
Chlorine in an Organic Molecule, a Universal Promoter—Workhorse—Of Reactions

Abstract
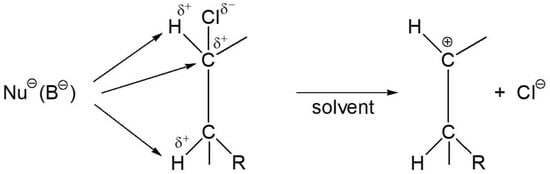
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Nucleophilic Substitution of Chlorine
2.1. In Aliphatic Compounds
2.2. Substitution of Chlorine in Aromatic Rings
3. β-Elimination
4. Chlorine Substituent as Promotor of CH Acidity
4.1. Generation and Reactions of Dihalocarbenes
4.2. Generation of α-Halocarbanions and Their Addition to Activated Carbon–Carbon Multiple Bonds
4.3. Reactions of α-Chlorocarbanions with Electron-Deficient Arenes
4.4. γ-Halocarbanions
5. Chlorine as Electrophilic Center
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mąkosza, M.; Fedoryński, M. Catalysis in Two-Phase Systems. Phase Transfer and Related Phenomena. Adv. Catal. 1987, 35, 375–422. [Google Scholar]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 3rd ed.; VCH: New York, NY, USA, 1993. [Google Scholar]
- Starks, C.M.; Liotta, C.L.; Halpern, M. Phase Transfer Catalysis. Fundamentals, Applications and Industrial Perspectives; Chapman & Hall: New York, NY, USA, 1994. [Google Scholar]
- Mąkosza, M. Two-phase reactions in organic chemistry. In Survey of Progress in Chemistry; Scott, A., Ed.; Academic Press: Cambridge, MA, USA, 1980; Volume 9, pp. 1–53. [Google Scholar]
- Jones, R.A. Quaternary Ammonium Salts: Their Use in Phase Transfer Catalysis; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Mąkosza, M.; Fedoryński, M. Phase Transfer Catalysis. In Interfacial Catalysis; Volkov, A.G., Ed.; Marcel Dekker: New York, NY, USA, 2003; pp. 159–201. [Google Scholar]
- Mąkosza, M.; Fedoryński, M. Phase Transfer Catalysis. In Encyclopedia of Catalysis; Horvath, I.T., Ed.; J. Wiley & Sons: New York, NY, USA, 2003; Volume 5, pp. 511–564. [Google Scholar] [CrossRef]
- Urbański, T.; Bełżecki, C.; Lange, J.; Makaruk, H.; Mąkosza, M.; Serafinowa, B. Sposób Wytwarzania Kwasu α-Fenylomasłowego do Celów Farmaceutycznych (Production of α-Phenylbutyric Acid for Pharmaceutical Purposes). Polish Patent 46,030, 19 June 1962. [Google Scholar]
- Mąkosza, M.; Serafinowa, B. Reakcje anionów organicznych. I. Katalityczne etylowanie fenyloacetronitrylu w środowisku wodnym (Reactions of organic anions. I. Catalytic ethylation of phenylacetonitrile in aqueous medium). Rocz. Chem. 1965, 39, 1223–1231. [Google Scholar]
- Mąkosza, M.; Jończyk, A. Phase-transfer alkylation of nitriles: 2-phenylbutyronitrile. Org. Synth. 1976, 55, 91–95. [Google Scholar]
- O’Donnell, M.J. Benzophenone Schiff bases of glycine derivatives: Versatile starting materials for the synthesis of amino acids and their derivatives. Tetrahedron 2019, 75, 3667–3696. [Google Scholar] [CrossRef]
- Maruoka, K. (Ed.) Asymmetric Phase Transfer Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Shirakawa, S.; Maruoka, K. Recent developments in asymmetric phase-transfer reactions. Angew. Chem. Int. Ed. 2013, 52, 2–39. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Gould, N.D.; Wolf, L.M. A systematic investigation of quaternary ammonium ions as asymmetric phase-transfer catalysts. Application of quantitative structure activity/selectivity relationships. J. Org. Chem. 2011, 76, 4337–4357. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M. Two-phase reactions in the chemistry of carbanions and halocarbenes. A useful tool in organic synthesis. Pure Appl. Chem. 1975, 43, 439–462. [Google Scholar] [CrossRef]
- Jończyk, A.; Kwast, A.; Mąkosza, M. Stereochemical control of the interfacial Darzens condensation. J. Chem. Soc. Chem. Commun. 1977, 902–903. [Google Scholar] [CrossRef]
- Jończyk, A.; Kwast, A.; Mąkosza, M. Stereochemical control of the interfacial cyclopropane derivatives formation. Tetrahedron Lett. 1979, 20, 541–544. [Google Scholar] [CrossRef]
- Hamkało, M.; Fita, P.; Fedoryński, M.; Mąkosza, M. Interfacial generation of a carbanion: The key step of PTC reaction directly observed by second harmonic generation. Chem. Eur. J. 2018, 24, 3975–3979. [Google Scholar] [CrossRef] [PubMed]
- Starks, C.M. Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts. J. Am. Chem. Soc. 1971, 93, 195–199. [Google Scholar] [CrossRef]
- Mąkosza, M.; Chesnokov, A. How iodide anions inhibit the phase-transfer catalyzed reactions of carbanions. Tetrahedron 2008, 64, 5925–5932. [Google Scholar] [CrossRef]
- Fedoryński, M.; Wojciechowski, K.; Matacz, Z.; Mąkosza, M. Sodium and potassium carbonates: Efficient strong bases in solid-liquid two-phase systems. J. Org. Chem. 1978, 43, 4682–4684. [Google Scholar] [CrossRef]
- Albanese, D.; Landini, D.; Maia, A.; Penso, M. Phase Transfer Catalysis: Some recent applications in organic synthesis. J. Mol. Catal. A Chem. 1999, 150, 113–131. [Google Scholar] [CrossRef]
- Khachatryan, D.S.; Matevosyan, K.R. Potassium carbonate as a base for generation of carbanions from CH-acids in organic synthesis. Russ. Chem. Bull. 2016, 65, 14–28. [Google Scholar] [CrossRef]
- Mąkosza, M. Reactions of organic anions. XVI. Catalytic nitroarylation of phenylacetonitrile derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 673–676. [Google Scholar] [CrossRef]
- Mąkosza, M.; Chesnokov, A. Cocatalysis in phase-transfer catalyzed base induced β-elimination. Model studies of dehydrobromination of bromocyclohexane. Tetrahedron 2000, 56, 3553–3558. [Google Scholar] [CrossRef]
- Mąkosza, M.; Fedoryński, M. Co-catalysis in phase transfer catalyzed reactions (a concept paper). Arkivoc 2006, 4, 7–17. [Google Scholar] [CrossRef]
- Parham, W.E.; Schweizer, E.E. Halocyclopropanes from halocarbenes. Org. React. 1963, 13, 55–90. [Google Scholar]
- Fedoryński, M. Syntheses of gem-dihalocyclopropanes and their use in organic synthesis. Chem. Rev. 2003, 103, 1099–1132. [Google Scholar] [CrossRef]
- Jończyk, A.; Fedoryński, M. Houben-Weyl Methods of Organic Chemistry; de Meijere, A., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1997; Volume E17a, pp. 589–734. [Google Scholar]
- Mąkosza, M.; Wawrzyniewicz, M. Reactions of organic anions. XXIV. Catalytic method for preparation of dichlorocyclopropane derivatives in aqueous medium. Tetrahedron Lett. 1969, 10, 4659–4662. [Google Scholar] [CrossRef]
- Dehmlow, E.V. Houben-Weyl Methoden der Organischen Chemie; Regitz, M., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 1989; Volume E19, pp. 1467–1505, 1521–1600, 1608–1622, and 1625–1627. [Google Scholar]
- Dehmlow, E.V.; Schönefeld, J. Umsetzung von Olefinen mit Dihalogencarbenen (Reactions of alkenes with dihalocarbenes). Liebigs Ann. Chem. 1971, 744, 42–50. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Fastabend, U. Do the structures of phase-transfer catalysts influence dihalogenocarbene-carbenoid selectivities? J. Chem. Soc. Chem. Commun. 1993, 1241–1242. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Lissel, M.; Heider, J. Anwendungen der phasentransfer-katalyse—4: Halogenaustausch und reaktivitäten bei dibrom-, chlorbrom- und dichlorcarben (Applications of phase transfer catalysis—4: Halogen interchange and reactivities of dibromo-, chlorobromo- and dichlorocarbenes). Tetrahedron 1977, 33, 363–366. [Google Scholar] [CrossRef]
- Isagawa, K.; Kimura, Y.; Kwon, S. Catalysis by certain amines in an aqueous phase. Preparation of dichlorocyclopropane derivatives. J. Org. Chem. 1974, 39, 3171–3172. [Google Scholar] [CrossRef]
- Mąkosza, M.; Kacprowicz, A.; Fedoryński, M. How do trialkylamines catalyze reactions of dichlorocarbene in a two-phase system. Tetrahedron Lett. 1975, 16, 2119–2122. [Google Scholar] [CrossRef]
- Jończyk, A.; Bańko, K.; Mąkosza, M. Some carbanionic reactions of halomethyl aryl sulfones. J. Org. Chem. 1975, 40, 266–267. [Google Scholar] [CrossRef]
- Jończyk, A.; Fedoryński, M.; Mąkosza, M. Reactions of organic anions. XLIII. Catalytic method for synthesis of glycidic nitriles in aqueous medium. Tetrahedron Lett. 1972, 13, 2395–2396. [Google Scholar] [CrossRef]
- Goliński, J.; Mąkosza, M. Reactions of organic anions. LXXXIV. A convenient method for synthesis of 1-chloro-, 1,1-dichloro-, and 1,2-epoxyalkanesulfonamides. Synthesis 1978, 1978, 823–825. [Google Scholar] [CrossRef]
- Jończyk, A.; Zomerfeld, T. Convenient synthesis of t-butyl Z-3-substituted glycidates under conditions of phase-transfer catalysis. Tetrahedron Lett. 2003, 44, 2359–2361. [Google Scholar] [CrossRef]
- Jończyk, A.; Mąkosza, M. Reactions of organic anions. LXVII. Catalytic synthesis of cyclopropane derivatives in aqueous medium. Synthesis 1976, 1976, 387–388. [Google Scholar] [CrossRef]
- Russell, G.A.; Mąkosza, M.; Hershberger, J. Synthesis of nitrocyclopropanes by cyclization of γ-chloro-γ-nitrocarboxylic esters and derivatives. J. Org. Chem. 1979, 44, 1195–1199. [Google Scholar] [CrossRef]
- Kryshtal, G.V.; Kulganek, V.V.; Kucherov, V.F.; Yanovskaya, L.A. Phase-transfer catalysis of the Michael addition to α,β-unsaturated aldehydes. Synthesis 1979, 1979, 107–109. [Google Scholar] [CrossRef]
- Kryshtal, G.V.; Serebryakov, E.P. Regio- and stereoselectivity in the addition reactions of CH-acids under the conditions of phase-transfer catalysis. Russ. Chem. Bull. 1995, 44, 1785–1804. [Google Scholar] [CrossRef]
- Mąkosza, M.; Winiarski, J. Vicarious nucleophilic substitution of hydrogen. Acc. Chem. Res. 1987, 20, 282–289. [Google Scholar] [CrossRef]
- Goliński, J.; Mąkosza, M. “Vicarious” nucleophilic substitution of hydrogen in aromatic nitro compounds. Tetrahedron Lett. 1978, 19, 3495–3498. [Google Scholar] [CrossRef]
- Mąkosza, M.; Owczarczyk, Z. Dihalomethylation of nitroarenes via vicarious nucleophilic substitution of hydrogen with trihalomethyl carbanions. J. Org. Chem. 1989, 54, 5094–5100. [Google Scholar] [CrossRef]
- Kisiel, K.; Brześkiewicz, J.; Loska, R.; Mąkosza, M. Transition metal free nucleophilic benzylation of nitroarenes. Umpolung of the Friedel Crafts reaction. Adv. Synth. Catal. 2019, 361, 1641–1646. [Google Scholar] [CrossRef]
- Czaban-Jóźwiak, J.; Loska, R.; Mąkosza, M. Synthesis of α-fluoro-α-nitroarylacetates via vicarious nucleophilic substitution of hydrogen. J. Org. Chem. 2016, 81, 11751–11757. [Google Scholar] [CrossRef]
- Khutorianskyi, V.V.; Klepetářová, B.; Beier, P. Vicarious nucleophilic chloromethylation of nitroarenes. Org. Lett. 2019, 21, 5443–5446. [Google Scholar] [CrossRef]
- Błażej, S.; Mąkosza, M. Substituents effects on the electrophilic activity of nitroarenes in the reactions with carbanions. Chem. Eur. J. 2008, 14, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How does nucleophilic aromatic substitution really proceed in nitroarenes? Computational prediction and experimental verification. J. Am. Chem. Soc. 2016, 138, 7276–7281. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M. How does nucleophilic aromatic substitution in nitroarenes really proceed: General mechanism. Synthesis 2017, 49, 3247–3254. [Google Scholar] [CrossRef]
- Mąkosza, M.; Lemek, T.; Kwast, A.; Terrier, F. Elucidation of the vicarious nucleophilic substitution of hydrogen mechanism via studies of competition between substitution of hydrogen, deuterium, and fluorine. J. Org. Chem. 2002, 67, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Błaziak, K.; Danikiewicz, W.; Mąkosza, M. How do aromatic nitro compounds react with nucleophiles? Theoretical description using aromaticity, nucleophilicity and electrophilicity indices. Molecules 2020, 25, 4819. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M.; Kwast, A. Vicarious nucleophilic substitution of hydrogen in electrophilic alkenes. Tetrahedron 1991, 47, 5001–5018. [Google Scholar] [CrossRef]
- Mąkosza, M.; Nizamov, S.; Kwast, A. Vicarious nucleophilic substitution of hydrogen versus vinylic substitution of halogen in the reactions of carbanions of halomethyl aryl sulfones with dialkyl halofumarates and halomaleates. Tetrahedron 2004, 60, 5413–5421. [Google Scholar] [CrossRef]
- Mąkosza, M.; Nizamov, S.; Kwast, A. Vicarious nucleophilic substitution of hydrogen in electrophilic aldimines: Synthesis of enamines substituted with electron-withdrawing groups. Mendeleev Commun. 1996, 6, 43–44. [Google Scholar] [CrossRef]
- Mąkosza, M.; Nizamov, S. Vicarious nucleophilic substitution of hydrogen (VNS) in 1,4-naphthoquinone derivatives—Competition between VNS and vinylic nucleophilic substitution (SNV). Tetrahedron 2001, 57, 9615–9621. [Google Scholar] [CrossRef]
- Mąkosza, M.; Judka, M. New reactions of γ-halocarbanions: Simple synthesis of substituted tetrahydrofurans. Chem. Eur. J. 2002, 8, 4234–4240. [Google Scholar] [CrossRef]
- Mąkosza, M.; Judka, M. New synthesis of substituted cyclopentanes via reactions of γ-chlorocarbanions with electron-deficient alkenes. Synlett 2004, 2004, 717–719. [Google Scholar] [CrossRef]
- Mąkosza, M.; Judka, M. New synthesis of pyrrolidines via reactions of γ-halocarbanions with imines. Helv. Chim. Acta 2005, 88, 1676–1681. [Google Scholar] [CrossRef]
- Barbasiewicz, M.; Mąkosza, M. Intermolecular reactions of chlorohydrine anions: Acetalization of carbonyl compounds under basic conditions. Org. Lett. 2006, 8, 3745–3748. [Google Scholar] [CrossRef]
- Judka, M.; Wojtasiewicz, A.; Danikiewicz, W.; Mąkosza, M. Halogens in γ-position enhance the acidity of alkyl aryl sulfones and alkane nitriles. Tetrahedron 2007, 63, 8902–8909. [Google Scholar] [CrossRef]
- Mąkosza, M.; Podraza, R.; Białecki, M. New simple method of synthesis of substituted haloalkenes via Knoevenagel type condensation of aldehydes with α-halosubstituted CH acids. Gazz. Chim. Ital. 1995, 125, 601–603. [Google Scholar]
- Mąkosza, M.; Serafinowa, B.; Gajos, I. Reakcje anionów organicznych. XXI. Katalityczne reakcje nitryli arylooctowych z chloroformem i czterochlorkiem węgla w środowisku wodnym (Reactions of organic anions. XXI. Catalytic reactions of aryloacetonitriles with chloroform and carbon tetrachloride in aqueous medium). Roczniki Chem. 1969, 43, 671–676. [Google Scholar]
- Jończyk, A.; Kwast, A.; Mąkosza, M. Reactions of carbon tetrachloride with carbon acids in catalytic two-phase system. J. Org. Chem. 1979, 44, 1192–1194. [Google Scholar] [CrossRef]
- Mąkosza, M.; Kwast, E.; Kwast, A.; Jończyk, A. Reactions of carbanions with carbon tetrachloride in two-phase systems. Chlorinated products as nucleophilic and electrophilic intermediates. J. Org. Chem. 1985, 50, 3722–3727. [Google Scholar] [CrossRef]
- Mąkosza, M.; Goliński, J. Vicarious nucleophilic substitution of hydrogen in nitroarenes using 1-chloroalkanesulfonic esters; A simple synthesis of 1-(nitrophenyl)-alkanesulfonic and (nitrophenyl)-methanesulfonic esters. Synthesis 1983, 1983, 1023–1025. [Google Scholar] [CrossRef]
- Jończyk, A.; Radwan-Pytlewski, T. Some carbanionic reactions of α-chloroallyl sulfones. Chem. Lett. 1983, 12, 1557–1560. [Google Scholar] [CrossRef]
- Meyers, C.Y.; Matthews, W.S.; Ho, L.L.; Kolb, V.M.; Parady, T.E. Catalysis in Organic Synthesis—1977; Smith, G.V., Ed.; Academic Press: New York, NY, USA, 1978; p. 197. [Google Scholar]
- Mąkosza, M.; Kwast, A. Dichloro(2,2-dimethylcyclopropyl)methyl phenyl sulfone—A radical probe for detecting single electron transfer processes. Bull. Soc. Chim. Belg. 1994, 103, 445–448. [Google Scholar] [CrossRef]
- Appel, R. Tertiary phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and P-N linkage. Angew. Chem. Int. Ed. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Le Corre, S.S.; Berchel, M.; Couthon-Gourvès, H.; Haelters, J.-P.; Jaffrès, P.-A. Atherton-Todd reaction mechanism, scope and applications. Beilst. J. Org. Chem. 2014, 10, 1166–1196. [Google Scholar] [CrossRef] [PubMed]
- Zwierzak, A. Phase-transfer-catalysed phosphorylation of amines in an aqueous system. Synthesis 1975, 1975, 507–509. [Google Scholar] [CrossRef]

































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mąkosza, M.; Fedoryński, M. Chlorine in an Organic Molecule, a Universal Promoter—Workhorse—Of Reactions. Molecules 2023, 28, 7957. https://doi.org/10.3390/molecules28247957
Mąkosza M, Fedoryński M. Chlorine in an Organic Molecule, a Universal Promoter—Workhorse—Of Reactions. Molecules. 2023; 28(24):7957. https://doi.org/10.3390/molecules28247957
Chicago/Turabian StyleMąkosza, Mieczysław, and Michał Fedoryński. 2023. "Chlorine in an Organic Molecule, a Universal Promoter—Workhorse—Of Reactions" Molecules 28, no. 24: 7957. https://doi.org/10.3390/molecules28247957
APA StyleMąkosza, M., & Fedoryński, M. (2023). Chlorine in an Organic Molecule, a Universal Promoter—Workhorse—Of Reactions. Molecules, 28(24), 7957. https://doi.org/10.3390/molecules28247957