

In Silico Investigation of the Molecular Mechanism of PARP1 Inhibition for the Treatment of BRCA-Deficient Cancers


Abstract
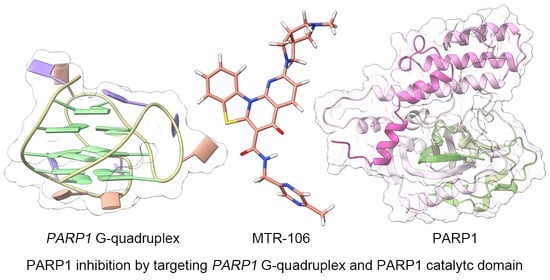
:
1. Introduction
2. Computational Procedures
2.1. Data
2.2. Molecular Docking
2.3. Molecular Dynamics
2.4. Principal Components Analysis
2.5. Noncovalent Interactions
2.6. Binding-Free-Energy Analysis
2.7. ADMET Analysis
3. Results
3.1. Conformational Characteristics of the Apo PARP1 G4/G4(K+)
3.2. Binding Modes of MTR-106 and Talazoparib with PARP1 G4/G4(K+)
3.3. Dynamic Features of the PARP1 G4/G4(K+)–MTR-106/Talazoparib Binding Complexes
3.4. Hydrogen-Bond Analysis of the PARP1 G4/G4(K+)–MTR-106/Talazoparib Binding Complexes
3.5. Binding-Free Energies between PARP1 G4 and MTR-106/Talazoparib
3.6. Binding Characteristics of PARP1 and MTR-106/Talazoparib
3.7. The ADMET Properties of MTR-106 and Talazoparib
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L. Transcriptional control by PARP-1: Chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Ferrara, R.; Simionato, F.; Ciccarese, C.; Grego, E.; Cingarlini, S.; Iacovelli, R.; Bria, E.; Tortora, G.; Melisi, D. The development of PARP as a successful target for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 161–175. [Google Scholar] [CrossRef]
- McGlynn, P.; Lloyd, R.G. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002, 3, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Pay, S.L.; Bhandare, S.B.; Arimpur, U.; Motea, E.A. Therapeutic strategies and biomarkers to modulate PARP activity for targeted cancer therapy. Cancers 2020, 12, 972. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef]
- Sengar, A.; Vandana, J.J.; Chambers, V.S.; Di Antonio, M.; Winnerdy, F.R.; Balasubramanian, S.; Phan, A.T. Structure of a (3+1) hybrid G-quadruplex in the PARP1 promoter. Nucleic Acids Res. 2019, 47, 1564–1572. [Google Scholar] [CrossRef]
- Dutta, D.; Debnath, M.; Müller, D.; Paul, R.; Das, T.; Bessi, I.; Schwalbe, H.; Dash, J. penetrating thiazole peptides inhibit c-MYC expression via site-specific targeting of c-MYC G-quadruplex. Nucleic Acids Res. 2018, 46, 5355–5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsen, R.C.; DeLeeuw, L.W.; Dean, W.L.; Gray, R.D.; Chakravarthy, S.; Hopkins, J.B.; Chaires, J.B.; Trent, J.O. Long promoter sequences form higher-order G-quadruplexes: An integrative structural biology study of c-Myc, k-Ras and c-Kit promoter sequences. Nucleic Acids Res. 2022, 50, 4127–4147. [Google Scholar] [CrossRef]
- Głuszyńska, A.; Juskowiak, B.; Kuta-Siejkowska, M.; Hoffmann, M.; Haider, S. Carbazole Derivatives’ Binding to c-KIT G-Quadruplex DNA. Molecules 2018, 23, 1134. [Google Scholar] [CrossRef] [Green Version]
- Jana, J.; Mondal, S.; Bhattacharjee, P.; Sengupta, P.; Roychowdhury, T.; Saha, P.; Kundu, P.; Chatterjee, S. Chelerythrine down regulates expression of VEGFA, BCL2 and KRAS by arresting G-Quadruplex structures at their promoter regions. Sci. Rep. 2017, 7, 40706. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- Dallavalle, S.; Princiotto, S.; Mattio, L.M.; Artali, R.; Musso, L.; Aviñó, A.; Eritja, R.; Pisano, C.; Gargallo, R.; Mazzini, S. Investigation of the complexes formed between PARP1 inhibitors and PARP1 G-quadruplex at the gene promoter region. Int. J. Mol. Sci. 2021, 22, 8737. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-Z.; Meng, T.; Song, S.-S.; Bao, X.-B.; Ma, L.-P.; Zhang, N.; Yu, T.; Zhang, Y.-L.; Xiong, B.; Shen, J.-K.; et al. Discovery of MTR-106 as a highly potent G-quadruplex stabilizer for treating BRCA-deficient cancers. Investig. New Drug. 2021, 39, 1213–1221. [Google Scholar] [CrossRef]
- Ryan, K.; Bolaňos, B.; Smith, M.; Palde, P.B.; Cuenca, P.D.; VanArsdale, T.L.; Niessen, S.; Zhang, L.; Behenna, D.; Ornelas, M.A.; et al. Dissecting the molecular determinants of clinical PARP1 inhibitor selectivity for tankyrase1. J. Biol. Chem. 2021, 296, 100251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, R.; Hou, L.; Li, J.; Liu, J.P. Molecular dynamics and principal components of potassium binding with human telomeric intra-molecular G-quadruplex. Protein Cell 2015, 6, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Liu, J.P. Characterization of potassium binding with human telomeres. Clin. Exp. Pharmacol. Physiol. 2015, 42, 902–909. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.P. Effects of the central potassium ions on the G-quadruplex and stabilizer binding. J. Mol. Graph. Model. 2017, 72, 168–177. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.; Meng, T.; Qi, L.; Yan, H.; Wang, Z. Molecular insights into the selective binding mechanism targeting parallel human telomeric G-quadruplex. J. Mol. Graph. Model 2022, 110, 108058. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Liu, J.; Wang, L.; Lu, Y.; Liu, J.-P. Molecular mechanism of anionic stabilizer for telomere G-quadruplex. Biophys. Rep. 2022. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, L.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Bhimaneni, S.; Kumar, A. Abscisic acid and aloe-emodin against NS2B-NS3A protease of Japanese encephalitis virus. Environ. Sci. Pollut. Res. Int. 2022, 29, 8759–8766. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-S.; Allen, B.K.; Giese, T.J.; Guo, Z.; Li, P.; Lin, C.; McGee, J.T.D.; Pearlman, D.A.; Radak, B.K.; Tao, Y.; et al. Alchemical binding free energy calculations in Amber20: Advances and best practices for drug discovery. J. Chem. Inf. Model. 2020, 60, 5595–5623. [Google Scholar] [CrossRef]
- Machireddy, B.; Kalra, G.; Jonnalagadda, S.; Ramanujachary, K.; Wu, C. Probing the binding pathway of BRACO19 to a parallel-stranded human telomeric G-quadruplex using molecular dynamics binding simulation with Amber DNA OL15 and Ligand GAFF2 force fields. J. Chem. Inf. Model. 2017, 57, 2846–2864. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbova, M.; Sponer, J.; Otyepka, M.; Jurecka, P.; Cheatham, T.E., III. Assessing the current state of Amber force field modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Wang, Z.; Li, G.; Tian, Z.; Lou, X.; Huang, Y.; Wang, L.; Li, J.; Hou, T.; Liu, J.-P. Insight derived from molecular dynamics simulation into the selectivity mechanism targeting c-MYC G-quadruplex. J. Phys. Chem. B. 2020, 124, 9773–9784. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Min, Z.; Zhang, X.; Wu, W.; Xin, Y.; Liu, M.; Wang, K.; Zhang, X.; He, Y.; Fan, C.; Wang, Z.; et al. Crystal structure of an intramolecular mesaconyl-coenzyme A transferase from the 3-hydroxypropionic acid cycle of Roseiflexus castenholzii. Front. Microbiol. 2022, 13, 923367. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Gilson, M.K.; Sharp, K.A.; Honig, B.H. Calculating the electrostatic potential of molecules in solution: Method and error assessment. J. Comput. Chem. 1988, 9, 327–335. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Hou, J.-Q.; Chen, S.-B.; Tan, J.-H.; Luo, H.-B.; Li, D.; Gu, L.-Q.; Huang, Z.-S. New insights from molecular dynamic simulation studies of the multiple binding modes of a ligand with G-quadruplex DNA. J. Comput. Aid. Mol. Des. 2012, 26, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 15, 1067–1069. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Jayaram, B.; Sprous, D.; Young, M.A.; Beveridge, D.L. Free energy analysis of the conformational preferences of A and B forms of DNA in solution. J. Am. Chem. Soc. 1998, 120, 10629–10633. [Google Scholar] [CrossRef]
- Kongsted, J.; Söderhjelm, P.; Ryde, U. How accurate are continuum solvation models for drug-like molecules? J. Comput. Aid. Mol. Des. 2009, 23, 395–409. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Gupta, M.; Sharma, R.; Kumar, A. Docking techniques in pharmacology: How much promising? Comput. Biol. Chem. 2018, 76, 210–217. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Nguyen, T.H.; Pham, T.N.H.; Huy, N.T.; Van Bay, M.; Pham, M.Q.; Nam, P.C.; Vu, V.V.; Ngo, S.T. Autodock Vina adopts more accurate binding poses but Autodock4 forms better binding affinity. J. Chem. Inf. Model 2020, 60, 204–211. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yu, Y.; Chakrabarti, J.; Piha-Paul, S.A.; Moroose, R.; Plotka, A.; Shi, H.; Durairaj, C.; Wang, D.D.; Wainberg, Z.A. Evaluation of pharmacokinetics and safety of talazoparib in patients with advanced cancer and varying degrees of hepatic impairment. Br. J. Clin. Pharmacol. 2022, 88, 3392–3403. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Conformation 1 | Hydrogen Bond | π–π Stacking | Affinity (G4/G4(K+)) 2 |
|---|---|---|---|---|
| MTR-106 | a | - | dG14 | −7.82/−7.60 |
| b | - | dT6 | −7.90/−7.73 | |
| c | - | dG3, dG12, dG14 | −8.46/−8.58 | |
| d | dG11 | dA10 | −8.01/−7.52 | |
| talazoparib | a | - | dT1, dG3, dC13 | −6.91/−6.84 |
| b | - | dT6, dG12 | −6.83/−6.67 | |
| c | - | dT6, dG12 | −6.06/−5.91 | |
| d | - | dT6 | −6.40/−6.55 |
| Model 1 | Acceptor | Donor | Ocpy 2 (%) | Dist 3 (Å) | Ang 4 (°) |
|---|---|---|---|---|---|
| G4(K+)–MTR-106 c | MTR-106@N1 | dG12@H22–N2 | 57.08 | 3.16 | 148.18 |
| G4(K+)–MTR-106 d | MTR-106@O1 | dG5@H22–N2 | 95.71 | 2.92 | 160.90 |
| MTR-106@N4 | dG4@H22–N2 | 49.24 | 3.16 | 145.38 | |
| G4–talazoparib a | dG14@O6 | talazoparib@H3–N3 | 65.24 | 2.97 | 159.49 |
| G4–talazoparib b | dG5@N3 | talazoparib@H1–N1 | 98.85 | 2.97 | 161.86 |
| talazoparib@O1 | dG5@H22–N2 | 96.25 | 2.99 | 161.02 | |
| talazoparib@N6 | dG12@H22–N2 | 92.44 | 3.03 | 150.94 | |
| talazoparib@N2 | dG4@H22–N2 | 83.98 | 3.10 | 140.54 | |
| G4–talazoparib d | talazoparib@O1 | dG12@H22–N2 | 32.88 | 2.98 | 160.17 |
| dG12@N3 | talazoparib@H1–N1 | 32.69 | 3.10 | 153.39 | |
| G4(K+)–talazoparib b | dG5@N3 | talazoparib@H1–N1 | 98.24 | 2.98 | 161.26 |
| talazoparib@O1 | dG5@ H22–N2 | 93.30 | 3.02 | 158.85 | |
| talazoparib@N6 | dG12@ H22–N2 | 88.54 | 3.04 | 149.11 | |
| talazoparib@N2 | dG4@H22–N2 | 65.31 | 3.04 | 135.88 | |
| G4(K+)–talazoparib c | dG5@N3 | talazoparib@H1–N1 | 95.68 | 2.97 | 161.89 |
| talazoparib@O1 | dG5@H22–N2 | 90.86 | 3.02 | 160.43 | |
| talazoparib@N6 | dG12@H22–N2 | 88.03 | 3.04 | 147.75 | |
| talazoparib@N2 | dG4@H22–N2 | 60.19 | 3.04 | 133.66 | |
| alazoparib@N6 | dG4@H22–N2 | 42.51 | 3.23 | 139.98 |
| Receptor | Ligand 1 | Energy Components 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEele | ΔEvdW | ΔGGB | ΔGSA | ΔH | TΔS | ΔGbind | ||
| G4 | MTR-106 a | 2.4 ± 1.5 | −54.1 ± 3.0 | 1.9 ± 1.3 | −6.0 ± 0.2 | −55.8 ± 3.0 | 21.8 ± 9.2 | −34.0 |
| MTR-106 b | 4.6 ± 1.3 | −59.6 ± 2.5 | 3.1 ± 1.1 | −5.8 ± 0.2 | −57.7 ± 2.4 | 22.1 ± 10.1 | −35.6 | |
| MTR-106 c | 0.3 ± 3.6 | −41.0 ± 7.8 | 3.1 ± 3.0 | −4.5 ± 0.7 | −42.0 ± 8.3 | 22.2 ± 10.4 | −19.8 | |
| MTR-106 d | −2.7 ± 2.3 | −41.2 ± 9.3 | 5.5 ± 4.1 | −4.3 ± 3.0 | −42.7 ± 7.4 | 21.5 ± 10.2 | −21.2 | |
| G4(K+) | MTR-106 a | 0.8 ± 1.7 | −55.2 ± 3.7 | 3.8 ± 1.6 | −6.2 ± 0.4 | −56.8 ± 3.8 | 22.1 ± 10.4 | −34.7 |
| MTR-106 b | −3.1 ± 1.4 | −58.0 ± 3.1 | 6.8 ± 1.3 | −5.9 ± 0.3 | −60.2 ± 3.0 | 23.8 ± 10.4 | −36.4 | |
| MTR-106 c | −4.7 ± 1.1 | −46.0 ± 3.5 | 8.2 ± 1.0 | −5.0 ± 0.3 | −47.6 ± 3.6 | 25.1 ± 11.1 | −22.5 | |
| MTR-106 d | −3.3 ± 1.2 | −60.4 ± 3.7 | 7.8 ± 1.0 | −6.6 ± 0.3 | −62.6 ± 3.6 | 27.5 ± 10.5 | −35.1 | |
| G4 | talazoparib a | −7.2 ± 1.3 | −31.5 ± 3.9 | 9.2 ± 1.1 | −4.0 ± 0.4 | −33.5 ± 4.0 | 18.0 ± 9.3 | −15.5 |
| talazoparib b | −1.7 ± 1.3 | −38.4 ± 3.1 | 3.6 ± 1.1 | −4.7 ± 0.3 | −41.2 ± 2.9 | 17.2 ± 9.4 | −24.0 | |
| talazoparib c | −5.4 ± 1.6 | −39.0 ± 3.2 | 8.2 ± 1.3 | −4.5 ± 0.3 | −40.8 ± 3.2 | 17.8 ± 8.7 | −23.0 | |
| talazoparib d | −2.0 ± 1.8 | −20.0 ± 2.6 | 3.3 ± 1.5 | −2.7 ± 0.2 | −21.2 ± 2.3 | 16.9 ± 8.9 | −4.3 | |
| G4(K+) | talazoparib a | −3.9 ± 1.4 | −36.0 ± 3.5 | 6.5 ± 1.1 | −4.1 ± 0.3 | −37.5 ± 2.8 | 18.0 ± 8.7 | −19.5 |
| talazoparib b | −2.1 ± 0.9 | −42.0 ± 3.0 | 4.2 ± 0.8 | −4.8 ± 0.2 | −44.7 ± 3.0 | 17.6 ± 9.5 | −27.1 | |
| talazoparib c | −2.2 ± 1.0 | −41.0 ± 2.9 | 4.2 ± 0.8 | −4.8 ± 0.2 | −43.7 ± 2.8 | 18.5 ± 9.2 | −25.2 | |
| talazoparib d | −2.3 ± 2.6 | −25.1 ± 8.3 | 4.5 ± 3.0 | −3.2 ± 1.1 | −26.1 ± 8.7 | 16.6 ± 9.1 | −9.5 | |
| Receptor | Ligand | Energy Components 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔEele | ΔEvdW | ΔGGB | ΔGSA | ΔH | TΔS | ΔGbind | ||
| PARP1 | MTR-106 | −4.0 ± 1.1 | −58.4 ± 2.7 | 8.3 ± 0.9 | −7.5 ± 0.2 | −61.6 ± 2.7 | 26.1 ± 13.0 | −35.5 |
| PARP1 | talazoparib | −7.0 ± 1.0 | −43.3 ± 2.8 | 8.7 ± 0.7 | −5.6 ± 0.1 | −47.2 ± 2.6 | 20.7 ± 11.4 | −26.5 |
| Property | MTR-106 | Talazoparib | ||
|---|---|---|---|---|
| AdmetSAR2 | ProTox-II | AdmetSAR2 | ProTox-II | |
| Human oral bioavailability | 0.5857 | - | 0.5429 | - |
| GI 1 absorption | Yes (0.9922) | - | Yes (0.9951) | - |
| Caco-2 permeability 2 | No (0.7863) | - | Yes (0.6579) | - |
| BBB 3 | Yes (0.9000) | - | Yes (0.8000) | - |
| P-gp 4 substrate | Yes (0.8047) | - | Yes (0.5664) | - |
| CYP1A2 5 inhibitor | No (0.5952) | - | Yes (0.8200) | - |
| CYP2C9 5 inhibitor | No (0.6741) | - | No (0.6486) | - |
| CYP2C19 5 inhibitor | No (0.5853) | - | No (0.5888) | - |
| CYP2D6 5 inhibitor | No (0.8830) | - | No (0.9394) | - |
| CYP3A4 5 inhibitor | No (0.6878) | - | No (0.6895) | - |
| CYP2C9 5 substrate | No (1.0000) | - | No (0.8038) | - |
| CYP2D6 5 substrate | No (0.8202) | - | No (0.8402) | - |
| CYP3A4 5 substrate | Yes (0.6977) | - | No (0.6895) | - |
| Oral LD50 6 | - | 1414 | - | 500 |
| Oral-toxicity class | III 7 | IV 8 | III 7 | IV 8 |
| Acute oral toxicity 9 | 2.008 | - | 2.632 | - |
| Hepatotoxicity | Yes (0.7176) | No (0.60) | Yes (0.5803) | Yes (0.63) |
| Carcinogenicity | No (0.8600) | No (0.58) | No (0.8857) | No (0.54) |
| Immunotoxicity | - | No (0.92) | - | No (0.73) |
| Mutagenicity | - | Yes (0.54) | - | No (0.57) |
| Cytotoxicity | - | No (0.60) | - | No (0.81) |
| Eye irritation and corrosion | No | - | No | - |
| Skin sensitization | No (0.8543) | - | No (0.9017) | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, F.; Fu, Z.; Li, G.; Wang, Z. In Silico Investigation of the Molecular Mechanism of PARP1 Inhibition for the Treatment of BRCA-Deficient Cancers. Molecules 2023, 28, 1829. https://doi.org/10.3390/molecules28041829
Yan F, Fu Z, Li G, Wang Z. In Silico Investigation of the Molecular Mechanism of PARP1 Inhibition for the Treatment of BRCA-Deficient Cancers. Molecules. 2023; 28(4):1829. https://doi.org/10.3390/molecules28041829
Chicago/Turabian StyleYan, Fengqin, Zhenfu Fu, Guo Li, and Zhiguo Wang. 2023. "In Silico Investigation of the Molecular Mechanism of PARP1 Inhibition for the Treatment of BRCA-Deficient Cancers" Molecules 28, no. 4: 1829. https://doi.org/10.3390/molecules28041829
APA StyleYan, F., Fu, Z., Li, G., & Wang, Z. (2023). In Silico Investigation of the Molecular Mechanism of PARP1 Inhibition for the Treatment of BRCA-Deficient Cancers. Molecules, 28(4), 1829. https://doi.org/10.3390/molecules28041829