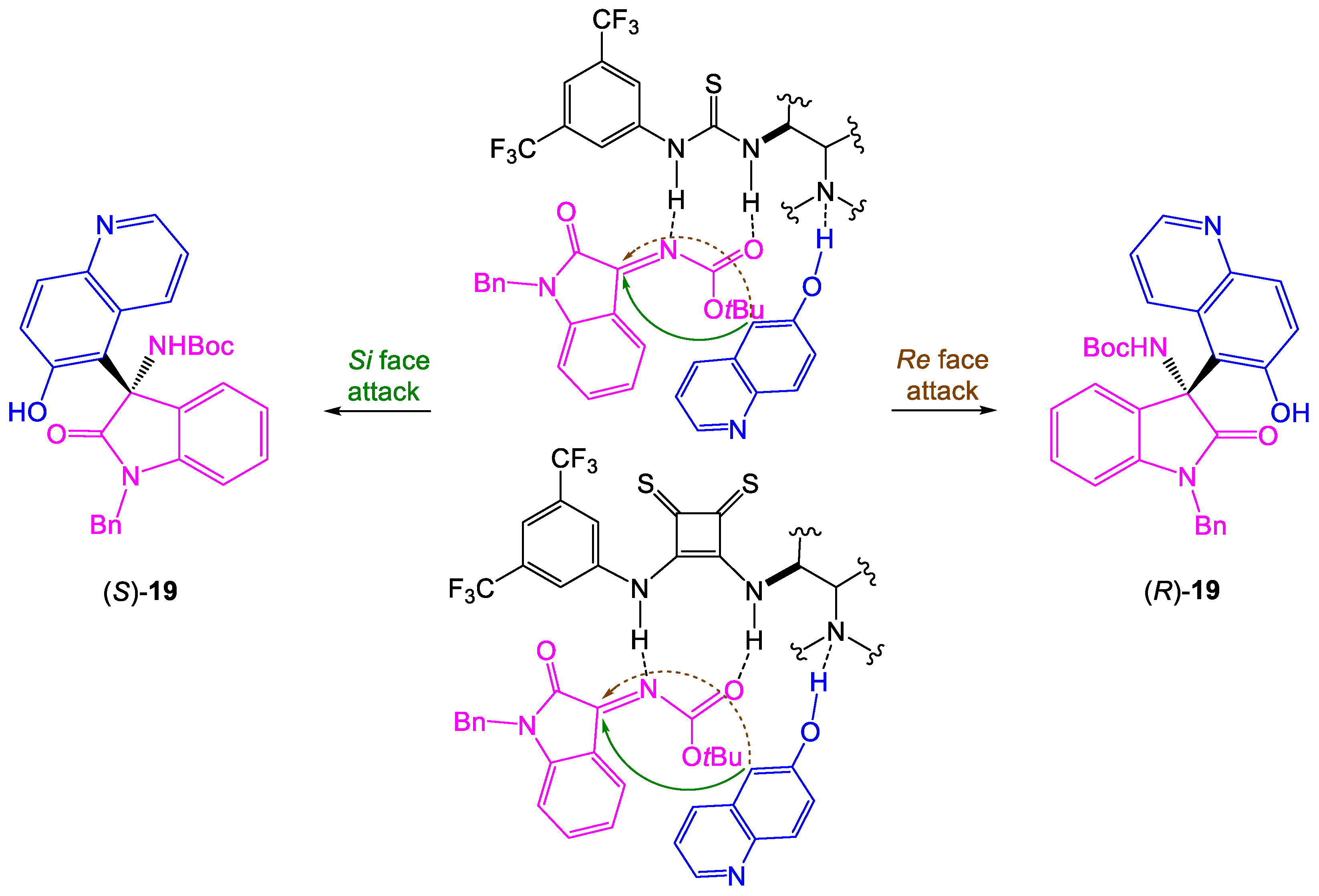
Taking into account the obtained results, we have proposed the possible transition state shown in
Scheme 5. Bifunctional organocatalysts are responsible for the preorientation and simultaneous activation of electrophile via H-bond donor thiourea/thiosquaramide moiety and the enhancement of the nucleophilicity of 6-hydroxyquinoline by the tertiary amine unit of catalyst [
16,
17,
18]. Generally, nucleophile attacks from C-5 of 6-hydroxyquinoline on the
Si face of ketimine results in the
S configuration of product in the case of Takemoto-type organocatalysts
1–
5 and
9, while in reactions catalyzed by
6–
7 and
10–
11 Si face of ketimine becomes less available and the formation of the
R isomer is favored.
Finally, the reactions of naphthols
12 and
15 with ketimine
18 were developed (
Scheme 6,
Table 7). For testing, we chose thiourea
7, which reacted best with 6-hydroxyquinoline, and its thiosquaramide counterpart
11. The obtained results, similarly to those obtained for the reaction 6-hydroxyquinoline (
17) with ketimine
18, indicated a higher activity of thiourea
7 compared to thiosquaramide
11. 3-Substituted 3-amino-2-oxindoles were obtained in moderate yield (53%) and enantioselectivity (78% ee) in the presence of
7, while thiosquaramide
11 led to racemic products.
3. Materials and Methods
Commercially available chemicals used in this work were purchased from Merck (Darmstadt, Germany) and were used as supplied, without additional purification. NMR spectra were recorded in CDCl3 on a Bruker Avance III (600 MHz for 1H NMR, 150 MHz for 13C NMR); coupling constants are reported in hertz (Hz). The chemical shift values were expressed in ppm (part per million) with tetramethylsilane (TMS) as an internal reference. The rotations were measured using an Anton Paar MCP 500 polarimeter. Melting points measured on the DigiMelt apparatus are uncorrected. The milling treatments were carried out in a vibrating Retsch Mixer Mill 400 (vbm). Milling load is defined as the sum of the mass of the reactants per free volume in the jar. All the reactions using a vibratory ball mill were performed at 25 Hz under air with no interruption of the milling. Chromatographic purification of compounds was achieved with 230–400 mesh size silica gel. The progress of reactions was monitored by silica gel thin-layer chromatography plates (Merck TLC Silicagel 60 F254). The enantiomeric excess was determined by HPLC (1260 Infinity, Agilent Technologies, Santa Clara, CA, USA), employing a Chiralpak AD-H or OD-H column (25 cm × 4.6 mm).
Copies of
1H and
13C NMR spectra of compounds
1–
3,
5–
11,
14,
16,
19–
21 and selected HPLC chromatograms of Betti bases are included in the
Supplementary Materials.
3.1. General Procedure for the Synthesis of Thiourea Catalysts
To the solution of corresponding diamine (0.4 mmol) in CH2Cl2 at 0 °C, isothiocyanate (0.48 mmol) was added dropwisely under argon atmosphere. The resulting mixture was stirred at room temperature for around 30 min. The progress of the reaction was monitored by silica gel thin-layer chromatography plates. After reaction completion, the crude product was concentrated and purified by column chromatography.
3.1.1. 1-[3,5-Bis(trifluoromethyl)phenyl]-3-[(1R,2R)-2-(piperidin-1-yl)cyclohexyl]thiourea (1)
Yellowish solid, 167 mg, 92% yield; R
f = 0.09 (hexane/ethyl acetate, 2:1); [α
= −6.0 (
c 0.4, CHCl
3), {Lit. [
32]: [α
= 0.60 (
c 0.5, CHCl
3)}; m.p. = 53.2–55.5 °C, {Lit. [
32]: m.p. = 57–59 °C}; δ
H (600 MHz, CDCl
3): 1.10–1.47 (m, 10H, H
aliph), 1.75 (d, 1H,
J = 13.1, H
aliph), 1.85 (d, 1H,
J = 11.2, H
aliph), 1.94 (d, 1H,
J = 11.5, H
aliph), 2.33–2.55 (m, 3H, H
aliph), 2.57–2.82 (m, 3H, H
aliph), 3.85 (bs, 1H, NH), 7.68 (s, 1H, H
ar), 7.87 (s, 2H, H
ar); δ
C (150 MHz, CDCl
3): 23.5, 23.8, 24.4, 25.1, 25.7, 27.2, 32.6 (C
aliph), 49.7 (CH
2N), 55.6 (CHNH), 68.6 (CHN), 118.7, 120.3, 122.1, 123.8, 124.1 (q,
1JC-F = 270.0 Hz), 132.5 (q,
2JC-F = 34.7 Hz), 140.0 (C
ar) 180.3 (CS); Elementar analysis: C
20H
25F
6N
3S (453.49 g/mol) calculated: C% 52.97, H% 5.56, N% 9.27, S% 7.07; found: C% 52.81, H% 5.61, N% 9.47, S% 6.97.
Spectral data matched that reported by Mukherjee [
32].
3.1.2. 1-(3,4,5-Trimetoxyphenyl)-3-[(1R,2R)-2-(piperidin-1-yl)cyclohexyl]thiourea (2)
Yellowish solid, 143 mg, 88% yield; Rf = 0.12 (hexane/ethyl acetate, 2:1); [α = −23.80 (c 0.5, CHCl3); m.p. = 99.3–103.1 °C; δH (600 MHz, CDCl3): 0.80–0.91 (m, 1H, Haliph), 1.05–1.14 (m, 1H, Haliph), 1.21–1.31 (m, 5H, Haliph), 1.32–1.42 (m, 6H, Haliph), 1.71 (d, 1H, J = 13.8 Haliph), 1.83 (d, 1H, J = 10.7, Haliph), 1.88 (d, 1H, J = 11.4, Haliph), 2.27 (bs, 2H, CHN), 2.42 (bs, 1H, NH), 2.63 (bs, 2H, CHN), 2.89 (bs, 1H, NH), 3.84 (s, 9H, OCH3), 6.57 (s, 2H, Har); δC (150 MHz, CDCl3): 14.2, 23.4, 24.6, 25.6, 29.8, 30.4, 32.7 (Caliph), 49.3 (CHN), 55.5 (CHN), 56.5 (2OCH3), 61.0 (OCH3), 68.3 (CHN), 103.5, 132.7, 137.0, 153.9 (Car), 181.0 (CS); Elementar analysis: C21H33N3O3S (407.57 g/mol) calculated: C% 61.89, H% 8.16, N% 10.31, S% 7.87; found: C% 61.79, H% 8.10, N% 10.10, S% 7.75.
3.1.3. 1-[3,5-Bis(trifluoromethyl)phenyl]-3-[(1R,2R)-2(dimethylamino)cyclohexyl]thiourea (3)
Commercially available catalyst (Merck).
3.1.4. 1-(3,4,5-Trimetoxyphenyl)-3-[(1R,2R)-2-(dimethylamino)cyclohexyl]thiourea (4)
Yellowish solid, 144 mg, 98% yield; Rf = 0.24 (hexane/ethyl acetate, 2:1); [α = −82.17 (c 0.5, CHCl3); m.p. = 135.5–136.5 °C; δH (600 MHz, CDCl3): 0.95–1.05 (m, 1H, Haliph), 1.11–1.27 (m, 3H, Haliph), 1.30–1.39 (m, 1H, Haliph), 1.68 (d, 1H, J = 13.5 Hz, Haliph), 1.82 (d, 1H, J = 12.8 Hz, Haliph), 1.86 (d, 1H, J = 12.8 Hz, Haliph), 2.23 (s, 6H, 2CH3), 2.32–2.41 (ddd, 1H, J = 5.5, 5.5, 2.88 Hz, CHN), 2.64–2.74 (m, 1H, CHN), 3.82 (s, 6H, 2OCH3), 3.83 (s, 3H, OCH3), 3.97 (bs, 1H, NH), 6.50 (s, 2H, Har), 6.91 (bs, 1H, NH); δC (150 MHz, CDCl3): 21.6, 24.6, 25.1, 32.9 (Caliph), 40.1 (CH3), 56.0 (CHN), 56.3 (2OCH3), 61.0 (OCH3), 66.8 (CHN), 102.4, 133.2, 136.3, 153.7 (Car), 179.9 (CS); Elementar analysis: C18H29N3O3S (367.51 g/mol) calculated: C% 58.83, H% 7.95, N% 11.43, S% 8.72; found: C% 58.64, H% 7.81, N% 11.68, S% 8.56.
Spectral data matched that reported by Steve Tse and Yeung [
36].
3.1.5. 1-(Perfluorophenyl)-3-[(1R,2R)-2-(dimethylamino)cyclohexyl]thiourea (5)
Yellowish solid, 144 mg, 78% yield; Rf = 0.44 (hexane/ethyl acetate, 2:1); [α = +101.68 (c 0.35, CHCl3); m.p. = 117.8–119.5 °C; δH (600 MHz, CDCl3): 0.96–1.30 (m, 5H, Haliph), 1.70 (d, 1H, J = 9.4 Hz, Haliph), 1.77 (m, 1H, J = 12.7 Hz, Haliph), 1.85 (d, 1H, J = 12.7 Hz, Haliph), 2.13–2.20 (m, 1H, Haliph), 2.28 (s, 6H, 2CH3), 2.38–2.47 (m, 1H, Haliph), 3.58 (bs, 1H, NH), 6.34 (bs, 1H, NH); δC (150 MHz, CDCl3): 20.9, 25.3, 25.8, 33.3 (Caliph), 40.3, 40.7 (CH3), 57.3, 69.9 (CHN), 137.2, 138.8 (Car), 178.3 (CS); Elementar analysis: C15H18F5N3S (367.38 g/mol) calculated: C% 49.04, H% 4.94, N% 11.44, S% 8.73; found: C% 48.89, H% 5.30, N% 10.59, S% 8.76.
3.1.6. 1-[3,5-Bis(trifluoromethyl)phenyl]-3-[(R)-4-methyl-1-(piperidin-1-yl)pentan-2-yl]thiourea (6)
Colorless solid, 138 mg, 76% yield; R
f = 0.21 (hexane/ethyl acetate, 2:1); [α
= −26.20 (
c 0.4, CHCl
3), {Lit. [
33]: [α
= −30.80 (
c 1.0, CHCl
3)}; m.p. = 43.5–45.5 °C; δ
H (600 MHz, CDCl
3): 0.96 (d, 3H,
J = 5.5 Hz, CH
3), 0.98 (d, 3H,
J = 6.7 Hz, CH
3), 1.29–1.35 (m, 1H, H
aliph), 1.41–1.67 (m, 7H, H
aliph), 1.72–1.80 (m, 1H, CH(CH
3)
2), 2.30–2.50 (m, 3H, H
aliph), 2.55–2.85 (m, 3H, H
aliph), 3.79 (bs, 1H, H
aliph), 6.23 (s, 1H), 7.64 (s, 1H, H
ar), 8.05 (s, 2H, H
ar), 12.77 (bs, 1H, NH); δC (150 MHz, CDCl
3): 22.2 (C
aliph), 22.8 (CH
3), 23.5 (C
aliph), 25.0 (CH
3), 25.7 (
CH(CH
3)
2), 31.0, 43.0 (
CH
2CH(CH
3)
2), 53.0 (CH
2N), 55.3 (CHN), 67.2 (CH
2N), 118.3, 122.4, 124.2, 124.8, 126.0 (C
6H
3(CF
3)
2), 131.8 (q,
2JC-F = 32.7 Hz), 141.9 (C
ar), 183.8 (CS); Elementar analysis: C
20H
27F
6N
3S (455.51 g/mol) calculated: C% 52.74, H% 5.97, N% 9.23, S% 7.04; found: C% 52.66, H% 6.02, N% 9.08, S% 7.02.
Spectral data matched that reported by Qu [
33].
3.1.7. 1-[3,5-Bis(trifluoromethyl)phenyl]-3-[(R)-1-phenyl-3-(piperidin-1-yl)propan-2-yl]thiourea (7)
Colorless solid, 161 mg, 82% yield; R
f = 0.17 (hexane/ethyl acetate, 2:1); [α
= −30.36 (c 0.4, CHCl
3); {Lit. [
34]: [α
= −32.90 (c 0.65, CH
2Cl
2)}; m.p. = 52.5–54.5 °C; δ
H (600 MHz, CDCl
3): 1.34–1.69 (m, 7H, H
aliph), 2.29 (bs, 2H, H
aliph), 2.42–2.66 (m, 4H, H
aliph), 2.72–2.82 (m, 1H, H
aliph), 2.92–3.04 (m, 1H, H
aliph), 4.02 (bs, 1H, NH), 6.35 (bs, 1H, NH), 7.22 (d, 2H,
J = 7.2, H
ar), 7.27–7.32 (m, 1H, H
ar), 7.34–7.38 (m, 2H, H
ar), 7.65 (s, 1H, H
ar), 8.01 (s, 2H, H
ar), 12.85 (bs, 1H, NH); δC (150 MHz, CDCl
3): 23.5, 25.6 (C
aliph), 40.0 (
CH
2C
6H
5), 54.8 (2CH
2N), 55.5 (CHN), 65.1 (CH
2N), 118.5, 120.6, 122.4, 124.2, 125.0, 126.0, 127.5, 128.9, 129.2, 129.3, 131.0, 131.9 (q,
2JC-F = 32.7 Hz), 136.2, 141.8 (C
ar), 183.5 (CS); Elementar analysis: C
23H
25F
6N
3S (489.52 g/mol) calculated: C% 6.43, H% 5.15, N% 8.58, S% 6.55; found: C% 56.49, H% 5.04, N% 8.64, S% 6.53.
Spectral data matched that reported by Zhu [
34].
3.2. General Procedure for the Synthesis of Thiosquaramides
Procedure A: The 3-[(3,5-bis(trifluoromethyl)phenyl)amino]-4-(cyclopentyloxy)cyclobut-3-ene-1,2-thione (124.0 mg, 0.3 mmol, 1 eq.) was dissolved under argon atmosphere in dry CH2Cl2 (1 mL), cooled to 0 °C, then the appropriate amine (0.3 mmol, 1 eq.) was added. The mixture was stirred for about 1 h at 0 °C, while the reaction was monitored by TLC tests. Then, the solvent was evaporated, and the obtained crude product was purified by silica gel column chromatography, using a solvent mixture of ethyl acetate:methanol = 7:1 as the eluent.
Procedure B: The 3-[(3,5-bis(trifluoromethyl)phenyl)amino]-4-(cyclopentyloxy)cyclobut-3-ene-1,2-thione [
31] (124.0 mg, 0.3 mmol, 1 eq.) and appropriate amine (0.3 mmol, 1 eq.) were introduced in a 5 mL stainless steel grinding jar with 3 stainless steel balls (5 mm diameter). The jar was closed and subjected to grinding for 35 min in the vibratory ball mill operated at 23 Hz. The progress of the reaction was monitored by silica gel thin-layer chromatography plates and the obtained crude product was purified by silica gel column chromatography, using a solvent mixture of ethyl acetate:methanol = 7:1 as the eluent.
A detailed analysis of the resulting structure was performed for the hydrochlorides.
3.2.1. 3-[(3,5-Bis(trifluoromethyl)phenyl)amino]-4-[((1R,2R)-2-(piperidin-1-yl)cyclohexyl)amino]cyclobut-3-ene-1,2-dithione (8)
Yellow–orange solid, 112 mg, 69% yield (procedure A), 138 mg, 85% yield (procedure B); R
f = 0.80 (ethyl acetate/methanol, 7:1); [α
= −34.14 (
c 0.1, CHCl
3); m.p. = 137.5–139.8 °C {Lit. [
31]: m.p. = 138–144 °C}; NMR characterization for 8·HCl: δ
H (600 MHz, CDCl
3): 1.35–1.63 (m, 4H, H
aliph), 1.84–2.11 (m, 7H, H
aliph), 2.15–2.25 (m, 1H, H
aliph), 2.48–2.59 (m, 2H, H
aliph), 2.76–2.87 (m, 1H, H
aliph), 3.06–3.16 (m, 1H, H
aliph), 3.34–3.45 (m, 2H, CHN), 3.46–3.54 (m, 1H, CHN), 5.09–5.19 (m, 1H, CHN), 7.63 (s, 1H, H
ar), 8.23 (s, 2H, H
ar), 9.26 (bs, 1H, NH), 9.46 (bs, 1H, NH), 10.51 (bs, 1H, NH); δ
C (150 MHz, CDCl
3): 22.3, 23.2, 23.6, 24.0, 24.5, 29.8, 35.7 (C
aliph), 47.4 (2CH
3), 53.2, 53.7 (CHN), 68.2, 118.4, 121.9, 122.2, 124.0, 125.8 (C
6H
3(CF
3)
2), 132.3 (q,
2JC-F = 33.6 Hz), 137.8 (C
ar), 169.4, 170.4 (C=C), 206.3, 209.4 (CS); TOF MS ES
+ calculated for C
23H
25N
3F
6S
2 [M]
+ 522.1472; found 522.1475.
Spectral data matched that reported by Rawal [
31].
3.2.2. 3-[(3,5)-Bis(trifluoromethyl)phenyl)amino]-4-[((1R,2R)-2-(dimethylamino)cyclohexyl)amino]cyclobut-3-ene-1,2-dithione (9)
Yellow–orange solid, 112 mg, 74% yield (procedure A), 134 mg, 89% yield (procedure B); R
f = 0.82 (ethyl acetate/methanol, 7:1); [α
= −28.82 (
c 0.3, CHCl
3); m.p. = 153.2–156.0 °C {Lit. [
31]: m.p. = 156–160 °C}; NMR characterization for 9·HCl: δ
H (600 MHz, CDCl
3): 0.85–0.91 (m, 1H, H
aliph), 1.83–1.91 (m, 2H, H
aliph), 2.00–2.04 (m, 1H, H
aliph), 2.09–2.14 (m, 1H, H
aliph), 2.40–2.47 (m, 1H, H
aliph), 2.74–2.82 (m, 2H, H
aliph), 2.85 (s, 3H, CH
3), 2.93 (m, 3H, CH
3), 3.28–3.36 (m, 1H, H
aliph), 5.23–5.29 (m, 1H, CHN), 7.63 (s, 1H, H
ar), 8.26 (s, 2H, H
ar), 9.03 (bs, 1H, NH), 10.30 (bs, 1H, NH), 10.79 (bs, 1H, NH); δ
C (150 MHz, CDCl
3): 23.0, 23.6, 23.7, 34.5, 37.5 (C
aliph), 43.4 (2CH
3), 52.9 (CHN), 68.1, 114.3, 118.1, 121.8, 122.0, 123.9, 125.7 (C
6H
3(CF
3)
2), 132.2 (q,
2JC-F = 33.5 Hz), 138.0 (C
ar), 169.6, 171.6 (C=C), 205.2, 209.1 (CS); TOF MS ES
+ calculated for C
26H
26N
3F
6S
2 [M]
+ 558.1472; found 558.1468. TOF MS ES
+ calculated for C
20H
21N
3F
6S
2 [M]
+ 482.1159; found 482.1160.
Spectral data matched that reported by Rawal [
31].
3.2.3. 3-[(3,5)-Bis(trifluoromethyl)phenyl)amino]-4-[((S)-4-methyl-1-(piperidin-1-yl)pentan-2-yl)amino]cyclobut-3-ene-1,2-dithione (10)
Yellow–orange solid, 114 mg, 70% yield (procedure A), 140 mg, 86% yield (procedure B); Rf = 0.75 (ethyl acetate/methanol, 7:1); [α = −81.0 (c 0.2, CHCl3); m.p. = 153.9–156.5 °C; NMR characterization for 10·HCl: δH (600 MHz, CDCl3): 0.97 (d, 3H, J = 7.0, CH3), 0.98 (d, 3H, J = 7.0, CH3), 1.37–1.49 (m, 2H, Haliph), 1.68–1.77 (m, 2H, Haliph), 1.83–1.93 (m, 4H, Haliph), 2.10–2.20 (m, 1H, Haliph), 2.78–2.91 (m, 2H, Haliph), 2.95–3.06 (m, 1H, Haliph), 3.15–3.32 (m, 1H, Haliph), 3.45–3.58 (m, 1H, Haliph), 4.00–4.12 (m, 1H, Haliph), 5.63 (bs, 1H, CHN), 7.62 (s, 1H, Har), 8.33 (s, 2H, Har), 9.15 (bs, 1H, NH), 9.84 (bs, 1H, NH), 11.17 (bs, 1H, NH); δC (150 MHz, CDCl3): 21.7, 22.3, 22.5 (Caliph), 22.7 (CH3), 22.9 (Caliph), 24.8 (CH3), 43.7 (CH2), 48.0 (CHN), 53.4, 56.1, 60.8 (CH2), 114.2, 118.2, 122.2, 123.9, 125.7, 129.0 (C6H3(CF3)2), 132.1 (q, 2JC-F = 34.0 Hz), 137.9 (Car), 169.1, 171.3, (C=C), 204.8, 209.0 (CS); TOF MS ES+ calculated for C23H27N3F6S2 [M]+ 524.1629; found 524.1631.
3.2.4. 3-[(3,5)-Bis(trifluoromethyl)phenyl)amino]-4-[((S)-1-phenyl-3-(piperidin-1-yl)propan-2-yl)amino]cyclobut-3-ene-1,2-dithione (11)
Yellow–orange solid, 132 mg, 76% yield (procedure A), 157 mg, 91% yield (procedure B); Rf = 0.70 (ethyl acetate/methanol, 7:1); [α = −35.9 (c 0.2, CHCl3); m.p. = 159.3–163.0 °C; NMR characterization for 11·HCl: δH (600 MHz, CDCl3): 1.38–1.44 (m, 1H, Haliph), 1.69–1.90 (m, 3H, Haliph), 1.94–2.02 (m, 1H, Haliph), 2.11–2.18 (m, 1H, Haliph), 2.63–2.70 (m, 1H, Haliph), 2.83–2.91 (m, 2H, Haliph), 2.96–3.04 (m, 1H, Haliph), 3.43–3.49 (m, 1H, Haliph), 3.50–3.57 (m, 1H, Haliph), 3.63–3.70 (m, 1H, Halph), 3.76–3.84 (m, 1H, Haliph), 5.77 (bs, 1H, CHN), 7.30–7.35 (m, 5H, Har), 7.58 (s, 1H, Har), 8.27 (s, 2H, Har), 9.65 (bs, 1H, NH), 9.81 (bs, 1H, NH), 11.04 (bs, 1H, NH); δC (150 MHz, CDCl3): 21.8, 22.9 (Caliph), 42.2 (CH2N), 50.8 (CHN), 53.2 (CH2N), 56.6 (CH2N), 59.4 (CH2N), 118.5, 121.9, 122.2, 124.0, 128.0, 129.3, 129.4 (Car), 132.2 (q, 2JC-F = 34.0 Hz), 135.0 (C6H3(CF3)2), 137.9, 169.0 (C=C), 171.2, 209.0, 209.8 (CS); TOF MS ES+ calculated for C26H26N3F6S2 [M]+ 558.1472; found 558.1468.
3.3. General Procedure for the Reaction of N-tosylimine (13) with 1- and 2-naphthol (12 or 15) in the Presence of Catalyst 1–11
22.0 mg of 1- or 2-naphthol (0.15 mmol, 3 eq.), 13.0 mg of N-benzylidene-para-toluenesulfonamide (13) (0.05 mmol, 1 eq.), 0.005 mmol (10 mol%) of a corresponding organocatalyst, 4 Å molecular sieves, and 1 mL of toluene were placed in the Schlenk flask. The reaction was carried out under an argon atmosphere at 0 °C and the progress of the reaction was controlled by TLC tests. After completion of the reaction, the solvent was evaporated, and the residue was purified by silica gel column chromatography using a solvent mixture of hexane:ethyl acetate = 4:1 as eluent.
3.3.1. N-[(1-hydroxynaphtalen-2-yl)(phenyl)methyl]-4-methylbenzene sulfonamide (14)
Colorless oil, 20 mg, 98% yield; R
f = 0.29 (hexane/ethyl acetate, 4:1); [α
= −30.2 (
c 0.9; CH
2Cl
2) for ee = 80% (
Table 1, entry 6) {Lit. [
14]: [α
= −38.8 (c 1.0; CH
2Cl
2 for ee = 94%)}; δ
H (600 MHz, CDCl
3): 2.18 (s, 3H, CH
3), 5.91 (s, 1H, CH), 6.89 (d, 2H,
J = 8.5), 6.97 (d, 2H,
J = 8.0), 7.18–7.21 (m, 2H), 7.22–7.28 (m, 3H), 7.45–7.48 (m, 2H), 7.53–7.56 (m, 2H), 7.71–7.74 (m, 1H), 8.03–8.07 (m, 1H); δ
C (150 MHz, CDCl
3): 21.3, 58.7, 119.5, 120.6, 121.3, 125.3, 125.6, 126.2, 126.6, 127.1, 127.2, 127.6, 127.8, 128.7, 129.2, 134.2, 136.1, 139.0, 143.7, 149.5.
Spectral data matched that reported by Wang [
14].
HPLC: Kromasil AD-H (250 × 4.6 mm), hexane/iPrOH = 90/10, flow = 1 mL/min, λ = 235 nm, tS = 26.2 min and tR = 31.7 min.
3.3.2. N-[(2-hydroxynaphtalen-1-yl)(phenyl)methyl]-4-methylobenzene sulfonamide (16)
Colorless solid, 20 mg, 98% yield; R
f = 0.32 (hexane/ethyl acetate, 4:1); m.p. = 139.0–140.9 °C {Lit. [
13]: m.p. = 140–142 °C}; [α
= +33.4 (
c 0.5, CHCl
3) for ee = 54% (
Table 3, entry 2) {Lit. [
13]: [α
= +50.0 (
c 0.26; CHCl
3 for ee = 84%)}; δ
H (600 MHz, CDCl
3): 2.07 (s. 3H, CH
3), 5.68 (s, 1H, CH), 6.23 (d, 1H,
J = 9.2), 6.38 (d, 1H,
J = 9.2), 6.63 (d, 2H,
J = 8.0), 6.73 (d, 1H,
J = 8.7), 7.12–7.18 (m, 3H), 7.22–7.29 (m, 5H), 7.32–7.37 (m, 1H), 7.49 (d, 1H,
J = 8.8), 7.60 (d,
J = 8.0 Hz), 7.68 (d, 1H,
J = 8.5); δ
C (150 MHz, CDCl
3): 21.2. 54.3, 117.7, 118.1, 122.0, 123.4, 126.7, 126.8, 127.2, 127.4, 128.4, 128.7, 129.0, 129.7, 132.4, 136.3, 140.0, 142.8, 151.1.
Spectral data matched that reported by Hui [
11].
HPLC: Chiralpak OD-H (250 × 4.6 mm), hexane/iPrOH = 94/6, flow = 1 mL/min, λ = 235 nm, tS = 17.3 min, and tR = 22.9 min.
3.4. General Procedure for the Reaction of Ketimine (18) with 6-hydroxyquinoline (17) and Ketimine (18) with 1- and 2-naphthol (12 or 15) in the Presence of Catalyst 1–11
14.5 mg (0.1 mmol, 1 eq.) of 6-hydroxyquinoline or 14.4 mg (0.1 mmol, 1 eq.) of corresponding naphthol, 33.6 mg (0.1 mmol, 1 eq.) of ketimine, 0.005 mmol (5 mol%) of the appropriate organocatalyst and 4 Å molecular sieves were placed in the Schlenk flask and diluted with 1 mL of toluene. The reaction was carried out under an argon atmosphere at room temperature (or 0 °C) and the progress of the reaction was controlled by TLC tests. After completion of the reaction, the solvent was evaporated, and the residue was purified by silica gel column chromatography using a solvent mixture of hexane:ethyl acetate = 1:5 as eluent.
3.4.1. [1-Benzyl-3-(6-hydroxyquinolin-5-yl)-2-oxoindolin-3-yl]carbamate tert-butyl (19)
Yellowish solid, 37.5 mg, 78% yield; R
f = 0.26 (hexane/ethyl acetate, 1:5); m.p. = 180.5–183.0 °C {Lit. [
17]: m.p. = 183–186 °C}; [α
= −366.3 (
c 0.3, MeOH) for ee = 98% (
Table 6, entry 9) {Lit. [
17]: [α
= −368.7 (
c 0.31; MeOH for ee = 98%)}; δ
H (600 MHz, CDCl
3): 1.32 (s, 9H, C(CH
3)
3), 4.86 (d, 1H,
J = 16.1, CH
2Ph), 5.21 (d, 1H,
J = 16.1, CH
2Ph), 5.82 (s, 1H), 6.71–6.80 (m, 2H), 6.85 (d, 1H,
J = 7.9), 7.08–7.12 (m, 1H), 7.19–7.25 (m, 4H), 7.27–7.32 (m, 2H), 7.34–7.38 (m, 1H), 7.48 (d, 1H,
J = 9.1), 8.00 (d, 1H,
J = 9.1), 8.57 (dd, 1H,
J = 4.0, 1.3), 8.77 (dd, 1H), 10.45 (s, 1H, OH); δ
C (150 MHz, CDCl
3): 28.2 (CH
3), 44.7 (CH
2), 65.1, 80.9, 110.1, 114.4, 119.9, 123.4, 124.8, 125.8, 127.2, 127.5, 127.7, 129.7, 130.1, 132.2, 132.7, 135.2, 143.2, 144.4, 146.4, 153.9, 155.8, 178.8.
Spectral data matched that reported by Pedro [
17].
HPLC: Kromasil AD-H (250 × 4,6 mm), eluent hexane/iPrOH = 80/20, flow = 1 mL/min, λ = 235 nm, tS = 12.9 min and tR = 31.7 min.
3.4.2. [1-Benzyl-3-(1-hydroxynaphthalen-2-yl)-2-oxoindolin-3-yl)carbamate tert-butyl (20)
Colorless solid, 25.4 mg, 53% yield; R
f = 0.55 (hexane/ethyl acetate, 2:1); m.p. = 102.4–103.9 °C {Lit. [
15]: m.p. = 104–105 °C}; [α
= +284.7 (
c 0.4, CHCl
3) for ee = 78% (
Table 7, entry 1) {Lit. [
15]: [α
= +359.6 (
c 0.44, CHCl
3 for ee = 99%)}; δ
H (600 MHz, CDCl
3): 1.32 (s, 9H, C(CH
3)
3), 4.84 (s, 1H, CH
2Ph), 5.06 (d, 1H,
J = 12.0, CH
2Ph), 5.79 (s, 1H), 6.77 (d, 1H,
J = 7.8), 6.81 (d, 1H,
J = 8.7), 7.16–7.27 (m, 7H), 7.28–7.34 (m, 1H), 7.41 (d, 1H,
J = 7.3), 7.48–7.55 (m, 2H), 7.68–7.75 (m, 1H), 8.44–8.50 (m, 1H), 10.79 (s, 1H, OH); δ
C (150 MHz, CDCl
3): 28.3 (CH
3), 44.6 (CH
2), 65.3, 80.9, 108.7, 110.5, 114.7, 119.8, 123.4, 123.6, 125.2, 125.5, 125.8, 127.2, 127.3, 127.6, 127.8, 128.9, 129.6, 135.0, 135.1, 142.8, 154.1, 155.4, 180.0.
Spectral data matched that reported by Pedro [
15].
HPLC: Kromasil AD-H (250 × 4.6 mm), eluent hexane/iPrOH = 80/20, flow = 1.5 mL/min, λ = 254 nm, tS = 10.5 min and tR = 49.8 min.
3.4.3. [1-Benzyl-3-(2-hydroxynaphthalen-1-yl)-2-oxoindolin-3-yl)carbamate tert-butyl (21)
Colorless solid, 23.5 mg, 49% yield; R
f = 0.42 (hexane/ethyl acetate, 2:1); m.p. = 140.2–141.7 °C {Lit. [
20]: m.p. = 140.5–142.0 °C}; [α
= −7.6 (
c 0.4, CHCl
3) for ee = 46% (
Table 7, entry 3) {Lit. [
20]: [α
= −15.6 (
c 0.50, CHCl
3 for ee = 93%)}; δ
H (600 MHz, CDCl
3): 1.31 (s, 9H, C(CH
3)
3), 4.87 (d,
J = 16.0 Hz, 1H, CH
2Ph), 5.11 (d,
J = 16.0 Hz, 1H, CH
2Ph), 5.90 (s, 1H), 6.76 (d, 1H,
J = 7.8), 7.00 (d, 1H,
J = 7.7 Hz), 7.05–7.12 (m, 2H), 7.18–7.27 (m, 6H), 7.31–7.37 (m, 3H), 7.53 (d, 1H,
J = 8.7), 7.66 (d, 1H,
J = 7.5), 9.76 (s, 1H, OH); δ
C (150 MHz, CDCl
3): 28.3 (CH
3), 44.8 (CH
2), 65.5, 80.8, 110.1, 114.8, 121.2, 123.0, 123.5, 124.6, 125.4, 125.8, 127.7, 128.9, 130.4, 130.7, 131.6, 132.0, 135.3, 143.2, 154.0, 155.3, 179.8.
Spectral data matched that reported by Jin [
20].
HPLC: Kromasil AD-H (250 × 4.6 mm), eluent hexane/iPrOH = 80/20, flow = 1.5 mL/min, λ = 254 nm, tS = 10.6 min, and tR = 21.7 min.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}