
Functionalized Sulfur-Containing Heterocyclic Analogs Induce Sub-G1 Arrest and Apoptotic Cell Death of Laryngeal Carcinoma In Vitro

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
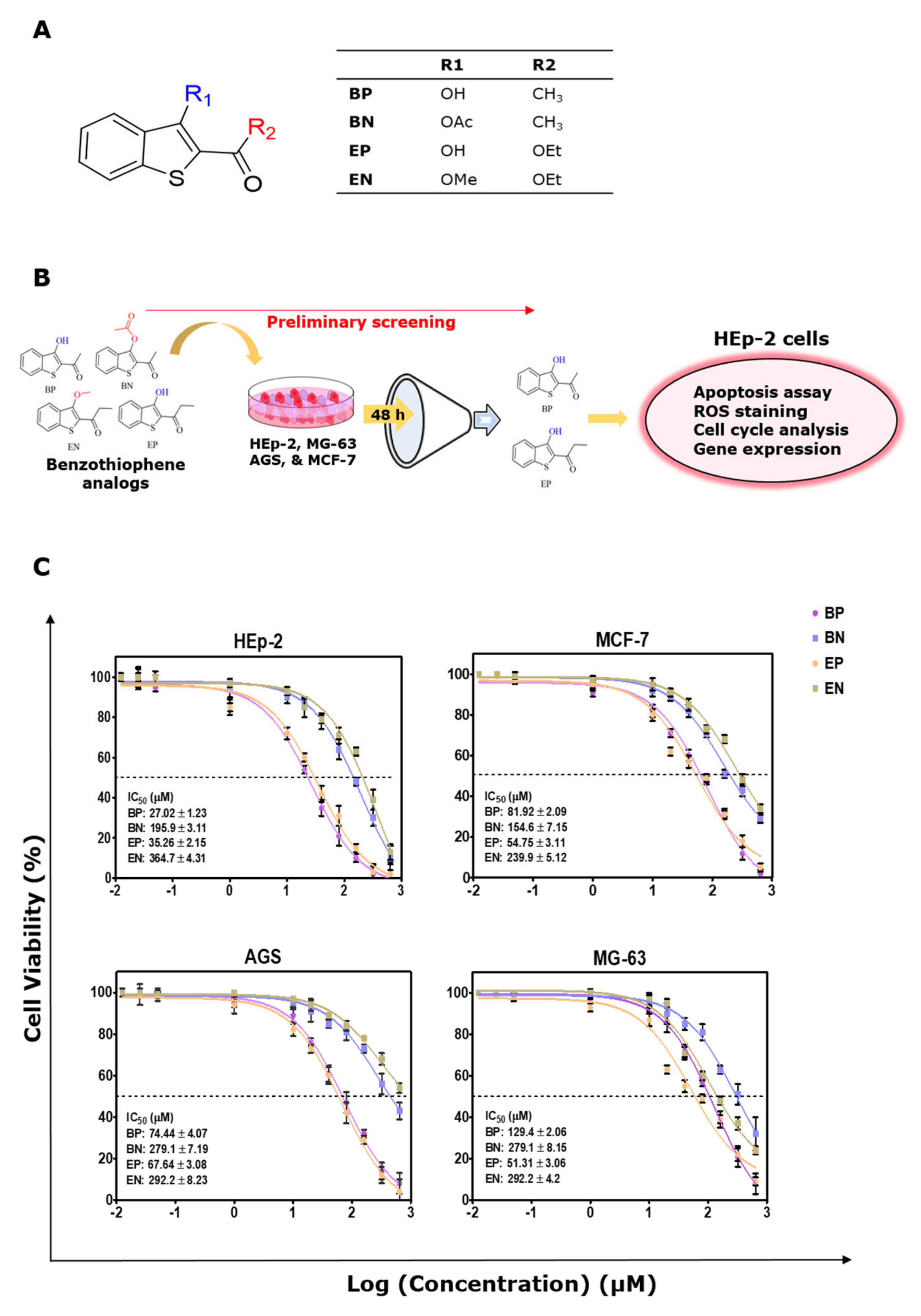
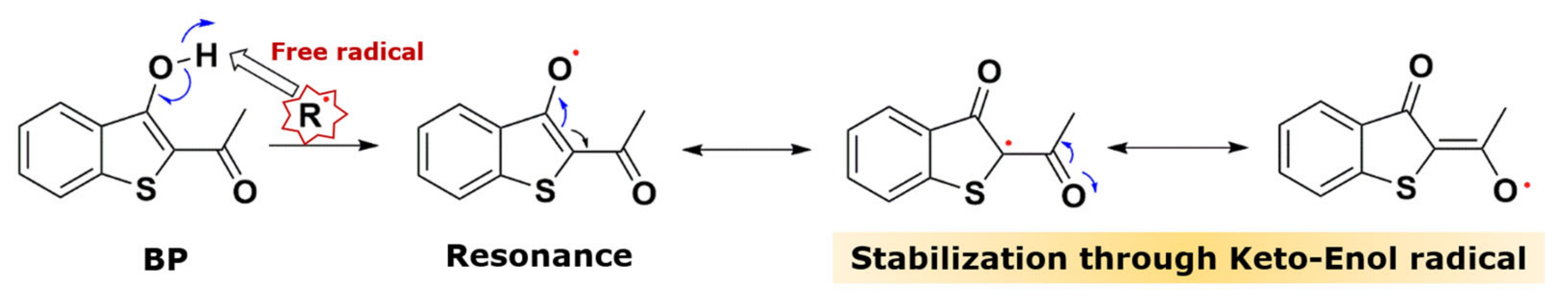
2.1. Cytotoxic Effect of the Benzo[b]thiophene Analogs
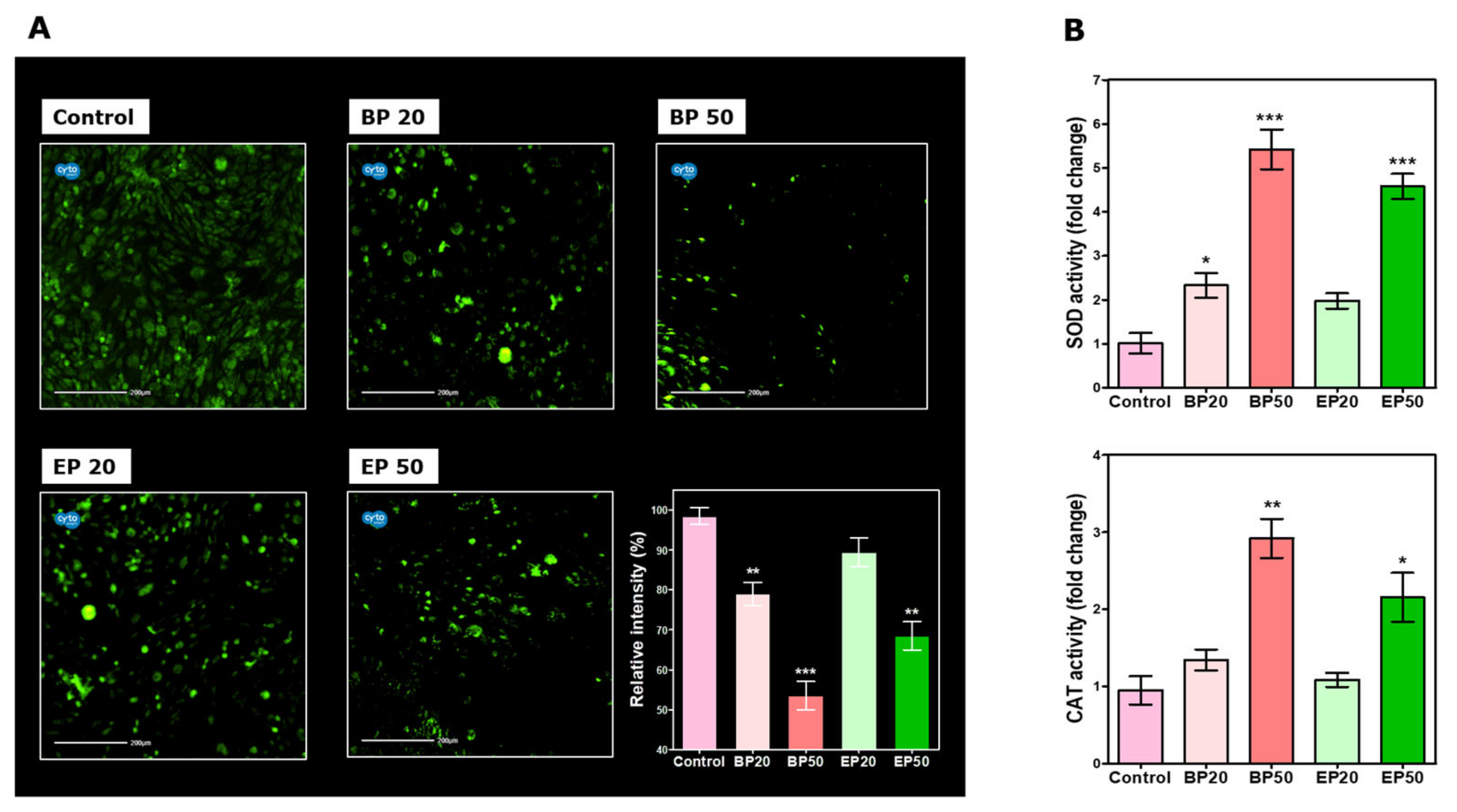
2.2. Alteration in ROS Status by BP and EP Analogs
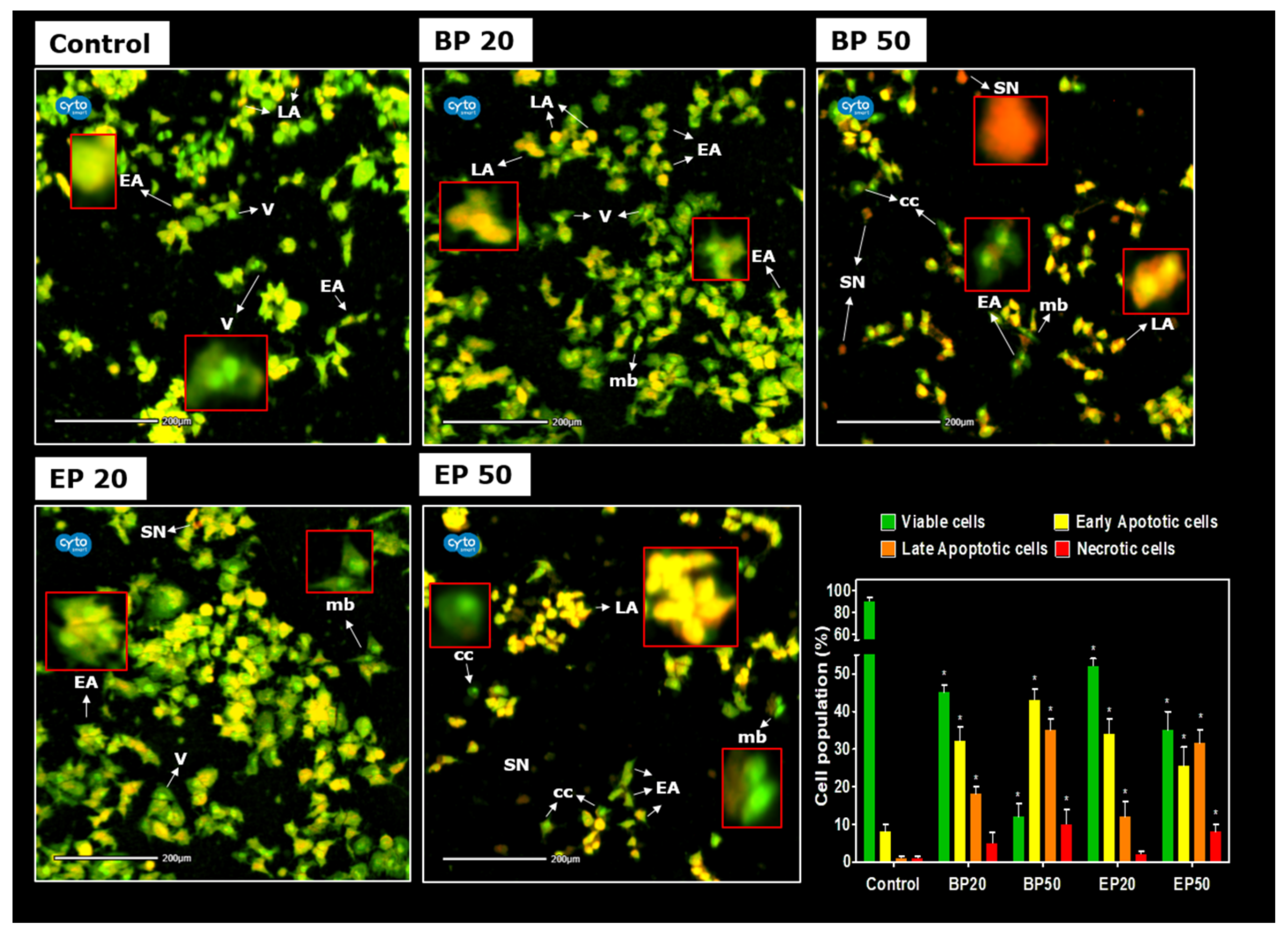
2.3. Activation of Cellular Apoptosis by Hydroxyl-Containing BP and EP Analogs
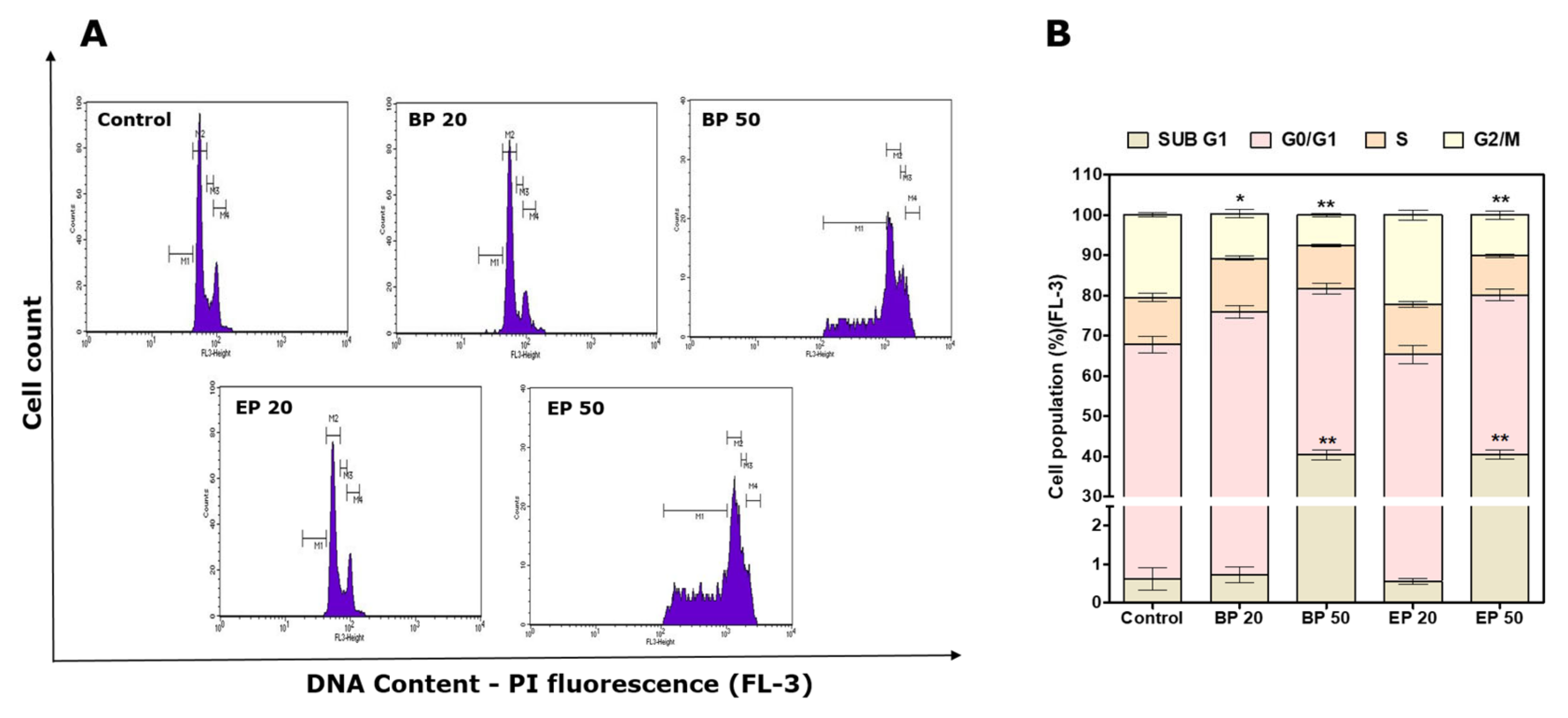
2.4. Sub-G1 Cell Cycle Arrest by Hydroxyl-Containing BP and EP Analogs
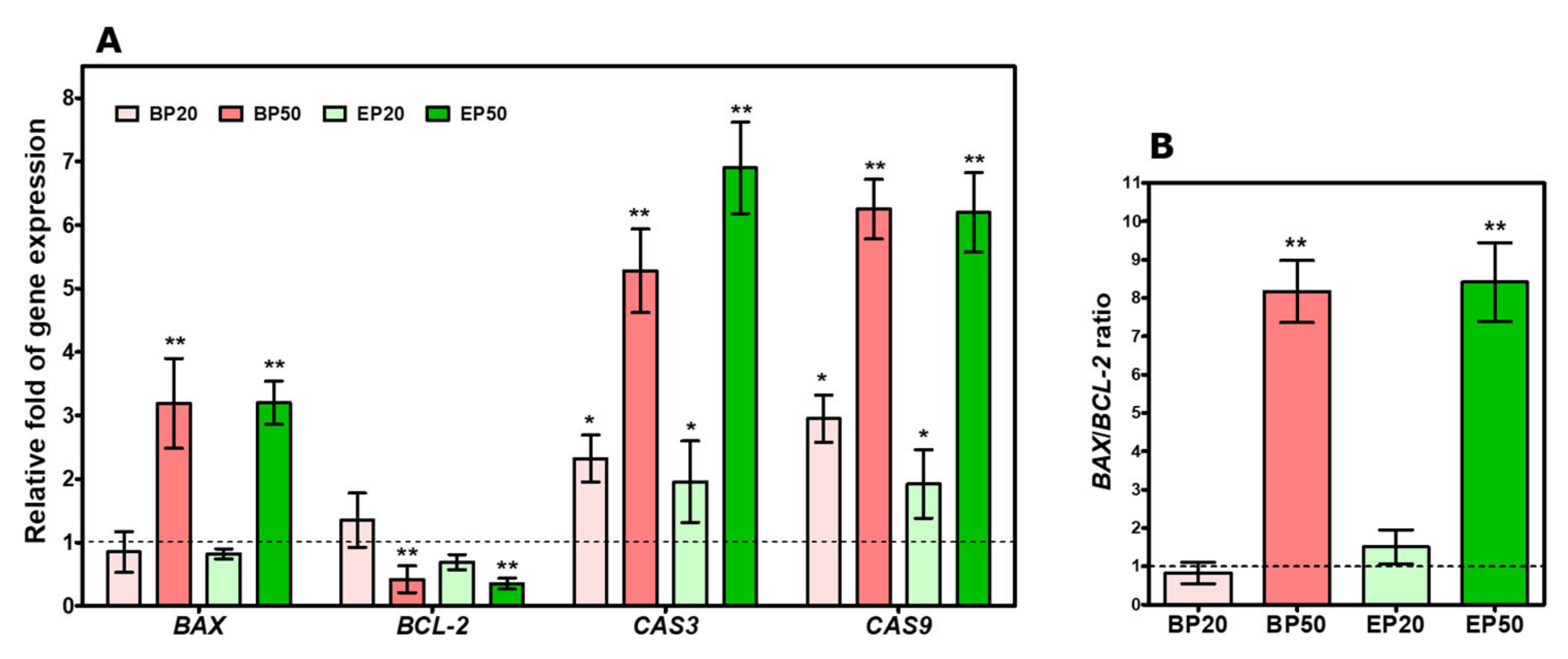
2.5. Increase in BAX/BCL-2 Transcript and Caspase Gene Expression by Hydroxyl-Containing BP and EP Analogs
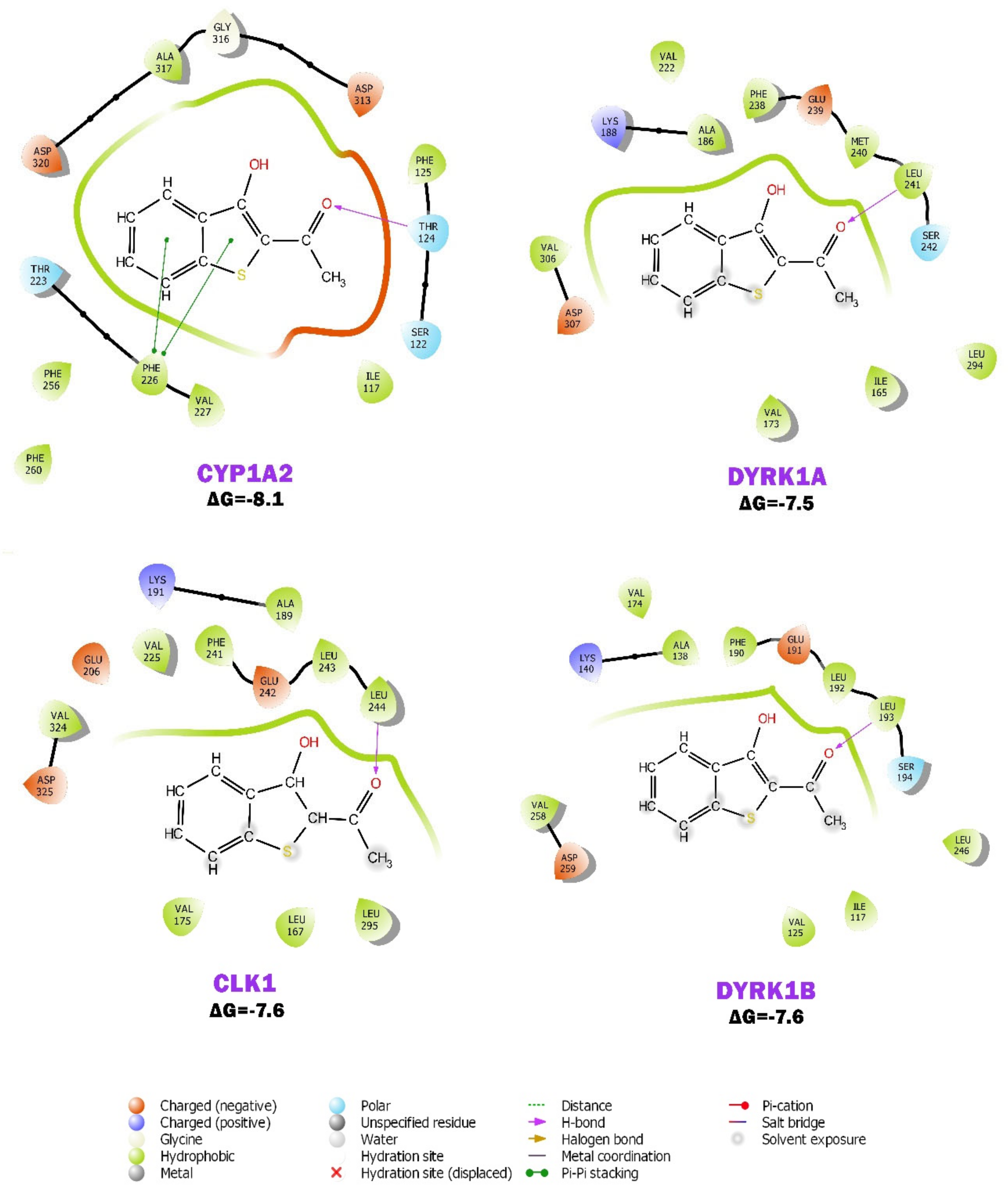
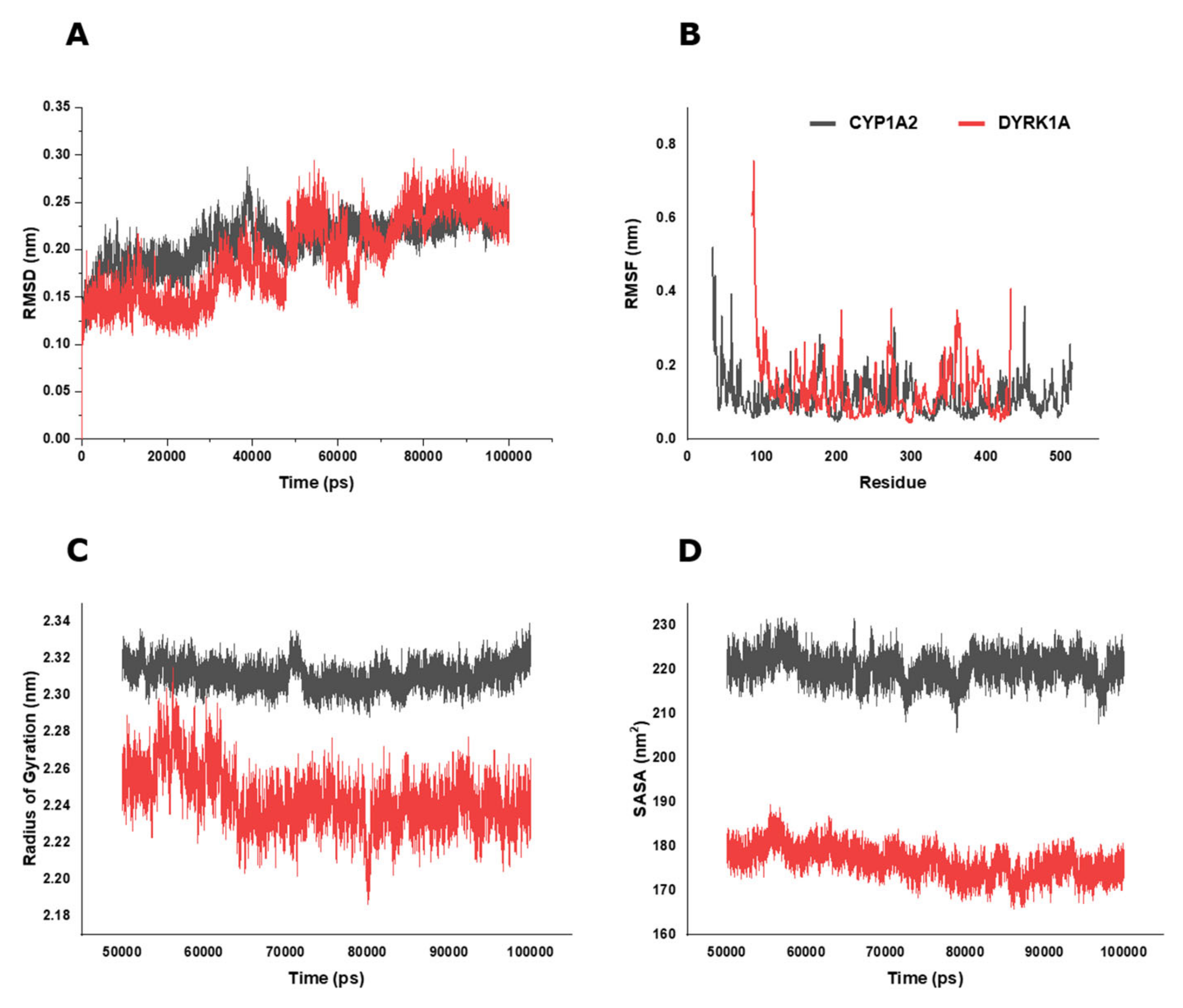
2.6. BP Possibly Interacts with Human CYP1A2 in MD Simulation
3. Materials and Methods
3.1. Cell Culture Maintenance
3.2. Assessment of Cell Growth Inhibition after Benzothiophene Analogs Treatment
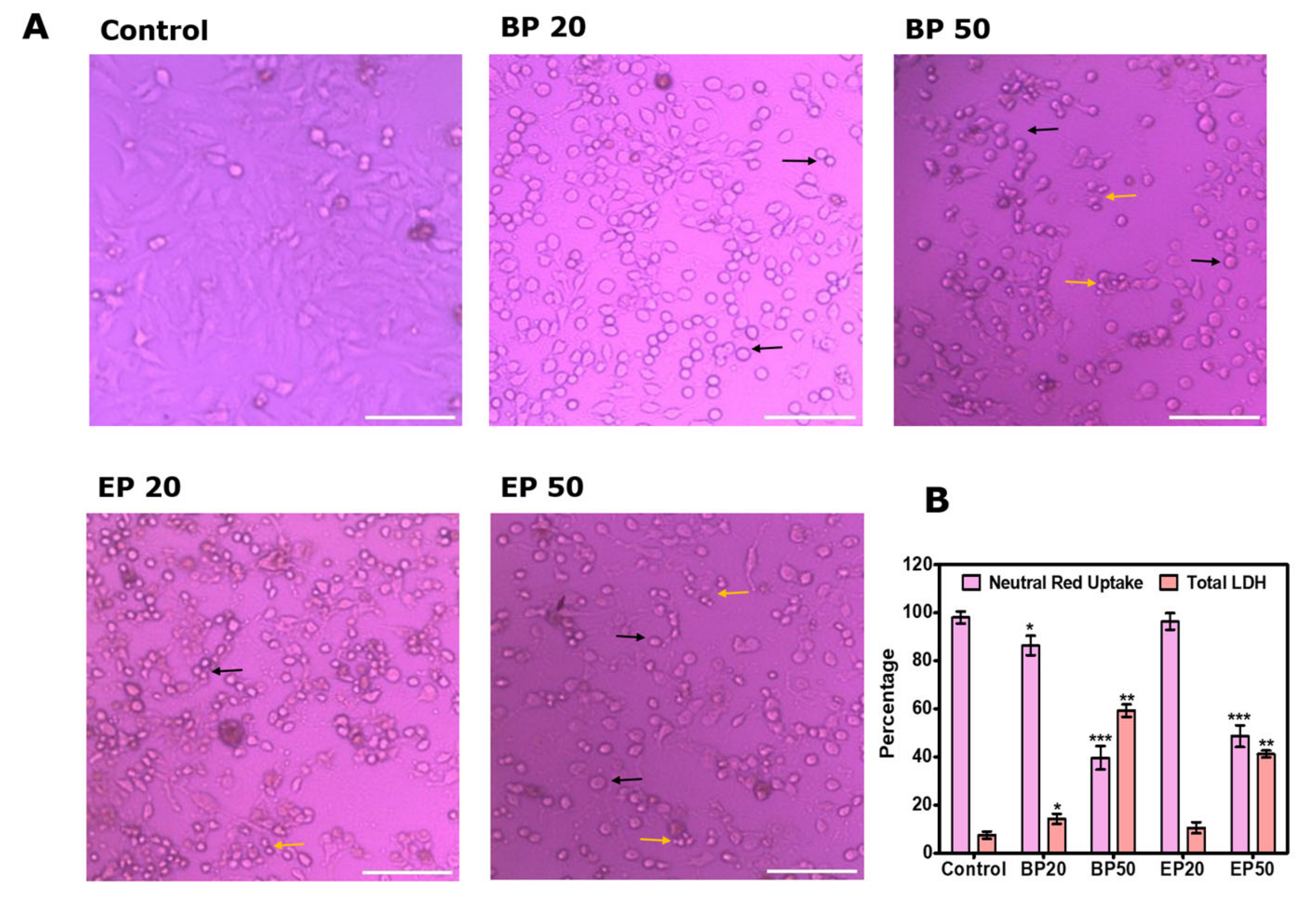
3.3. Neutral Red Uptake after Treatment with BP and EP
3.4. Total Lactate Dehydrogenase (LDH) after Treatment with BP and EP
3.5. Visual Morphological Changes after Treatment with BP and EP
3.6. Intracellular ROS Measurement after Treatment with BP and EP
3.7. SOD and CAT Activity of HEp-2 Cells
3.8. Ultramorphological Assessment of Apoptosis by Live/Dead Double Staining after Treatment with BP and EP
3.9. Change in Cell Cycle Analysis after Treatment with BP and EP
3.10. Apoptotic Gene Expression after Treatment with BP and EP
3.11. Molecular Docking and Dynamic Simulation
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck by Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Nelson, V.k.; Sahoo, N.K.; Sahu, M.; Sudhan, H.h.; Pullaiah, C.P.; Muralikrishna, K.S. In Vitro Anticancer Activity of Eclipta Alba Whole Plant Extract on Colon Cancer Cell HCT-116. BMC Complement. Med. Ther. 2020, 20, 355. [Google Scholar] [CrossRef]
- Reuter, C.W.M.; Morgan, M.A.; Eckardt, A. Targeting EGF-Receptor-Signalling in Squamous Cell Carcinomas of the Head and Neck. Br. J. Cancer 2007, 96, 408–416. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory. In Cancer Today; International Agency for Research on Cancer: Lyon, France, 2020; Available online: https://gco.iarc.fr/today (accessed on 27 March 2022).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Mastronikolis, N.; Ragos, V.; Fotiades, P.; Papanikolaou, V.; Kyrodimos, E.; Chrysovergis, A.; Mastronikolis, S.; Tsiambas, E. Mdm2 Oncogene in Laryngeal Squamous Cell Carcinoma. J. BUON 2020, 25, 594–596. [Google Scholar]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef]
- Hofmeister, V.; Vetter, C.; Schrama, D.; Bröcker, E.-B.; Becker, J.C. Tumor Stroma-Associated Antigens for Anti-Cancer Immunotherapy. Cancer Immunol. Immunother. 2006, 55, 481–494. [Google Scholar] [CrossRef]
- Pathania, S.; Bhatia, R.; Baldi, A.; Singh, R.; Rawal, R.K. Drug Metabolizing Enzymes and Their Inhibitors’ Role in Cancer Resistance. Biomed. Pharmacother. 2018, 105, 53–65. [Google Scholar] [CrossRef]
- Wiman, K.G.; Zhivotovsky, B. Understanding Cell Cycle and Cell Death Regulation Provides Novel Weapons against Human Diseases. J. Intern. Med. 2017, 281, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Plasencia, C.; Padilla-Benavides, T.; López-Urrutia, E.; Campos-Parra, A.D. Editorial: Repurposed Drugs Targeting Cancer Signaling Pathways: Clinical Insights to Improve Oncologic Therapies. Front. Oncol. 2021, 11, 1078–1088. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Abdallah, A.E.M.; Helal, M.H.E.; Shaloof, S.M.H. Synthesis and Structure Elucidation of Some Novel Thiophene and Benzothiophene Derivatives as Cytotoxic Agents. Acta Pharm. 2016, 66, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Waters, E.A.; McNeel, T.S.; Stevens, W.M.; Freedman, A.N. Use of Tamoxifen and Raloxifene for Breast Cancer Chemoprevention in 2010. Breast Cancer Res. Treat. 2012, 134, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Keri, R.S.; Chand, K.; Budagumpi, S.; Somappa, S.B.; Patil, S.A.; Nagaraja, B.M. An Overview of Benzo [b] Thiophene-Based Medicinal Chemistry. Eur. J. Med. Chem. 2017, 138, 1002–1033. [Google Scholar] [CrossRef]
- Penthala, N.R.; Sonar, V.N.; Horn, J.; Leggas, M.; Yadlapalli, J.S.K.B.; Crooks, P.A. Synthesis and Evaluation of a Series of Benzothiophene Acrylonitrile Analogs as Anticancer Agents. MedChemComm 2013, 4, 1073. [Google Scholar] [CrossRef] [Green Version]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Preti, D.; Fruttarolo, F.; Pavani, M.G.; Tabrizi, M.A.; Tolomeo, M.; Grimaudo, S.; et al. Synthesis and Biological Evaluation of 2- and 3-Aminobenzo[ b ]Thiophene Derivatives as Antimitotic Agents and Inhibitors of Tubulin Polymerization. J. Med. Chem. 2007, 50, 2273–2277. [Google Scholar] [CrossRef]
- Marastoni, E.; Bartoli, S.; Berettoni, M.; Cipollone, A.; Ettorre, A.; Fincham, C.I.; Mauro, S.; Paris, M.; Porcelloni, M.; Bigioni, M.; et al. Benzofused Hydroxamic Acids: Useful Fragments for the Preparation of Histone Deacetylase Inhibitors. Part 1: Hit Identification. Bioorg. Med. Chem. Lett. 2013, 23, 4091–4095. [Google Scholar] [CrossRef]
- Ester, K.; Hranjec, M.; Piantanida, I.; Ćaleta, I.; Jarak, I.; Pavelić, K.; Kralj, M.; Karminski-Zamola, G. Novel Derivatives of Pyridylbenzo[b]Thiophene-2-Carboxamides and Benzo[b]Thieno[2,3-c]Naphthyridin-2-Ones: Minor Structural Variations Provoke Major Differences of Antitumor Action Mechanisms. J. Med. Chem. 2009, 52, 2482–2492. [Google Scholar] [CrossRef]
- Gopinath, P.; Nilaya, S.; Debi, T.R.; Ramkumar, V.; Muraleedharan, K.M. As Many as Six Tandem Reactions in One Step! Unprecedented Formation of Highly Functionalized Benzothiophenes. Chem. Commun. 2009, 46, 7131–7133. [Google Scholar] [CrossRef] [Green Version]
- Haridevamuthu, B.; Manjunathan, T.; Guru, A.; Kumar, R.S.; Rajagopal, R.; Kuppusamy, P.; Juliet, A.; Gopinath, P.; Arockiaraj, J. Hydroxyl Containing Benzo[b]Thiophene Analogs Mitigates the Acrylamide Induced Oxidative Stress in the Zebrafish Larvae by Stabilizing the Glutathione Redox Cycle. Life Sci. 2022, 298, 120507. [Google Scholar] [CrossRef]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and Anticancer Activity of Brefeldin A Ester Derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef]
- Wang, X.; Matta, R.; Shen, G.; Nelin, L.D.; Pei, D.; Liu, Y. Mechanism of Triptolide-Induced Apoptosis: Effect on Caspase Activation and Bid Cleavage and Essentiality of the Hydroxyl Group of Triptolide. J. Mol. Med. 2006, 84, 405–415. [Google Scholar] [CrossRef]
- Dai, J.; Mumper, R.J. Plant Phenolics: Extraction, Analysis and Their Antioxidant and Anticancer Properties. Molecules 2010, 15, 7313–7352. [Google Scholar] [CrossRef]
- Grigalius, I.; Petrikaite, V. Relationship between Antioxidant and Anticancer Activity of Trihydroxyflavones. Molecules 2017, 22, 2169. [Google Scholar] [CrossRef] [Green Version]
- Kathiravan, A.; Manjunathan, T.; Ramasubramanian, K.; Gopinath, P. An Efficient Turn-ON Fluorescent Probe for Fluoride Ions—Meticulous Investigations and Development of Arduino Microcomputer Integrated Smartphone Device. J. Mol. Liq. 2022, 345, 117042. [Google Scholar] [CrossRef]
- Radi, S.; Tighadouini, S.; Feron, O.; Riant, O.; Bouakka, M.; Benabbes, R.; Mabkhot, Y. Synthesis of Novel β-Keto-Enol Derivatives Tethered Pyrazole, Pyridine and Furan as New Potential Antifungal and Anti-Breast Cancer Agents. Molecules 2015, 20, 20186–20194. [Google Scholar] [CrossRef] [Green Version]
- Shehab, W.S.; El-Bassyouni, G.T. Synthesis and Cyclization of β-Keto-Enol Derivatives Tethered Indole and Pyrazole as Potential Antimicrobial and Anticancer Activity. J. Iran. Chem. Soc. 2018, 15, 1639–1645. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Li, K.-M.; Balu, N.; Saeed, M.; Devanesan, P.; Higginbotham, S.; Zhao, J.; Gross, M.L.; Rogan, E.G. Catechol Ortho-Quinones: The Electrophilic Compounds That Form Depurinating DNA Adducts and Could Initiate Cancer and Other Diseases. Carcinogenesis 2002, 23, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-F.; Ke, C.-Y.; Wu, Y.-C.; Chuang, T.-H. Structure-Activity Relationship of Synthetic 2-Phenylnaphthalenes with Hydroxyl Groups That Inhibit Proliferation and Induce Apoptosis of MCF-7 Cancer Cells. PLoS ONE 2015, 10, e0141184. [Google Scholar] [CrossRef]
- Asadi-Samani, M.; Kaffash Farkhad, N.; Reza Mahmoudian-Sani, M.; Shirzad, H. Antioxidants as a Double-Edged Sword in the Treatment of Cancer. In Antioxidants; IntechOpen: London, UK, 2019. [Google Scholar]
- Helson, L. Curcumin (Diferuloylmethane) Delivery Methods: A Review. BioFactors 2013, 39, 21–26. [Google Scholar] [CrossRef]
- Huang, C.-H.; Jayakumar, T.; Chang, C.-C.; Fong, T.-H.; Lu, S.-H.; Thomas, P.; Choy, C.-S.; Sheu, J.-R. Hinokitiol Exerts Anticancer Activity through Downregulation of MMPs 9/2 and Enhancement of Catalase and SOD Enzymes: In Vivo Augmentation of Lung Histoarchitecture. Molecules 2015, 20, 17720–17734. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef]
- Stewart, Z.A.; Westfall, M.D.; Pietenpol, J.A. Cell-Cycle Dysregulation and Anticancer Therapy. Trends Pharmacol. Sci. 2003, 24, 139–145. [Google Scholar] [CrossRef]
- Nunez, R. DNA Measurement and Cell Cycle Analysis by Flow Cytometry. Curr. Issues Mol. Biol. 2001, 3, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.-C.; Tzao, C.; Chen, B.-H.; Liu, C.-W.; Yu, C.-P.; Jin, J.-S. Suberoylanilide Hydroxamic Acid Induces Apoptosis and Sub-G1 Arrest of 320 HSR Colon Cancer Cells. J. Biomed. Sci. 2010, 17, 76. [Google Scholar] [CrossRef] [Green Version]
- Haridevamuthu, B.; Manjunathan, T.; Guru, A.; Ranjith Wilson Alphonse, C.; Boopathi, S.; Murugan, R.; Gatasheh, M.K.; Atef Hatamleh, A.; Juliet, A.; Gopinath, P.; et al. Amelioration of Acrylamide Induced Neurotoxicity by Benzo[b]Thiophene Analogs via Glutathione Redox Dynamics in Zebrafish Larvae. Brain Res. 2022, 1788, 147941. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, Q.; Xiang, H.; Liu, M.; Zhang, Q.; Xue, W.; Song, B.; Yang, S. Antiproliferative Activity and Apoptosis-Inducing Mechanism of Constituents from Toona Sinensis on Human Cancer Cells. Cancer Cell Int. 2013, 13, 12. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Olsson, M.; Zhivotovsky, B. Caspases and Cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Tasyriq, M.; Najmuldeen, I.A.; In, L.L.A.; Mohamad, K.; Awang, K.; Hasima, N. 7α-Hydroxy-α-Sitosterol from Chisocheton Tomentosus Induces Apoptosis via Dysregulation of Cellular Bax/Bcl-2 Ratio and Cell Cycle Arrest by Downregulating ERK1/2 Activation. Evid.-Based Complement. Altern. Med. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Huang, G.; Zhang, Q.; Wang, Z.; Cui, J.; Wu, Y.; Meng, Q.; Li, S. Development of Benzochalcone Derivatives as Selective CYP1B1 Inhibitors and Anticancer Agents. MedChemComm 2019, 10, 1606–1614. [Google Scholar] [CrossRef]
- Dutour, R.; Cortés-Benítez, F.; Roy, J.; Poirier, D. Structure-Based Design and Synthesis of New Estrane–Pyridine Derivatives as Cytochrome P450 (CYP) 1B1 Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1159–1164. [Google Scholar] [CrossRef]
- Murray, G.I.; Taylor, M.C.; McFadyen, M.C.E.; McKay, J.A.; Greenlee, W.F.; Burke, M.D.; Melvin, W.T. Tumor-Specific Expression of Cytochrome P450 CYP1B11. Cancer Res. 1997, 57, 3026–3031. [Google Scholar]
- Ma, Q.; Lu, A.Y.H. CYP1A Induction and Human Risk Assessment: An Evolving Tale of in Vitro and in Vivo Studies: TABLE 1. Drug Metab. Dispos. 2007, 35, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Li, R.; Wang, C.; Zhou, S.; Fan, Y.; Ma, S.; Gao, D.; Gai, N.; Yang, J. Pinocembrin Inhibits the Proliferation and Metastasis of Breast Cancer via Suppression of the PI3K/AKT Signaling Pathway. Front. Oncol. 2021, 11, 661184. [Google Scholar] [CrossRef]
- Bhatt, S.; Dhiman, S.; Kumar, V.; Gour, A.; Manhas, D.; Sharma, K.; Ojha, P.K.; Nandi, U. Assessment of the CYP1A2 Inhibition-Mediated Drug Interaction Potential for Pinocembrin Using In Silico, In Vitro, and In Vivo Approaches. ACS Omega 2022, 7, 20321–20331. [Google Scholar] [CrossRef]
- Bruno, R.D.; Njar, V.C.O. Targeting Cytochrome P450 Enzymes: A New Approach in Anti-Cancer Drug Development. Bioorg. Med. Chem. 2007, 15, 5047–5060. [Google Scholar] [CrossRef]
- Velayutham, M.; Guru, A.; Gatasheh, M.K.; Hatamleh, A.A.; Juliet, A.; Arockiaraj, J. Molecular Docking of SA11, RF13 and DI14 Peptides from Vacuolar Protein Sorting Associated Protein 26B Against Cancer Proteins and In Vitro Investigation of Its Anticancer Potency in Hep-2 Cells. Int. J. Pept. Res. Ther. 2022, 28, 87. [Google Scholar] [CrossRef]
- Abdullah, A.-S.H.; Mohammed, A.S.; Abdullah, R.; Mirghani, M.E.S.; Al-Qubaisi, M. Cytotoxic Effects of Mangifera indica L. Kernel Extract on Human Breast Cancer (MCF-7 and MDA-MB-231 Cell Lines) and Bioactive Constituents in the Crude Extract. BMC Complement. Altern. Med. 2014, 14, 199. [Google Scholar] [CrossRef] [Green Version]
- Velayutham, M.; Guru, A.; Arasu, M.V.; Al-Dhabi, N.A.; Choi, K.C.; Elumalai, P.; Harikrishnan, R.; Arshad, A.; Arockiaraj, J. GR15 Peptide of S-Adenosylmethionine Synthase (SAMe) from Arthrospira Platensis Demonstrated Antioxidant Mechanism against H2O2 Induced Oxidative Stress in in-Vitro MDCK Cells and in-Vivo Zebrafish Larvae Model. J. Biotechnol. 2021, 342, 79–91. [Google Scholar] [CrossRef]
- Durak, I.; Yurtarslanl, Z.; Canbolat, O.; Akyol, Ö. A Methodological Approach to Superoxide Dismutase (SOD) Activity Assay Based on Inhibition of Nitroblue Tetrazolium (NBT) Reduction. Clin. Chim. Acta 1993, 214, 103–104. [Google Scholar] [CrossRef]
- Ellerby, L.M.; Bredesen, D.E. Measurement of Cellular Oxidation, Reactive Oxygen Species, and Antioxidant Enzymes during Apoptosis. Methods Enzymol. 2000, 322, 413–421. [Google Scholar] [CrossRef]
- Haridevamuthu, B.; Guru, A.; Murugan, R.; Sudhakaran, G.; Pachaiappan, R.; Almutairi, M.H.; Almutairi, B.O.; Juliet, A.; Arockiaraj, J. Neuroprotective Effect of Biochanin a against Bisphenol A-Induced Prenatal Neurotoxicity in Zebrafish by Modulating Oxidative Stress and Locomotory Defects. Neurosci. Lett. 2022, 790, 136889. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2-ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Al-Harbi, L.N.; Subash-Babu, P.; Binobead, M.A.; Alhussain, M.H.; AlSedairy, S.A.; Aloud, A.A.; Alshatwi, A.A. Potential Metabolite Nymphayol Isolated from Water Lily (Nymphaea Stellata) Flower Inhibits MCF-7 Human Breast Cancer Cell Growth via Upregulation of Cdkn2a, PRb2, P53 and Downregulation of PCNA MRNA Expressions. Metabolites 2020, 10, 280. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P.; Waterman, M.R.; Egli, M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed Automated Docking of Flexible Ligands to Proteins: Parallel Applications of AutoDock 2.4. J. Comput. Aided. Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Khan, S.; Khan, F.I.; Mohammad, T.; Khan, P.; Hasan, G.M.; Lobb, K.A.; Islam, A.; Ahmad, F.; Imtaiyaz Hassan, M. Exploring Molecular Insights into the Interaction Mechanism of Cholesterol Derivatives with the Mce4A: A Combined Spectroscopic and Molecular Dynamic Simulation Studies. Int. J. Biol. Macromol. 2018, 111, 548–560. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | BP (µM) | BN (µM) | EP (µM) | EN (µM) | ||||
|---|---|---|---|---|---|---|---|---|
| IC20 | IC50 | IC20 | IC50 | IC20 | IC50 | IC20 | IC50 | |
| HEp2 Laryngeal carcinoma | 6.01 ± 0.98 | 27.02 ± 1.23 | 35.28 ± 2.4 ¶ | 195.9 ± 3.11 ¶ | 3.89 ± 1.06 | 35.26 ± 2.15 * | 36.05 ± 2.56 § | 364.7 ± 4.31 § |
| AGS Gastric adenocarcinoma | 16.5 ± 1.23 | 74.44 ± 4.07 | 91.7 ± 3.36 ¶ | 279.1 ± 7.19 ¶ | 11.91 ± 1.64 | 67.64 ± 3.08 * | 131.2 ± 4.54 § | 292.2 ± 8.23 § |
| MG63 Osteosarcoma | 27.48 ± 2.05 | 129.4 ± 2.06 | 84.07 ± 3.57 ¶ | 279.1 ± 8.15 ¶ | 12.17 ± 1.24 | 51.31 ± 3.06 ** | 30.97 ± 2.8 § | 292.2 ± 4.2 § |
| MCF7 Breast adenocarcinoma | 11.41 ± 1.36 | 81.92 ± 2.09 | 47.29 ± 2.74 ¶ | 154.6 ± 7.15 ¶ | 9.9 ± 2.14 | 54.75 ± 3.11 * | 52.14 ± 3.26 § | 239.9 ± 5.12 § |
| Vero Kidney epithelial cells (non-tumorogenic) | - | 138.57 ± 7.9 | - | 202.5 ± 5.68 | - | 103.58 ± 8.57 | - | 324.2 ± 6.85 |
| Selectivity Index (SI) for HEp2 cells | 5.13 | >1 | 2.94 ** | >1 | ||||
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Reference |
|---|---|---|---|
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA | [59] |
| BAX | TCAGGATGCGTCCACCAAGAAG | TCAGGATGCGTCCACCAAGAAG | [59] |
| BCL-2 | GTGGATGACTGAGTACCT | CCAGGAGAAATCAAACAGAG | [59] |
| CASPASE 3 | ACATGGAAGCGAATCAATGGACTC | AAGGACTCAAATTCTGTTGCCACC | [59] |
| CASPASE 9 | GCTCTTCCTTTGTTCATC | CTCTTCCTCCACTGTTCA | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haridevamuthu, B.; Manjunathan, T.; Wilson Alphonse, C.R.; Kumar, R.S.; Thanigaivel, S.; Chandra Kishore, S.; Sundaram, V.; Gopinath, P.; Arockiaraj, J.; Bellucci, S. Functionalized Sulfur-Containing Heterocyclic Analogs Induce Sub-G1 Arrest and Apoptotic Cell Death of Laryngeal Carcinoma In Vitro. Molecules 2023, 28, 1856. https://doi.org/10.3390/molecules28041856
Haridevamuthu B, Manjunathan T, Wilson Alphonse CR, Kumar RS, Thanigaivel S, Chandra Kishore S, Sundaram V, Gopinath P, Arockiaraj J, Bellucci S. Functionalized Sulfur-Containing Heterocyclic Analogs Induce Sub-G1 Arrest and Apoptotic Cell Death of Laryngeal Carcinoma In Vitro. Molecules. 2023; 28(4):1856. https://doi.org/10.3390/molecules28041856
Chicago/Turabian StyleHaridevamuthu, B., Tamilvelan Manjunathan, Carlton Ranjith Wilson Alphonse, Rajendran Saravana Kumar, Sundaram Thanigaivel, Somasundaram Chandra Kishore, Vickram Sundaram, Pushparathinam Gopinath, Jesu Arockiaraj, and Stefano Bellucci. 2023. "Functionalized Sulfur-Containing Heterocyclic Analogs Induce Sub-G1 Arrest and Apoptotic Cell Death of Laryngeal Carcinoma In Vitro" Molecules 28, no. 4: 1856. https://doi.org/10.3390/molecules28041856
APA StyleHaridevamuthu, B., Manjunathan, T., Wilson Alphonse, C. R., Kumar, R. S., Thanigaivel, S., Chandra Kishore, S., Sundaram, V., Gopinath, P., Arockiaraj, J., & Bellucci, S. (2023). Functionalized Sulfur-Containing Heterocyclic Analogs Induce Sub-G1 Arrest and Apoptotic Cell Death of Laryngeal Carcinoma In Vitro. Molecules, 28(4), 1856. https://doi.org/10.3390/molecules28041856