


Three-Dimensional-QSAR and Relative Binding Affinity Estimation of Focal Adhesion Kinase Inhibitors



Abstract
:1. Introduction
2. Results and Discussion
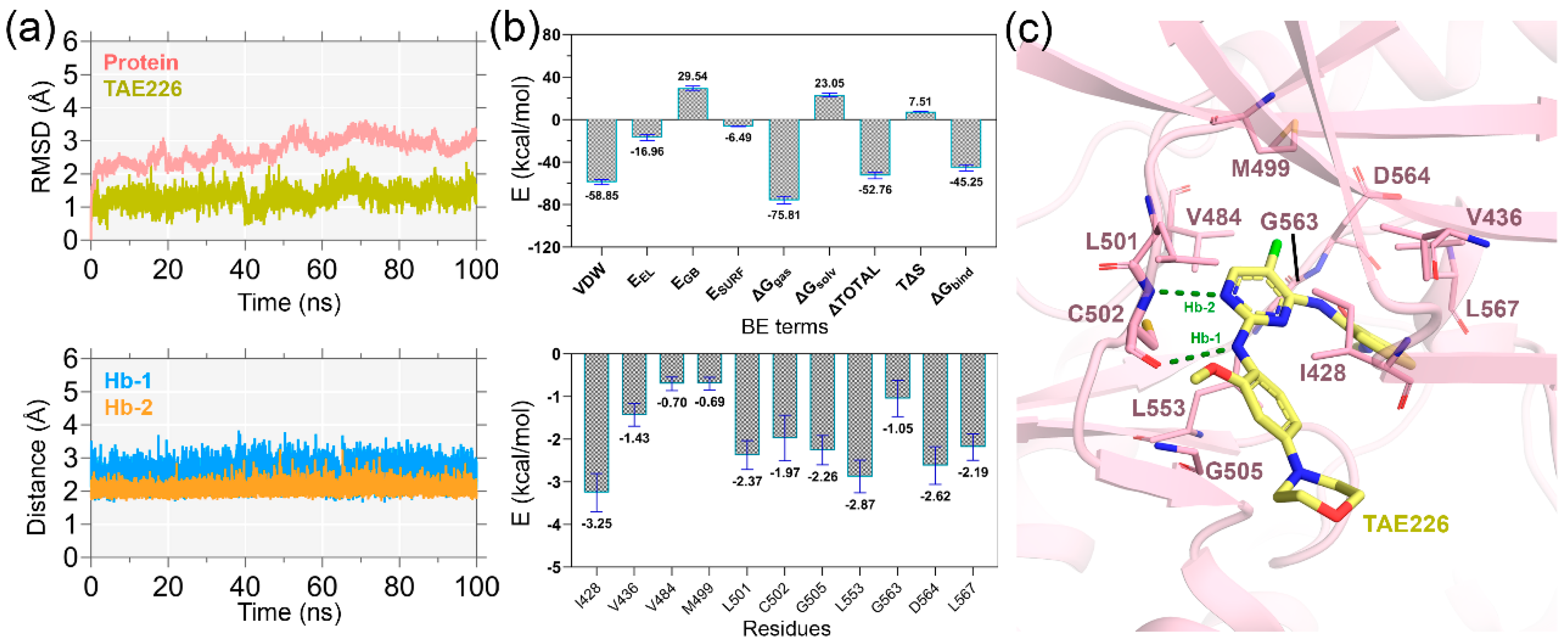
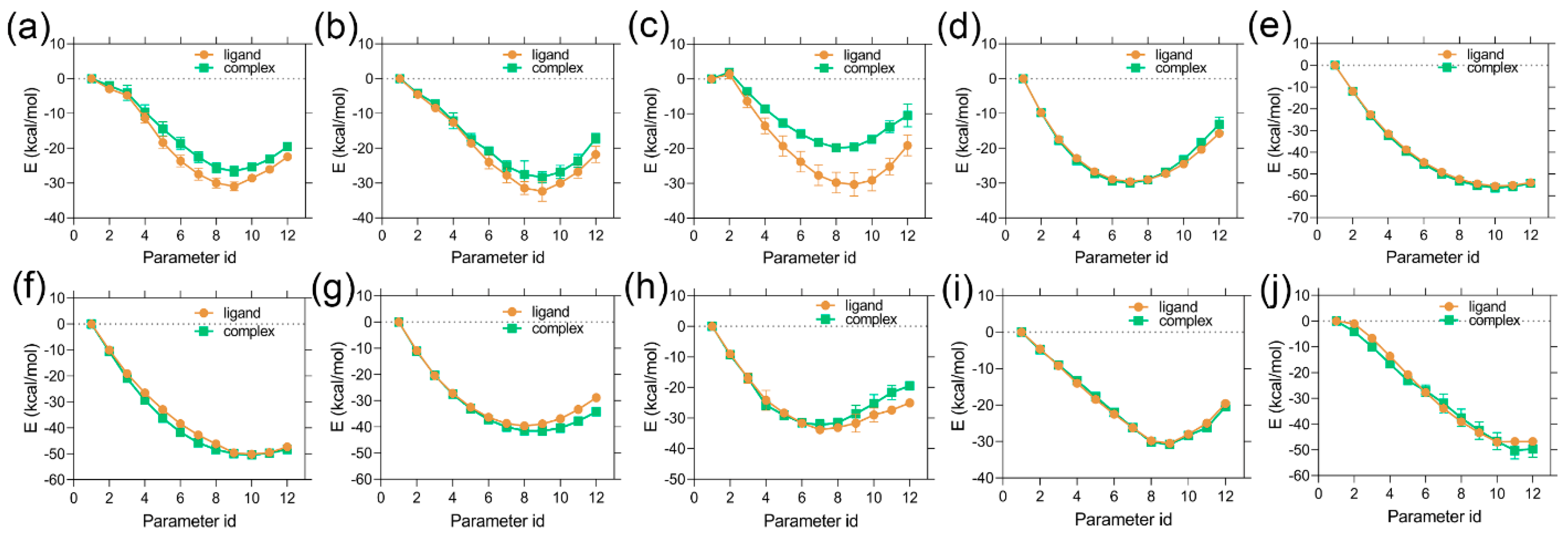
2.1. MD Simulation Analysis and Binding Energy Calculation
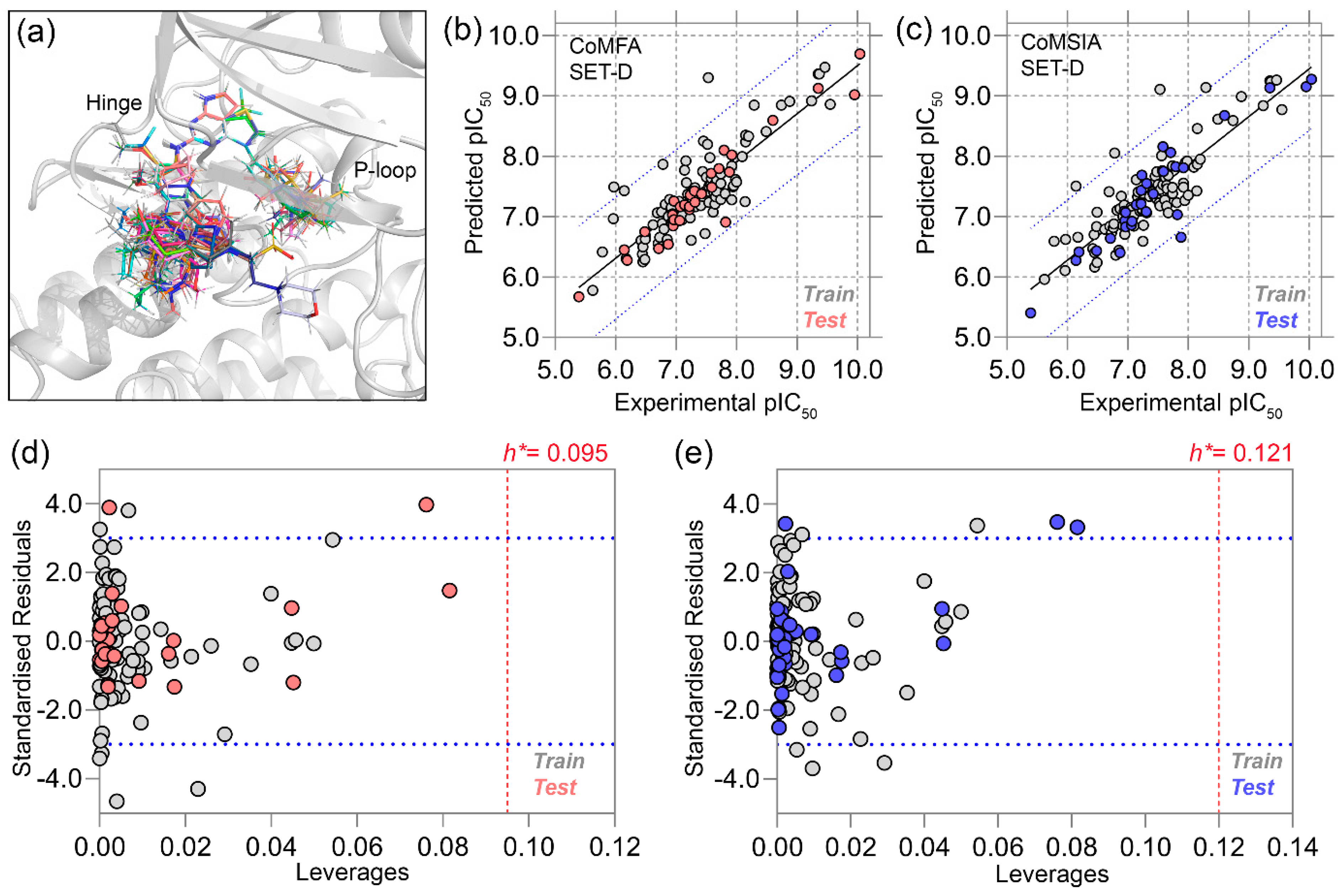
2.2. Statistical Analysis of 3D-QSAR Models
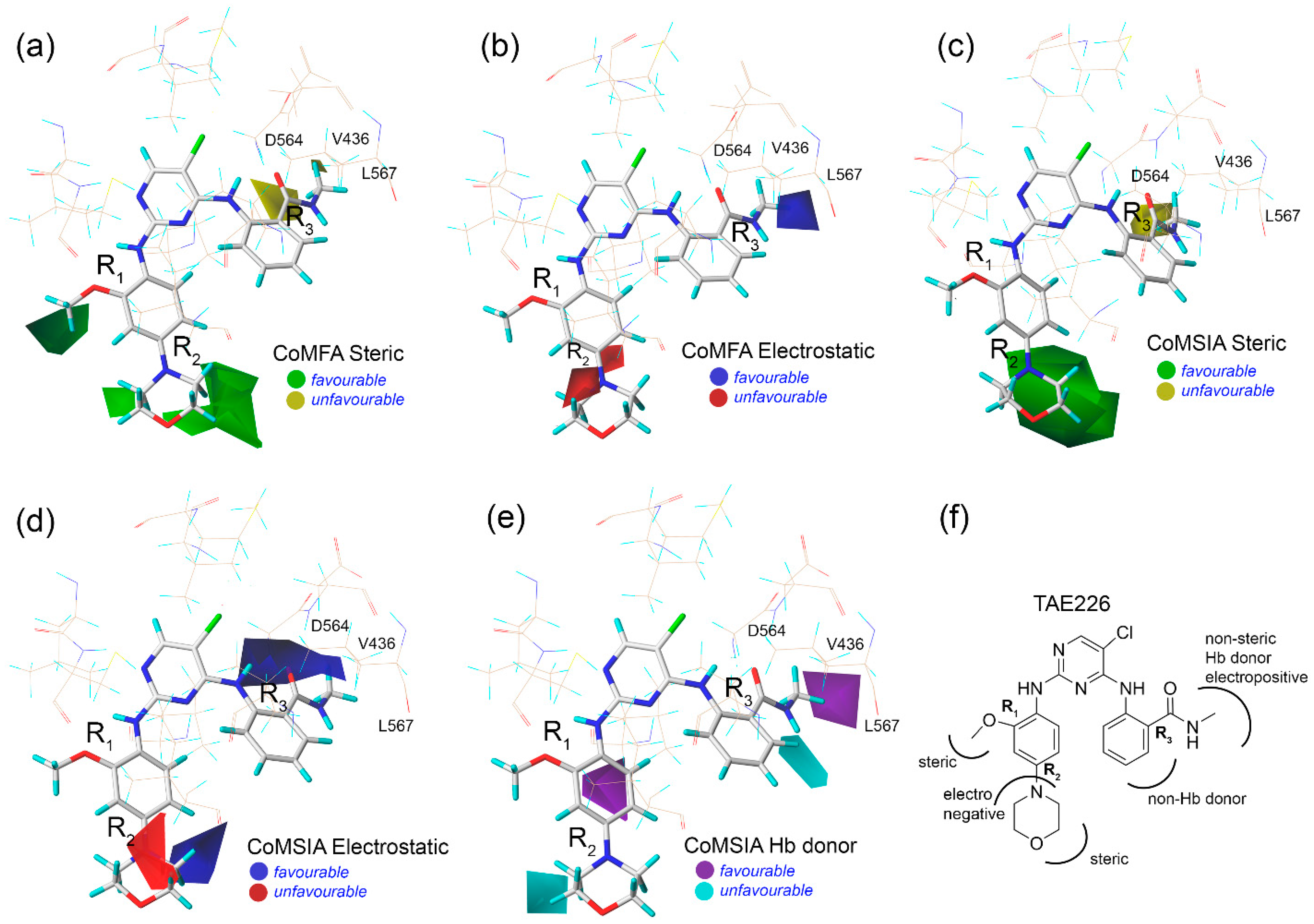
2.3. Contour Map Analysis
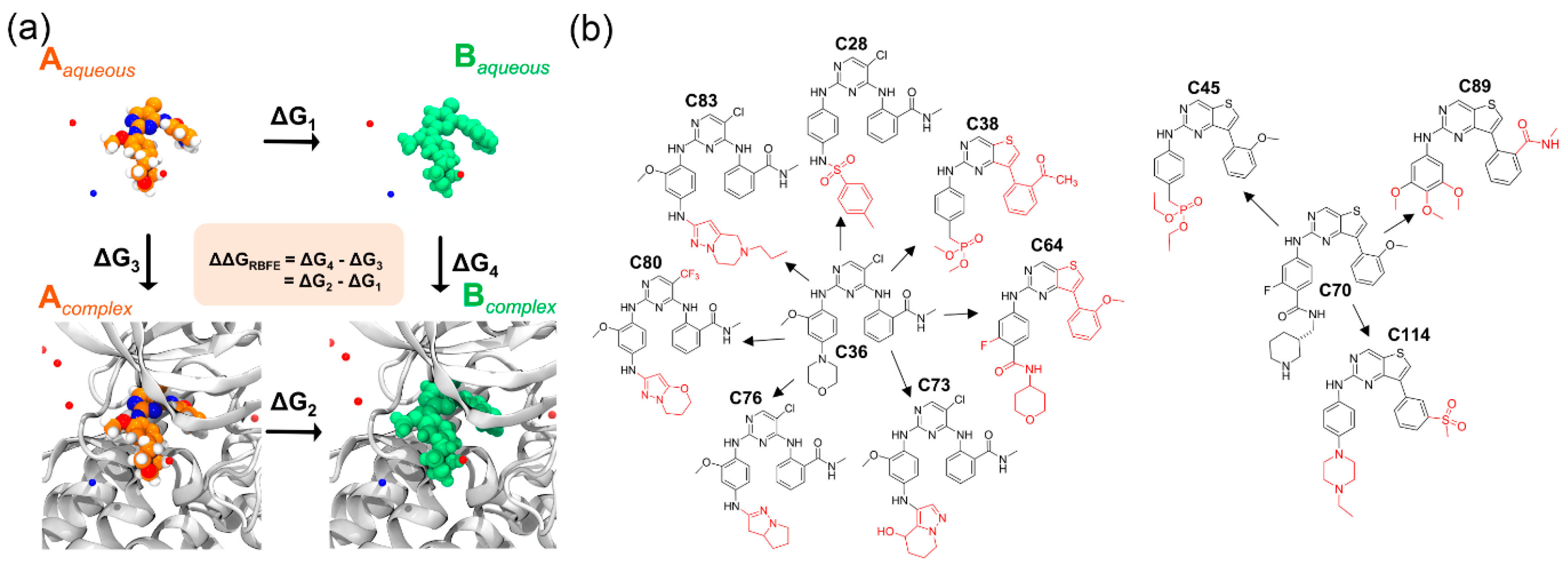
2.4. Relative Binding Affinity Estimation
3. Methodology
3.1. Structure Preparation
3.2. MD Simulation and Binding Energy Calculation
3.3. Dataset Preparation and Molecular Modeling
3.4. Development of 3D-QSAR Models
3.5. Relative Binding Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SBDD | Structure-based Drug Discovery |
| FAK | Focal Adhesion Kinase |
| MD | Molecular Dynamics |
| 3D-QSAR | Three-dimensional Quantitative Structure-Activity Relationship |
| MM-PB/GBSA | Molecular Mechanics-Poison Boltzmann/Generalized Born Surface Area |
| FEP | Free Energy Perturbation |
| CoMFA | Comparative Molecular Field Analysis |
| CoMSIA | Comparative Molecular Similarity Indices Analysis |
| ML | Machine Learning |
| SAR | Structure-Activity Relationship |
| LJ | Lennard-Jones |
| BAR | Bennet Acceptance Ratio |
| ACEPYPE | AnteChamber Python Parser interfacE |
| AD | Applicability Domain |
References
- Liao, Y.; Liu, L.; Yang, J.; Shi, Z. ATX/LPA axis regulates FAK activation, cell proliferation, apoptosis, and motility in human pancreatic cancer cells. In Vitro Cell. Dev. Biol. Anim. 2022, 58, 307–315. [Google Scholar]
- Pomella, S.; Cassandri, M.; Braghini, M.R.; Marampon, F.; Alisi, A.; Rota, R. New Insights on the Nuclear Functions and Targeting of FAK in Cancer. Int. J. Mol. Sci. 2022, 23, 1998. [Google Scholar]
- Le Coq, J.; Acebrón, I.; Martin, B.R.; Navajas, P.L.; Lietha, D. New insights into FAK structure and function in focal adhesions. J. Cell Sci. 2022, 135, jcs259089. [Google Scholar]
- Zhai, C.; Zhang, N.; Wang, J.; Cao, M.; Luan, J.; Liu, H.; Zhang, Q.; Zhu, Y.; Xue, Y.; Li, S. Activation of autophagy induces monocrotaline-induced pulmonary arterial hypertension by FOXM1-mediated FAK phosphorylation. Lung 2022, 200, 619–631. [Google Scholar] [CrossRef]
- Yang, J.Y.; Woo, H.J.; Lee, P.; Kim, S.-H. Induction of Apoptosis and Effect on the FAK/AKT/mTOR Signal Pathway by Evodiamine in Gastric Cancer Cells. Curr. Issues Mol. Biol. 2022, 44, 4339–4349. [Google Scholar] [CrossRef]
- Spallarossa, A.; Tasso, B.; Russo, E.; Villa, C.; Brullo, C. The Development of FAK Inhibitors: A Five-Year Update. Int. J. Mol. Sci. 2022, 23, 6381. [Google Scholar]
- Wankowicz, S.A.; de Oliveira, S.H.; Hogan, D.W.; Bedem, H.V.D.; Fraser, J.S. Ligand binding remodels protein side-chain conformational heterogeneity. Elife 2022, 11, e74114. [Google Scholar] [CrossRef]
- Singh, V.K.; Chaurasia, H.; Mishra, R.; Srivastava, R.; Naaz, F.; Kumar, P.; Singh, R.K. Docking, ADMET prediction, DFT analysis, synthesis, cytotoxicity, antibacterial screening and QSAR analysis of diarylpyrimidine derivatives. J. Mol. Struct. 2022, 1247, 131400. [Google Scholar] [CrossRef]
- Castelli, M.; Serapian, S.A.; Marchetti, F.; Triveri, A.; Pirota, V.; Torielli, L.; Collina, S.; Doria, F.; Freccero, M.; Colombo, G. New perspectives in cancer drug development: Computational advances with an eye to design. RSC Med. Chem. 2021, 12, 1491–1502. [Google Scholar] [PubMed]
- Wang, R.; Chen, Y.; Yang, B.; Yu, S.; Zhao, X.; Zhang, C.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrrolo[2,3-b]pyridine derivatives as potential anti-tumor agents. Bioorg. Chem. 2020, 94, 103474. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhao, X.; Yu, S.; Chen, Y.; Cui, H.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Discovery of 7H-pyrrolo[2,3-d]pyridine derivatives as potent FAK inhibitors: Design, synthesis, biological evaluation and molecular docking study. Bioorg. Chem. 2020, 102, 104092. [Google Scholar]
- Qu, M.; Liu, Z.; Zhao, D.; Wang, C.; Zhang, J.; Tang, Z.; Liu, K.; Shu, X.; Yuan, H.; Ma, X. Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (Sul-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with antitumor activity. Bioorg. Med. Chem. 2017, 25, 3989–3996. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yu, S.; Zhao, X.; Chen, Y.; Yang, B.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 2020, 188, 112024. [Google Scholar] [PubMed]
- Xie, H.; Lin, X.; Zhang, Y.; Tan, F.; Chi, B.; Peng, Z.; Dong, W.; An, D. Design, synthesis and biological evaluation of ring-fused pyrazoloamino pyridine/pyrimidine derivatives as potential FAK inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127459. [Google Scholar] [PubMed]
- Halder, J.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Honda, T.; Kamat, A.A.; Han, L.Y.; Kim, T.J.; Pluquet, O.; et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007, 67, 10976–10983. [Google Scholar]
- Shi, Q.; Hjelmeland, A.B.; Keir, S.T.; Song, L.; Wickman, S.; Jackson, D.; Ohmori, O.; Bigner, D.D.; Friedman, H.S.; Rich, J.N. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol. Carcinog. 2007, 46, 488–496. [Google Scholar] [PubMed]
- Lietha, D.; Eck, M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE 2008, 3, e3800. [Google Scholar]
- Zhou, J.; Bronowska, A.; Le Coq, J.; Lietha, D.; Gräter, F. Allosteric regulation of focal adhesion kinase by PIP2 and ATP. Biophys. J. 2015, 108, 698–705. [Google Scholar] [PubMed]
- Ghosh, S.; Cho, S.J. Structural Insights from Molecular Modeling of Isoindolin-1-One Derivatives as PI3Kγ Inhibitors against Gastric Carcinoma. Biomedicines 2022, 10, 813. [Google Scholar] [CrossRef] [PubMed]
- Keretsu, S.; Ghosh, S.; Cho, S.J. Computer aided designing of novel pyrrolopyridine derivatives as JAK1 inhibitors. Sci. Rep. 2021, 11, 23051. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.J.; Cho, S.J. Comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) study of mutagen X. Bull. Korean Chem. Soc. 2004, 25, 1525–1530. [Google Scholar]
- San Juan, A.A.; Cho, S.J. 3D-QSAR study of microsomal prostaglandin E2 synthase (mPGES-1) inhibitors. J. Mol. Model. 2007, 13, 601–610. [Google Scholar] [CrossRef]
- Ghosh, S.; Cho, S.J. Structure–activity relationship and in silico development of c-Met kinase inhibitors. Bull. Korean Chem. Soc. 2022, 43, 882. [Google Scholar]
- Ghosh, S.; Cho, S.J. Binding Studies and Lead Generation of Pteridin-7 (8H)-one Derivatives Targeting FLT3. Int. J. Mol. Sci. 2022, 23, 7696. [Google Scholar] [CrossRef]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Computational Modeling of Novel Phosphoinositol-3-kinase γ Inhibitors Using Molecular Docking, Molecular Dynamics, and 3D-QSAR. Bull. Korean Chem. Soc. 2021, 42, 1093. [Google Scholar] [CrossRef]
- Ghosh, S.; Keretsu, S.; Cho, S.J. 3D-QSAR, Docking and Molecular Dynamics Simulation Study of C-Glycosylflavones as GSK-3β Inhibitors. J. Chosun Nat. Sci. 2020, 13, 170–180. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Molecular Modeling Studies of N-phenylpyrimidine-4-amine Derivatives for Inhibiting FMS-like Tyrosine Kinase-3. Int. J. Mol. Sci. 2021, 22, 12511. [Google Scholar]
- Ghosh, S.; Cho, S.J. Comparative binding affinity analysis of dual CDK2/FLT3 inhibitors. Bull. Korean Chem. Soc. 2022, 43, 1320. [Google Scholar]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Keretsu, S.; Cho, S.J. Designing of the N-ethyl-4-(pyridin-4-yl)benzamide based potent ROCK1 inhibitors using docking, molecular dynamics, and 3D-QSAR. PeerJ 2021, 9, e11951. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Ballabio, D.; Grisoni, F. Beware of unreliable Q 2! A comparative study of regression metrics for predictivity assessment of QSAR models. J. Chem. Inf. Model. 2016, 56, 1905–1913. [Google Scholar] [CrossRef]
- Veerasamy, R.; Rajak, H.; Jain, A.; Sivadasan, S.; Varghese, C.P.; Agrawal, R.K. Validation of QSAR models-strategies and importance. Int. J. Drug Des. Discov. 2011, 3, 511–519. [Google Scholar]
- Abdizadeh, R.; Hadizadeh, F.; Abdizadeh, T. QSAR analysis of coumarin-based benzamides as histone deacetylase inhibitors using CoMFA, CoMSIA and HQSAR methods. J. Mol. Struct. 2020, 1199, 126961. [Google Scholar]
- Cournia, Z.; Allen, B.; Sherman, W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar]
- Jung, J.; Mori, T.; Kobayashi, C.; Matsunaga, Y.; Yoda, T.; Feig, M.; Sugita, Y. GENESIS: A hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput Mol. Sci. 2015, 5, 310–323. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36: An improved force field for folded and intrinsically disordered proteins. Biophys. J. 2017, 112, 175a–176a. [Google Scholar]
- Kim, S.; Oshima, H.; Zhang, H.; Kern, N.R.; Re, S.; Lee, J.; Roux, B.; Sugita, Y.; Jiang, W.; Im, W. CHARMM-GUI free energy calculator for absolute and relative ligand solvation and binding free energy simulations. J. Chem. Theory Comput. 2020, 16, 7207–7218. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Parameters | CoMFA | CoMSIA (SED) SET-D | Threshold Values | Statistical Parameters | CoMFA | CoMSIASET-D | Threshold Values | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SET-A | SET-B | SET-C | SET-D | SET-A | SET-B | SET-C | SET-D | ||||||
| q2 | 0.593 | 0.541 | 0.505 | 0.633 | 0.656 | >0.5 | kTest | 0.994 | 0.979 | 1.009 | 1.007 | 1.011 | 0.85 ≤ k or k′ ≤ 1.15 |
| ONC | 5 | 2 | 2 | 6 | 6 | k′Test | 1.002 | 1.015 | 0.985 | 0.991 | 0.985 | ||
| SEP | 0.559 | 0.554 | 0.612 | 0.521 | 0.510 | r2Test | 0.578 | 0.422 | 0.767 | 0.922 | 0.850 | ||
| r2 | 0.839 | 0.666 | 0.643 | 0.897 | 0.862 | >0.6 | Test | 0.494 | 0.377 | 0.735 | 0.915 | 0.854 | ≈r2 |
| SEE | 0.352 | 0.473 | 0.277 | 0.277 | 0.323 | <<1 | Test | 0.540 | 0.240 | 0.417 | 0.886 | 0.816 | |
| F-value | 91.487 | 90.592 | 81.911 | 125.822 | 89.719 | Test | 0.046 | 0.137 | 0.317 | 0.028 | 0.037 | <0.3 | |
| BS-r2 | 0.895 | 0.712 | 0.699 | 0.934 | 0.940 | Test | 0.144 | 0.104 | 0.317 | 0.007 | −0.003 | <0.1 | |
| BS-SD | 0.025 | 0.051 | 0.050 | 0.017 | 0.016 | Test | 0.064 | 0.430 | 0.041 | 0.038 | 0.039 | ||
| χ2 | 0.285 | 0.537 | 0.507 | 0.387 | 0.325 | <1.0 | Test | 0.410 | 0.333 | 0.630 | 0.846 | N/A | > 0.5 |
| RMSE | 0.333 | 0.437 | 0.430 | 0.382 | 0.356 | <0.5 | Test | 0.466 | 0.242 | 0.313 | 0.748 | 0.694 | |
| MAE | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 | ≈0 | 0.495 | 0.361 | 0.724 | 0.911 | 0.843 | >0.6 | |
| RSS | 14.275 | 24.554 | 23.748 | 15.253 | 16.28 | 0.495 | 0.361 | 0.724 | 0.911 | 0.843 | |||
| kTrain | 0.996 | 1.003 | 0.998 | 0.991 | 0.997 | 0.85 ≤ k or k′ ≤ 1.15 | 0.493 | 0.353 | 0.723 | 0.910 | 0.842 | ||
| k′Train | 1.000 | 0.991 | 0.996 | 1.005 | 0.999 | 0.495 | 0.361 | 0.724 | 0.911 | 0.843 | |||
| Train | 0.814 | 0.665 | 0.597 | 0.667 | 0.718 | ≈r2 | 0.759 | 0.655 | 0.811 | 0.950 | 0.916 | ||
| Train | 0.785 | 0.396 | 0.467 | 0.635 | 0.662 | S (%) | 47.1 | 47.0 | 46.9 | 39.4 | 18.7 | ||
| Train | 0.028 | 0.269 | 0.129 | 0.041 | 0.055 | <0.3 | E (%) | 52.9 | 53.0 | 53.1 | 60.6 | 46.1 | |
| Train | 0.029 | 2.53 × 10−5 | 0.071 | 0.245 | 0.167 | <0.1 | H (%) | ||||||
| Train | 0.063 | 0.404 | 0.273 | 0.291 | 0.231 | A (%) | |||||||
| Train | 0.706 | 0.663 | 0.505 | 0.476 | 0.534 | > 0.5 | D (%) | 35.2 | |||||
| Train | 0.644 | 0.320 | 0.373 | 0.438 | 0.477 | ||||||||
| C36 (−11.33) | C28 (−9.89) | 1.44 | −19.51 ± 0.87 | −22.45 ± 0.99 | 2.94 |
| C38 (−8.33) | 3.00 | −17.12 ± 2.35 | −21.77 ± 1.41 | 4.58 | |
| C64 (−9.67) | 1.66 | −19.11 ± 3.26 | −10.48 ± 2.95 | −8.63 | |
| C73 (−10.40) | 0.93 | −13.09 ± 0.64 | −15.73 ± 2.06 | 2.64 | |
| C76 (−12.76) | −1.43 | −54.20 ± 0.31 | −53.97 ± 0.77 | −0.23 | |
| C80 (−14.03) | −2.70 | −34.22 ± 0.13 | −28.88 ± 1.16 | −5.34 | |
| C83 (−11.86) | −0.53 | −48.29 ± 0.63 | −47.34 ± 0.58 | −0.97 | |
| C70 (−9.53) | C45 (−9.50) | 0.03 | −19.44 ± 1.02 | −25.01 ± 0.26 | 5.57 |
| C89 (−10.13) | −0.60 | −20.41 ± 0.32 | −19.62 ± 0.60 | −0.79 | |
| C114 (−10.82) | −1.29 | −49.62 ± 1.06 | −46.76 ± 3.29 | −2.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, S.; Cho, S.J. Three-Dimensional-QSAR and Relative Binding Affinity Estimation of Focal Adhesion Kinase Inhibitors. Molecules 2023, 28, 1464. https://doi.org/10.3390/molecules28031464
Ghosh S, Cho SJ. Three-Dimensional-QSAR and Relative Binding Affinity Estimation of Focal Adhesion Kinase Inhibitors. Molecules. 2023; 28(3):1464. https://doi.org/10.3390/molecules28031464
Chicago/Turabian StyleGhosh, Suparna, and Seung Joo Cho. 2023. "Three-Dimensional-QSAR and Relative Binding Affinity Estimation of Focal Adhesion Kinase Inhibitors" Molecules 28, no. 3: 1464. https://doi.org/10.3390/molecules28031464
APA StyleGhosh, S., & Cho, S. J. (2023). Three-Dimensional-QSAR and Relative Binding Affinity Estimation of Focal Adhesion Kinase Inhibitors. Molecules, 28(3), 1464. https://doi.org/10.3390/molecules28031464