

A Survey on the Synthesis of Variolins, Meridianins, and Meriolins—Naturally Occurring Marine (aza)Indole Alkaloids and Their Semisynthetic Derivatives †


Abstract
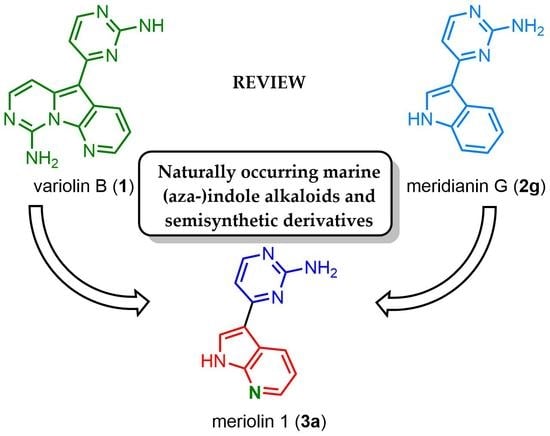
:
1. Introduction
2. Synthesis
2.1. Syntheses of Variolins
2.1.1. First Total Synthesis by Morris and Anderson
2.1.2. Synthesis by Molina and Fresneda
2.1.3. Variolin B Approach by Alvarez
2.1.4. Synthesis of Variolin B by Burgos and Vaquero
2.2. Syntheses of Meridianins
2.2.1. First Total Synthesis of Meridianins D and G by Jiang and Yang
2.2.2. Synthesis of Meridianins by Fresneda and Molina
2.2.3. Meridianin Synthesis by Müller via Carbonylative Alkynylation
2.2.4. Meridianin Synthesis by Penoni via Indolozation of Nitrosoarenes
2.2.5. Synthesis of Meridianins via One-Pot Masuda Borylation-Suzuki Coupling Sequence by Müller
2.2.6. Synthesis of Meridianin F by Grainger
2.2.7. Domino Amino-Palladation Reaction for the Synthesis of Meridianins C and G by Morris
2.3. Syntheses of Meriolins
2.3.1. First Synthesis of Meriolin 1 by Molina and Fresneda
2.3.2. Synthesis of Meriolin Derivatives by Joseph and Meijer
2.3.3. Meriolin Syntheses by Müller via Carbonylative Alkynylation
2.3.4. Three-Component Glyoxylation Decarbonylative Alkynylation Synthesis of Alkynones by Müller
2.3.5. Synthesis of Meriolins with a Suzuki Coupling as a Key Reaction by Huang
2.3.6. Meriolin Synthesis via the Masuda borylation-Suzuki Coupling Sequence by Müller
2.3.7. Domino Amino-Palladation Reaction for the Synthesis of Meriolins by Morris
2.3.8. Metal-Free CH-Activation of a Pyrimidine and an Indolylboronic Ester by Singh
2.3.9. Functionalization of Meriolins via Suzuki Coupling or Nucleophilic Substitution Reactions by Singh and Malik
2.3.10. Meriolin Synthesis via Friedel Crafts Acylation by Grädler
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- França, P.H.B.; Barbosa, D.; da Silva, D.; Ribeiro, Ê.N.; Santana, A.; Santos, B.; Barbosa-Filho, J.; Quintans, J.; Barreto, R.; Quintans-Júnior, L.J.; et al. Indole Alkaloids from Marine Sources as Potential Leads against Infectious Diseases. BioMed Res. Int. 2014, 2014, 375423. [Google Scholar] [CrossRef] [PubMed]
- König, G.M.; Wright, A.; Sticher, O.; Angerhofer, C.; Pezzuto, J. Biological Activities of Selected Marine Natural Products. Planta Med. 1994, 60, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, P.M.; Cintra, L.S.; Braguine, C.G.; Filho, A.A.D.S.; Silva, M.L.A.E.; Cunha, W.R.; Januário, A.H. Halogenated Indole Alkaloids from Marine Invertebrates. Mar. Drugs 2010, 8, 1526–1549. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.I.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Takuma, S. Hyrtiosins A and B, new indole alkaloids from the Okinawan marine sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Ban, Y.; Murakami, Y.; Iwasawa, Y.; Tsuchiya, M.; Takano, N. Indole alkaloids in medicine. Med. Res. Rev. 1988, 8, 231–308. [Google Scholar] [CrossRef]
- Abdelmohsen, U.R.; Balasubramanian, S.; Oelschlaeger, T.; Grkovic, T.; Pham, N.; Quinn, R.; Hentschel, U. Potential of marine natural products against drug-resistant fungal, viral, and parasitic infections. Lancet Infect. Dis. 2017, 17, e30–e41. [Google Scholar] [CrossRef]
- Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.; Munro, M.; Parkin, S.; Hope, H. Alkaloids from the antarctic sponge Kirkpatrickia varialosa: Part 1: Variolin b, a new antitumour and antiviral compound. Tetrahedron 1994, 50, 3987–3992. [Google Scholar] [CrossRef]
- Trimurtulu, G.; Faulkner, D.; Perry, N.; Ettouati, L.; Litaudon, M.; Blunt, J.; Munro, M.; Jameson, G. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin A and N(3′)-methyl tetrahydrovariolin B. Tetrahedron 1994, 50, 3993–4000. [Google Scholar] [CrossRef]
- Franco, L.H.; Joffé, E.K.; Puricelli, L.; Tatian, M.; Seldes, A.; Palermo, J. Indole Alkaloids from the Tunicate Aplidium meridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef]
- Seldes, A.M.; Brasco, M.R.; Franco, L.H.; Palermo, J. Identification of two meridianins from the crude extract of the tunicate Aplidium meridianum by tandem mass spectrometry. Nat. Prod. Res. 2007, 21, 555–563. [Google Scholar] [CrossRef]
- Gompel, M.; Leost, M.; De Kier Joffe, E.; Puricelli, L.; Franco, L.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.; Khan, S.; Tekwani, B.; Jacob, M.; Khan, I.; Vishwakarma, R. Meridianin G and its analogs as antimalarial agents. MedChemComm 2013, 4, 1042–1048. [Google Scholar] [CrossRef]
- Yadav, R.R.; Khan, S.; Singh, S.; Khan, I.; Vishwakarma, R.; Bharate, S. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur. J. Med. Chem. 2015, 98, 160–169. [Google Scholar] [CrossRef]
- Bettayeb, K.; Tirado, O.; Marionneau-Lambot, S.V.; Ferandin, Y.; Lozach, O.; Morris, J.; Mateo-Lozano, S.; Drueckes, P.; Schächtele, C.; Kubbutat, M.; et al. Meriolins, a New Class of Cell Death–Inducing Kinase Inhibitors with Enhanced Selectivity for Cyclin-Dependent Kinases. Cancer Res. 2007, 67, 8325–8334. [Google Scholar] [CrossRef] [Green Version]
- Fresneda, P.M.; Molina, P.; Bleda, J. Synthesis of the indole alkaloids meridianins from the tunicate Aplidium meridianum. Tetrahedron 2001, 57, 2355–2363. [Google Scholar] [CrossRef]
- Echalier, A.; Bettayeb, K.; Ferandin, Y.; Lozach, O.; Clément, M.; Valette, A.; Liger, F.; Marquet, B.; Morris, J.; Endicott, J.; et al. Meriolins (3-(Pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/Cyclin A/Meriolin Complex. J. Med. Chem. 2008, 51, 737–751. [Google Scholar] [CrossRef]
- Jarry, M.; Lecointre, C.; Malleval, C.; Desrues, L.; Schouft, M.-T.; Lejoncour, V.; Liger, F.; Lyvinec, G.; Joseph, B.; Loaëc, N.; et al. Impact of meriolins, a new class of cyclin-dependent kinase inhibitors, on malignant glioma proliferation and neo-angiogenesis. Neuro-Oncology 2014, 16, 1484–1498. [Google Scholar] [CrossRef] [Green Version]
- Drießen, D.; Stuhldreier, F.; Frank, A.; Stark, H.; Wesselborg, S.; Stork, B.; Müller, T. Novel meriolin derivatives as rapid apoptosis inducers. Bioorg. Med. Chem. 2019, 27, 3463–3468. [Google Scholar] [CrossRef]
- Alexander, A.; Karakas, C.; Chen, X.; Carey, J.; Yi, M.; Bondy, M.; Thompson, P.; Cheung, K.; Ellis, I.; Gong, Y.; et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget 2017, 8, 14897–14911. [Google Scholar] [CrossRef] [Green Version]
- Akli, S.; Van Pelt, C.; Bui, T.; Meijer, L.; Keyomarsi, K. Cdk2 is Required for Breast Cancer Mediated by the Low-Molecular-Weight Isoform of Cyclin E. Cancer Res. 2011, 71, 3377–3386. [Google Scholar] [CrossRef]
- Chashoo, G.; Singh, U.; Singh, P.; Mondhe, D.; Vishwakarma, R. A Marine-Based Meriolin (3-Pyrimidinylazaindole) Derivative (4ab) Targets PI3K/AKT /mTOR Pathway Inducing Cell Cycle Arrest and Apoptosis in Molt-4 Cells. Clin. Cancer Drugs 2019, 6, 33–40. [Google Scholar] [CrossRef]
- Mou, J.; Chen, D.; Deng, Y. Inhibitors of Cyclin-Dependent Kinase 1/2 for Anticancer Treatment. Med. Chem. 2020, 16, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Dorsch, D.; Sirrenberg, C.; Müller, T. Preparation of 4-(Pyrrolopyridinyl)pyrimidinyl-2-amines as Antitumor Agents. PCT International Application WO 2007/107221 A1, 27 September 2007. [Google Scholar]
- Dorsch, D.; Wuchrer, M.; Burgdorf, L.; Sirrenberg, C.; Esdar, C.; Mueller, T.J.J.; Merkul, E. 6-(Pyrrolopyridinyl)-pyrimidine-2-yl-amine Derivatives and Their Use for the Treatment of Cancers and Aids. PCT International Application WO 2008155000 A1, 24 December 2008. [Google Scholar]
- Dorsch, D.; Sirrenberg, C.; Müller, T.; Merkul, E. Preparation of 3-(4-Pyridinyl)-1H-pyrrolo[2,3-b]pyridines as Anti-Tumor Agents. Ger. Offen. DE 102008025751 A1, 3 December 2009. [Google Scholar]
- Dorsch, D.; Sirrenberg, C.; Müller, T.; Merkul, E.; Karapetyan, G. 7-Azaindole Derivatives as Kinase Inhibitors and Their Preparation and Use in the Treatment of Tumors. PCT International Application WO 2012104007 A2, 9 August 2012. [Google Scholar]
- Łukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef]
- Bharate, S.B.; Sawant, S.; Singh, P.; Vishwakarma, R. Kinase Inhibitors of Marine Origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef]
- Bharate, S.B.; Yadav, R.; Battula, S.; Vishwakarma, R. Meridianins: Marine-Derived Potent Kinase Inhibitors. Mini-Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef]
- Sandtorv, A.H. Chapter 5 - Chemical Synthesis of Meridianins and Related Derivatives. Stud. Nat. Prod. Chem. 2017, 53, 143–166. [Google Scholar] [CrossRef]
- Stanovnik, B.; Svete, J. The Synthesis Aplysinopsins, Meridianines, and Related Compounds. Mini-Rev. Org. Chem. 2005, 2, 211–224. [Google Scholar] [CrossRef]
- Popowycz, F.; Routier, S.; Joseph, B.; Mérour, J.-Y. Synthesis and reactivity of 7-azaindole (1H-pyrrolo[2,3-b]pyridine). Tetrahedron 2007, 63, 1031–1064. [Google Scholar] [CrossRef]
- Xiao, L. A Review: Meridianins and Meridianins Derivatives. Molecules 2022, 27, 8714. [Google Scholar] [CrossRef]
- Walker, S.R.; Carter, E.; Huff, B.; Morris, J. Variolins and Related Alkaloids. Chem. Rev. 2009, 109, 3080–3098. [Google Scholar] [CrossRef]
- Alvarez, M.; Fernández, D.; Joule, J. Synthesis of 3-Aryl- and 3-Heteroaryl-7-azaindoles. Synthesis 1999, 1999, 615–620. [Google Scholar] [CrossRef]
- Alvarez, M.; Fernández, D.; Joule, J. Synthesis of 1,2-dihydropyrrolo [1,2-c] pyrimidin-1-ones. J. Chem. Soc. Perkin Trans. 1 1999, 1999, 249–256. [Google Scholar] [CrossRef]
- Álvarez, M.; Fernández, D.; Joule, J. Synthesis of deoxyvariolin B. Tetrahedron Lett. 2001, 42, 315–317. [Google Scholar] [CrossRef]
- Mendiola, J.; Minguez, J.; Alvarez-Builla, J.; Vaquero, J. Reaction of 2-Bromomethylazoles and TosMIC: A Domino Process to Azolopyrimidines. Synthesis of Core Tricycle of the Variolins Alkaloids. Org. Lett. 2000, 2, 3253–3256. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, J.; Baeza, A.; Alvarez-Builla, J.; Vaquero, J. Reaction of Bromomethylazoles and Tosylmethyl Isocyanide. A Novel Heterocyclization Method for the Synthesis of the Core of Marine Alkaloids Variolins and Related Azolopyrimidines. J. Org. Chem. 2004, 69, 4974–4983. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P.; Delgado, S.; Bleda, J. Synthetic studies towards the 2-aminopyrimidine alkaloids variolins and meridianins from marine origin. Tetrahedron Lett. 2000, 41, 4777–4780. [Google Scholar] [CrossRef]
- Anderson, R.J.; Morris, J. Studies toward the total synthesis of the variolins: Rapid entry to the core structure. Tetrahedron Lett. 2001, 42, 311–313. [Google Scholar] [CrossRef]
- Anderson, R.J.; Morris, J. Total synthesis of variolin B. Tetrahedron Lett. 2001, 42, 8697–8699. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.J.; Hill, J.; Morris, J. Concise Total Syntheses of Variolin B and Deoxyvariolin B. J. Org. Chem. 2005, 70, 6204–6212. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.; Delgado, S.; Bleda, J. Synthesis of the potent antitumoral marine alkaloid variolin B. Tetrahedron Lett. 2002, 43, 1005–1007. [Google Scholar] [CrossRef]
- Molina, P.; Fresneda, P.; Delgado, S. Carbodiimide-Mediated Preparation of the Tricyclic Pyrido[3′,2′:4,5]pyrrolo[1,2-c]pyrimidine Ring System and Its Application to the Synthesis of the Potent Antitumoral Marine Alkaloid Variolin B and Analog. J. Org. Chem. 2003, 68, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Bredereck, H.; Effenberger, F.; Botsch, H.; Rehn, H. Synthesen in der heterocyclischen Reihe, V: Umsetzungen von vinylogen Carbonsäureamiden zu Heterocyclen. Chem. Ber. 1965, 98, 1081–1086. [Google Scholar] [CrossRef]
- Ahaidar, A.; Fernández, D.; Pérez, O.; Danelón, G.; Cuevas, C.; Manzanares, I.; Albericio, F.; Joule, J.; Álvarez, M. Synthesis of variolin B. Tetrahedron Lett. 2003, 44, 6191–6194. [Google Scholar] [CrossRef]
- Ahaidar, A.; Fernández, D.; Danelón, G.; Cuevas, C.; Manzanares, I.; Albericio, F.; Joule, J.; Álvarez, M. Total Syntheses of Variolin B and Deoxyvariolin B1. J. Org. Chem. 2003, 68, 10020–10029. [Google Scholar] [CrossRef]
- Fernández, D.; Ahaidar, A.; Danelón, G.; Cironi, P.; Marfil, M.; Pérez, O.; Cuevas, C.; Albericio, F.; Joule, J.; Álvarez, M. Synthesis of Polyheterocyclic Nitrogen-Containing Marine Natural Products. Monatsh. Chem. 2004, 135, 615–627. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Akutagawa, K. Carbon dioxide: A reagent for simultaneous protection of nucleophilic centers and the activation of alternative locations to electrophilic attack. V. Activation of the 2-alkyl group of a 2-alkylindole toward proton loss and subsequent electrophilic substitution. J. Am. Chem. Soc. 1986, 108, 6808–6809. [Google Scholar] [CrossRef]
- Baeza, A.; Mendiola, J.; Burgos, C.; Alvarez-Builla, J.; Vaquero, J. Palladium-mediated C–N, C–C, and C–O functionalization of azolopyrimidines: A new total synthesis of variolin B. Tetrahedron Lett. 2008, 49, 4073–4077. [Google Scholar] [CrossRef] [Green Version]
- Baeza, A.; Mendiola, J.; Burgos, C.; Alvarez-Builla, J.; Vaquero, J. Application of Selective Palladium-Mediated Functionalization of the Pyrido[3′,2′:4,5]pyrrolo[1,2-c]pyrimidine Heterocyclic System for the Total Synthesis of Variolin B and Deoxyvariolin B. Eur. J. Org. Chem. 2010, 2010, 5607–5618. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.-G. Synthesis of Indolylpyrimidiness via Cross-Coupling of Indolylboronic Acid with Choropyrimidines: Facile Synthesis of Meridianin D. Heterocycles 2000, 53, 1489–1498. [Google Scholar] [CrossRef]
- Radwan, M.A.A.; El-Sherbiny, M. Synthesis and antitumor activity of indolylpyrimidines: Marine natural product meridianin D analogues. Bioorg. Med. Chem. 2007, 15, 1206–1211. [Google Scholar] [CrossRef]
- Corbel, B.; Michaud, F.; Meijer, L.; Simon, G.; Couthon-Gourves, H.; Haelters, J.-P.; Kervarec, N. Towards the syntheses of N-H and N-alkylated derivatives of meridianins. J. Heterocycl. Chem. 2007, 44, 793–801. [Google Scholar] [CrossRef]
- Sperry, J. A concise synthesis of meridianin F. Tetrahedron Lett. 2011, 52, 4537–4538. [Google Scholar] [CrossRef]
- Lebar, M.D.; Hahn, K.; Mutka, T.; Maignan, P.; McClintock, J.; Amsler, C.; van Olphen, A.; Kyle, D.; Baker, B. CNS and antimalarial activity of synthetic meridianin and psammopemmin analogs. Bioorg. Med. Chem. 2011, 19, 5756–5762. [Google Scholar] [CrossRef] [PubMed]
- More, K.N.; Jang, H.; Hong, V.; Lee, J. Pim kinase inhibitory and antiproliferative activity of a novel series of meridianin C derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2424–2428. [Google Scholar] [CrossRef]
- Dong, J.; Huang, S.-S.; Hao, Y.-N.; Wang, Z.-W.; Liu, Y.-X.; Li, Y.-Q.; Wang, Q.-M. Marine-natural-products for biocides development: First discovery of meridianin alkaloids as antiviral and anti-phytopathogenic-fungus agents. Pest Manag. Sci. 2020, 76, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhuang, C.; Zhou, W.; Chen, F. Structural-Based Optimizations of the Marine-Originated Meridianin C as Glucose Uptake Agents by Inhibiting GSK-3β. Mar. Drugs 2021, 19, 149. [Google Scholar] [CrossRef]
- Karpov, A.S.; Merkul, E.; Rominger, F.; Müller, T. Concise Syntheses of Meridianins by Carbonylative Alkynylation and a Four-Component Pyrimidine Synthesis. Angew. Chem. Int. Ed. 2005, 44, 6951–6956. [Google Scholar] [CrossRef]
- Tibiletti, F.; Simonetti, M.; Nicholas, K.; Palmisano, G.; Parravicini, M.; Imbesi, F.; Tollari, S.; Penoni, A. One-pot synthesis of meridianins and meridianin analogues via indolization of nitrosoarenes. Tetrahedron 2010, 66, 1280–1288. [Google Scholar] [CrossRef]
- Merkul, E.; Schäfer, E.; Müller, T. Rapid synthesis of bis(hetero)aryls by one-pot Masuda borylation-Suzuki coupling sequence and its application to concise total syntheses of meridianins A and G. Org. Biomol. Chem. 2011, 9, 3139–3141. [Google Scholar] [CrossRef]
- Kruppa, M.; Sommer, G.; Müller, T. Concise Syntheses of Marine (Bis)indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence. Molecules 2022, 27, 2233. [Google Scholar] [CrossRef]
- Parsons, T.B.; Ghellamallah, C.; Male, L.; Spencer, N.; Grainger, R. Regioselective dibromination of methyl indole-3-carboxylate and application in the synthesis of 5,6-dibromoindoles. Org. Biomol. Chem. 2011, 9, 5021–5023. [Google Scholar] [CrossRef] [PubMed]
- Cacchi, S.; Fabrizi, G.; Marinelli, F.; Moro, L.; Pace, P. 3-Aryl-2-Unsubstituted Indoles through the Palladium-Catalysed Reaction of o-Ethynyltrifluoroacetanilide with Aryl Iodides. Synlett 1997, 12, 1363–1366. [Google Scholar] [CrossRef]
- Walker, S.R.; Czyz, M.; Morris, J. Concise Syntheses of Meridianins and Meriolins Using a Catalytic Domino Amino-Palladation Reaction. Org. Lett. 2014, 16, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Wucherer-Plietker, M.; Merkul, E.; Müller, T.; Esdar, C.; Knöchel, T.; Heinrich, T.; Buchstaller, H.-P.; Greiner, H.; Dorsch, D.; Finsinger, D.; et al. Discovery of novel 7-azaindoles as PDK1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3073–3080. [Google Scholar] [CrossRef] [PubMed]
- Merkul, E.; Oeser, T.; Müller, T. Consecutive Three-Component Synthesis of Ynones by Decarbonylative Sonogashira Coupling. Eur. J. Chem. 2009, 15, 5006–5011. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Connolly, P.; Emanuel, S.; Middleton, S. Synthesis of 2-amino-4-(7-azaindol-3-yl)pyrimidines as cyclin dependent kinase 1 (CDK1) inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4818–4821. [Google Scholar] [CrossRef] [PubMed]
- Rehberg, N.; Sommer, G.; Drießen, D.; Kruppa, M.; Adeniyi, E.; Chen, S.; Wang, L.; Wolf, K.; Tasch, B.; Ioerger, T.; et al. Nature-Inspired (di)Azine-Bridged Bisindole Alkaloids with Potent Antibacterial In Vitro and In Vivo Efficacy against Methicillin-Resistant Staphylococcus aureus. J. Med. Chem. 2020, 63, 12623–12641. [Google Scholar] [CrossRef]
- Thatikonda, T.; Singh, U.; Ambala, S.; Vishwakarma, R.; Singh, P. Metal free C-H functionalization of diazines and related heteroarenes with organoboron species and its application in the synthesis of a CDK inhibitor, meriolin 1. Org. Biomol. Chem. 2016, 14, 4312–4320. [Google Scholar] [CrossRef]
- Singh, U.; Chashoo, G.; Khan, S.; Mahajan, P.; Nargotra, A.; Mahajan, G.; Singh, A.; Sharma, A.; Mintoo, M.; Guru, S.; et al. Design of Novel 3-Pyrimidinylazaindole CDK2/9 Inhibitors with Potent In Vitro and In Vivo Antitumor Efficacy in a Triple-Negative Breast Cancer Model. J. Med. Chem. 2017, 60, 9470–9489. [Google Scholar] [CrossRef]






































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Azaindole 99 | R1 | (hetero)aryl R2 62 | Meriolins 3 (Yield) |
|---|---|---|---|---|
| 1 | 99a | H | 4-pyrimidin-2-amine (62b) | 3a, meriolin 1 (63%) |
| 2 | 99a | H | 6-pyrazin-2-amine (62c) | 3ao (53%) |
| 3 | 99a | H | 5-pyrimidin-2-amine (62d) | 3ap (66%) |
| 4 | 99a | H | 2-pyrimidin-4-amine (62e) | 3aq (37%) |
| 5 | 99a | H | 6-pyridin-2-amine (62f) | 3ar (81%) |
| 6 | 99a | H | 4-pyridin-2-amine (62g) | 3as (64%) |
| 7 | 99a | H | 2-aniline (62h) | 3at (74%) |
| 8 | 99a | H | 4-phenol (62i) | 3au (57%) |
| 9 | 99c | Bn | 4-pyrimidin-2-amine (62b) | 3av (96%) |
| 10 | 99c | Bn | 4-pyridin-2-amine (62g) | 3aw (93%) |
| 11 | 99b | H | 4-pyrimidin-2-amine (62b) | 3a, meriolin 1 (81%) |
| 12 | 99b | H | 5-pyridin-2-amine (62j) | 3ax (91%) |
| 13 | 99b | H | 4-pyridin-2-amine (62g) | 3ay (75%) |
| 14 | 99b | H | N-benzyl-5-pyridin-2-amine (62k) | 3az (77%) |
| 15 | 99b | H | 4-(2-methoxypyrimidine) (62l) | 3ba (40%) |
| 16 | 99b | H | 4-pyridin-2,6-diamine (62m) | 3bb (67%) |
| 17 | 99b | H | 5-pyrimidin-2-amine (62d) | 3bc (47%) |
| 18 | 99b | H | 4-(2-methylthiopyrimidine) (62n) | 3bd (83%) |
| 19 | 99b | H | 4-(6-methoxypyrimidin-2-amine) (62o) | 3be (62%) |
| 20 | 99b | H | 4-pyrimidin-2,6-diamine (62p) | 3bf (53%) |
| 21 | 99b | H | N-benzyl-4-pyridin-2-amine (62q) | 3bg (83%) |
| 22 | 99b | H | 2-pyrimidin-4-amine (62e) | 3bh (75%) |
| 23 a | 99b | H | 5-isoquinolin (62r) | 3bi (82%) |
| Entry | Boronic Acid R1-B(OH)2 (126) | Meriolins 3 (Yield) |
|---|---|---|
| 1 | (4-(trifluoromethyl)phenyl)boronic acid (126a) | 3bk (63%) |
| 2 | (4-fluorophenyl)boronic acid (126b) | 3bl (65%) |
| 3 | (4-chlorophenyl)boronic acid (126c) | 3bm (62%) |
| 4 | (4-(trifluoromethoxy)phenyl)boronic acid (126d) | 3bn (60%) |
| 5 | (4-methoxyphenyl)boronic acid (126e) | 3bo (59%) |
| 6 | (4-(methylthio)phenyl)boronic acid (126f) | 3bp (54%) |
| 7 | (3-fluorophenyl)boronic acid (126g) | 3bq (55%) |
| 8 | m-tolylboronic acid (126h) | 3br (50%) |
| 9 | (3-(trifluoromethyl)phenyl)boronic acid (126i) | 3bs (58%) |
| 10 | (2-(methylthio)phenyl)boronic acid (126j) | 3bt (59%) |
| 11 | (2-ethylphenyl)boronic acid (126k) | 3bu (48%) |
| 12 | naphthalen-1-ylboronic acid (126l) | 3bv (60%) |
| 13 | (2-methoxynaphthalen-1-yl)boronic acid (126m) | 3bw (55%) |
| 14 | furan-3-ylboronic acid (126n) | 3bx (48%) |
| 15 | thiophen-3-ylboronic acid (126o) | 3by (44%) |
| 16 | pyridin-3-ylboronic acid (126p) | 3bz (45%) |
| 17 | benzo[b]thiophen-2-ylboronic acid (126q) | 3ca (40%) |
| 18 | benzofuran-2-ylboronic acid (126r) | 3cb (39%) |
| 19 | (5-methoxy-1H-indol-2-yl)boronic acid (126s) | 3cc (45%) |
| Entry | Amine R2-NH2 (131) or R2NHR3 (132) | Meriolins 3 (Yield) |
|---|---|---|
| 1 | 2-phenylethan-1-amine (131a) | 3cf (65%) |
| 2 | 2-(4-methoxyphenyl)ethan-1-amine (131b) | 3cg (60%) |
| 3 | 2-(3,4-dimethoxyphenyl)ethan-1-amine (131c) | 3ch (68%) |
| 4 | 2-(1H-indol-3-yl)ethan-1-amine (131d) | 3ci (50%) |
| 5 | pyrrolidine (132a) | 3cj (52%) |
| 6 | piperidine (132b) | 3ck (52%) |
| 7 | morpholine (132c) | 3cl (60%) |
| 8 | 1-methylpiperazine (132d) | 3cm (54%) |
| Entry | Sulfonyl Chloride R4-SO2Cl (133) | Meriolins 3 (Yield) |
|---|---|---|
| 1 | 4-fluorobenzenesulfonyl chloride (133a) | 3cn (70%) |
| 2 | 4-bromobenzenesulfonyl chloride (133b) | 3co (70%) |
| 3 | 4-(trifluoromethyl)benzenesulfonyl chloride (133c) | 3cp (77%) |
| 4 | 4-(trifluoromethoxy)benzenesulfonyl chloride (133d) | 3cq (80%) |
| 5 | 4-acetamidobenzenesulfonyl chloride (133e) | 3cr (71%) |
| 6 | 2,3-dihydrobenzo[b][1,4]dioxine-6-sulfonyl chloride (133f) | 3cs (79%) |
| 7 | 1-methyl-1H-imidazole-5-sulfonyl chloride (133g) | 3ct (65%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruppa, M.; Müller, T.J.J. A Survey on the Synthesis of Variolins, Meridianins, and Meriolins—Naturally Occurring Marine (aza)Indole Alkaloids and Their Semisynthetic Derivatives. Molecules 2023, 28, 947. https://doi.org/10.3390/molecules28030947
Kruppa M, Müller TJJ. A Survey on the Synthesis of Variolins, Meridianins, and Meriolins—Naturally Occurring Marine (aza)Indole Alkaloids and Their Semisynthetic Derivatives. Molecules. 2023; 28(3):947. https://doi.org/10.3390/molecules28030947
Chicago/Turabian StyleKruppa, Marco, and Thomas J. J. Müller. 2023. "A Survey on the Synthesis of Variolins, Meridianins, and Meriolins—Naturally Occurring Marine (aza)Indole Alkaloids and Their Semisynthetic Derivatives" Molecules 28, no. 3: 947. https://doi.org/10.3390/molecules28030947
APA StyleKruppa, M., & Müller, T. J. J. (2023). A Survey on the Synthesis of Variolins, Meridianins, and Meriolins—Naturally Occurring Marine (aza)Indole Alkaloids and Their Semisynthetic Derivatives. Molecules, 28(3), 947. https://doi.org/10.3390/molecules28030947