
Synthesis and Biological Evaluation of Octahydroquinazolinones as Phospholipase A2, and Protease Inhibitors: Experimental and Theoretical Exploration

 , ,
, , 
 ,
,  ,
, 
Abstract
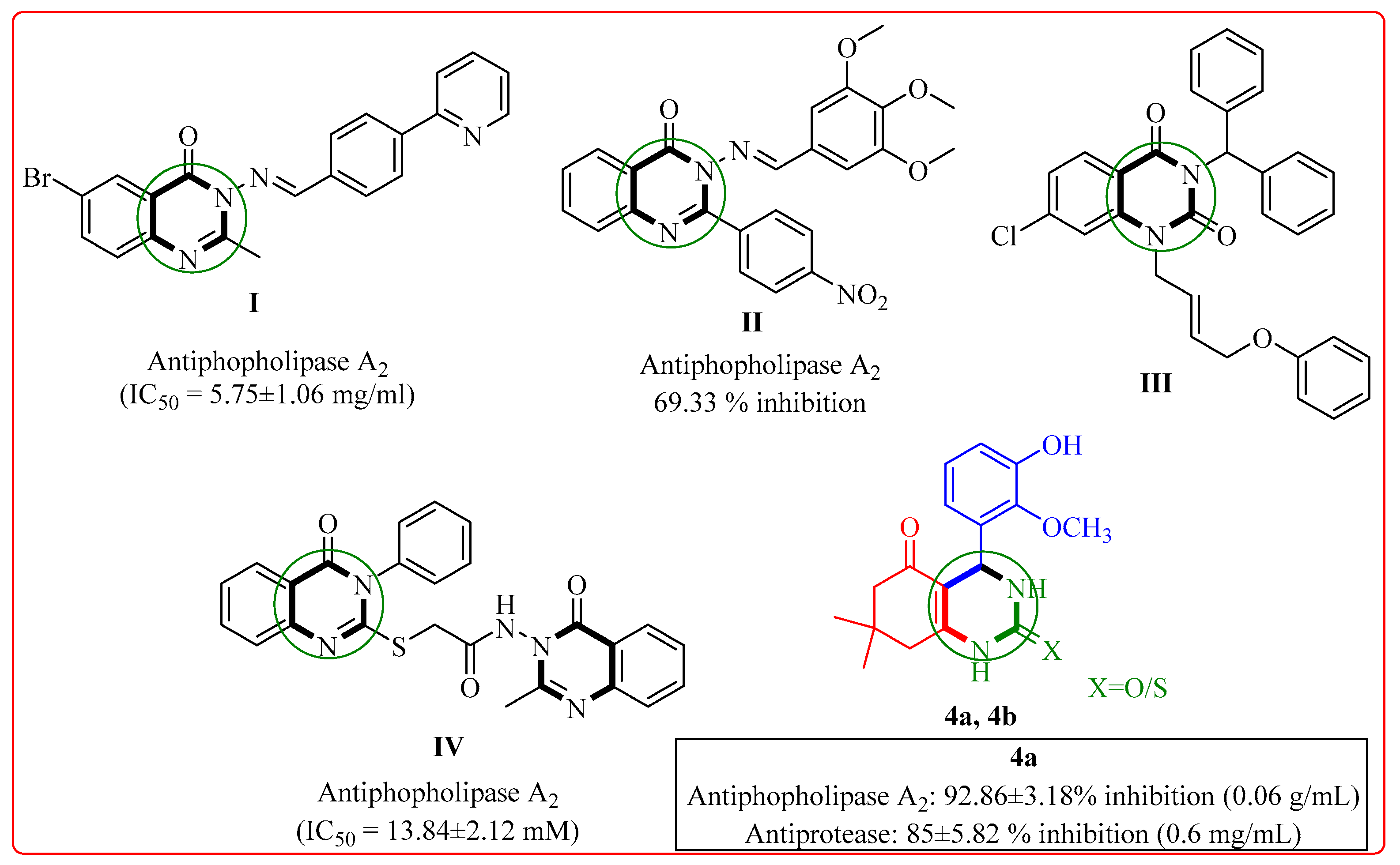
:1. Introduction
2. Results
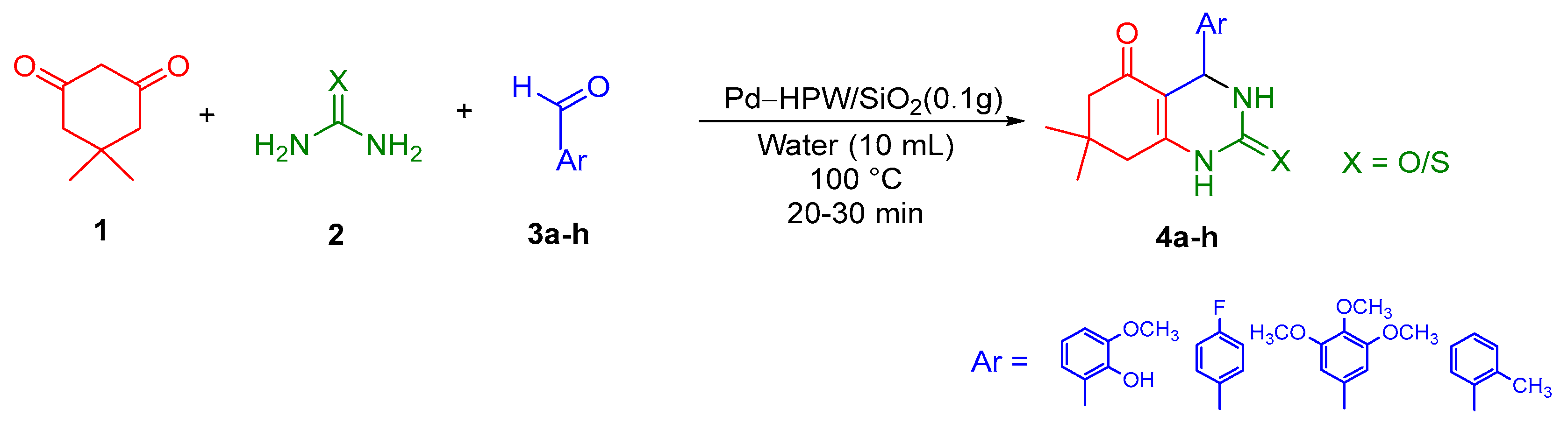
2.1. Chemistry
2.1.1. Spectroscopic Characterization of Synthesized Compounds
FT-IR
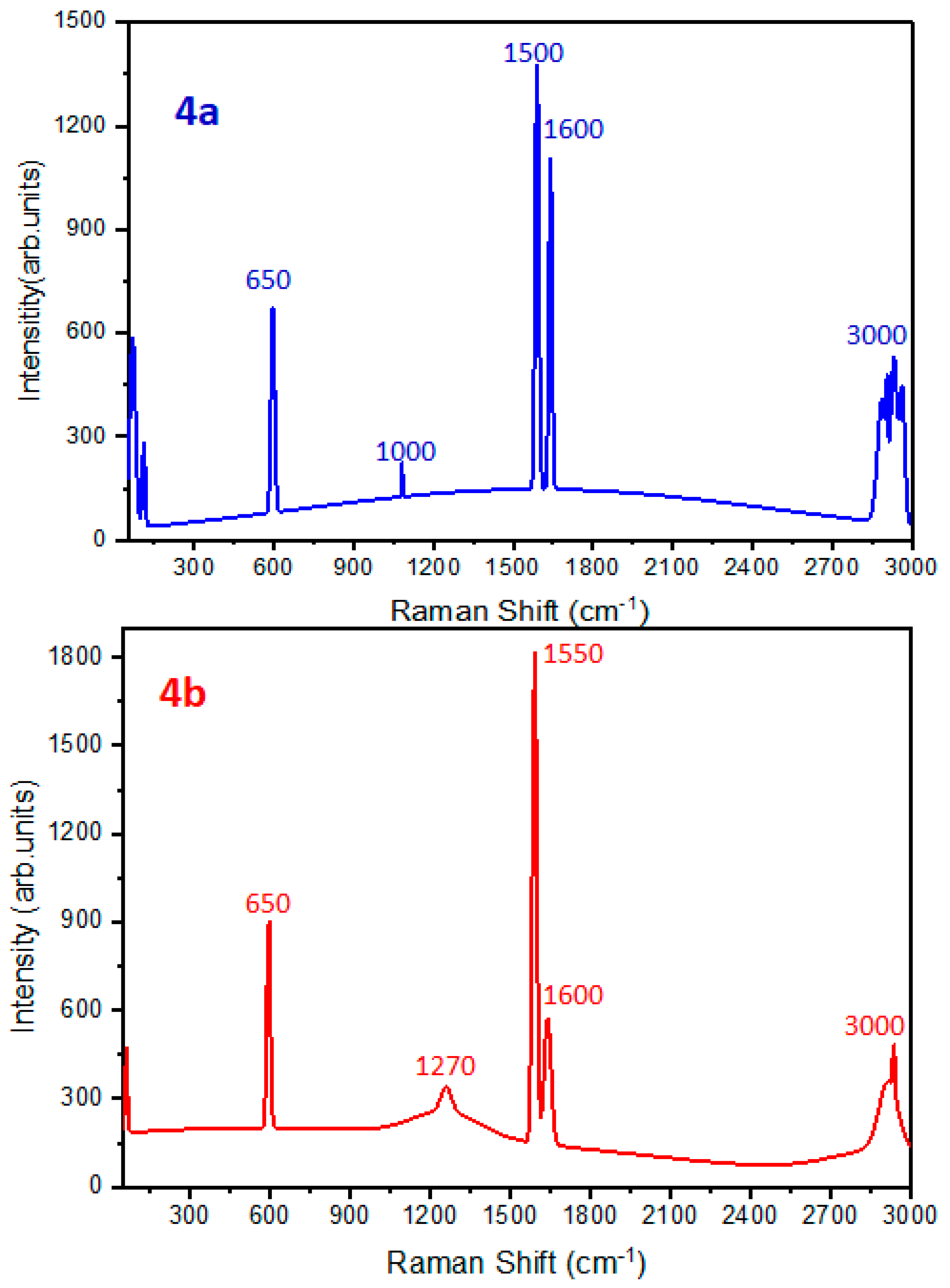
Raman Spectroscopy
Spectroscopic Results of Novel Compounds
NMR Spectrum

2.2. Biological Evaluation
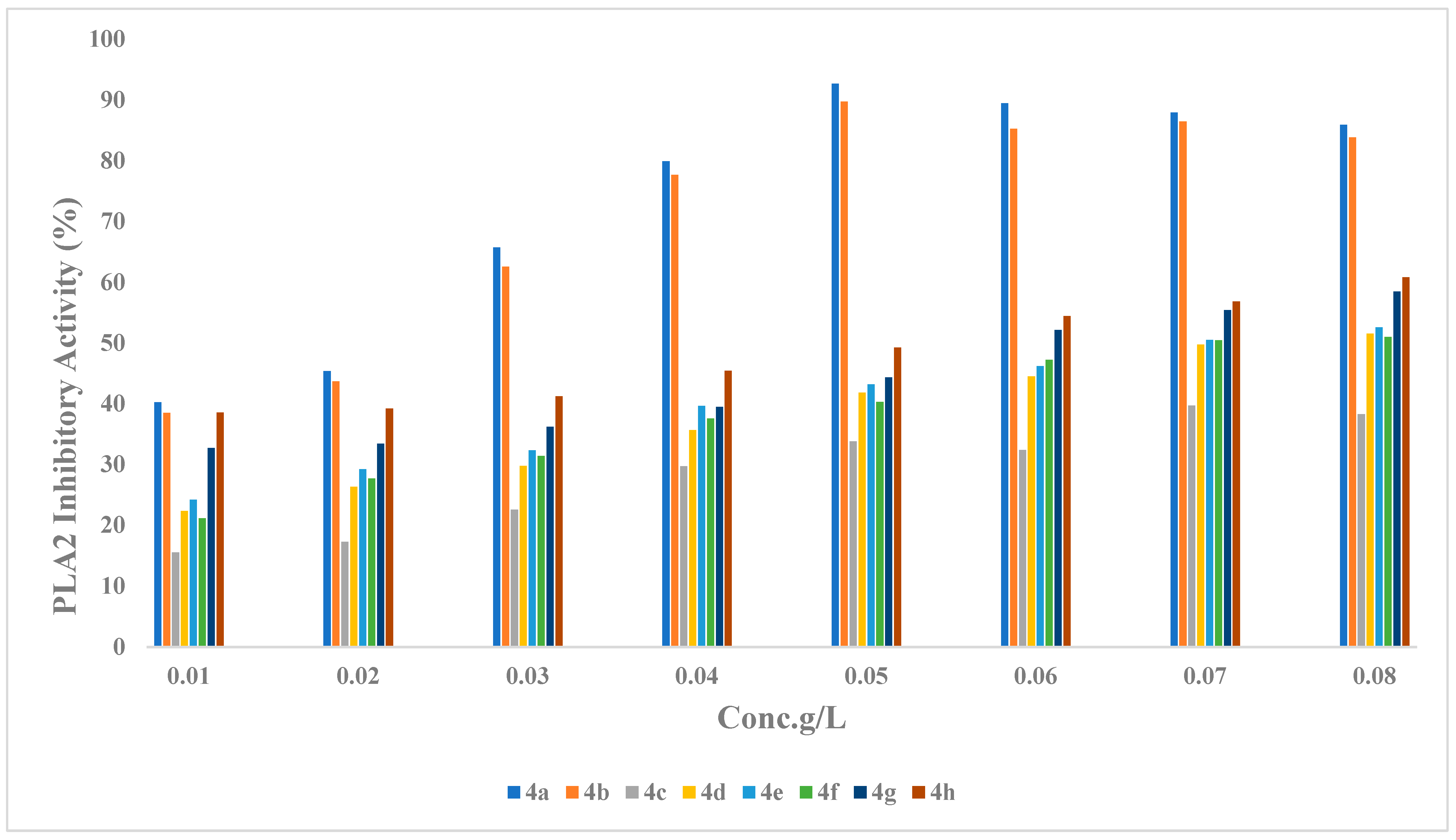
2.2.1. Phospholipase (PLA2) Inhibitory Activity
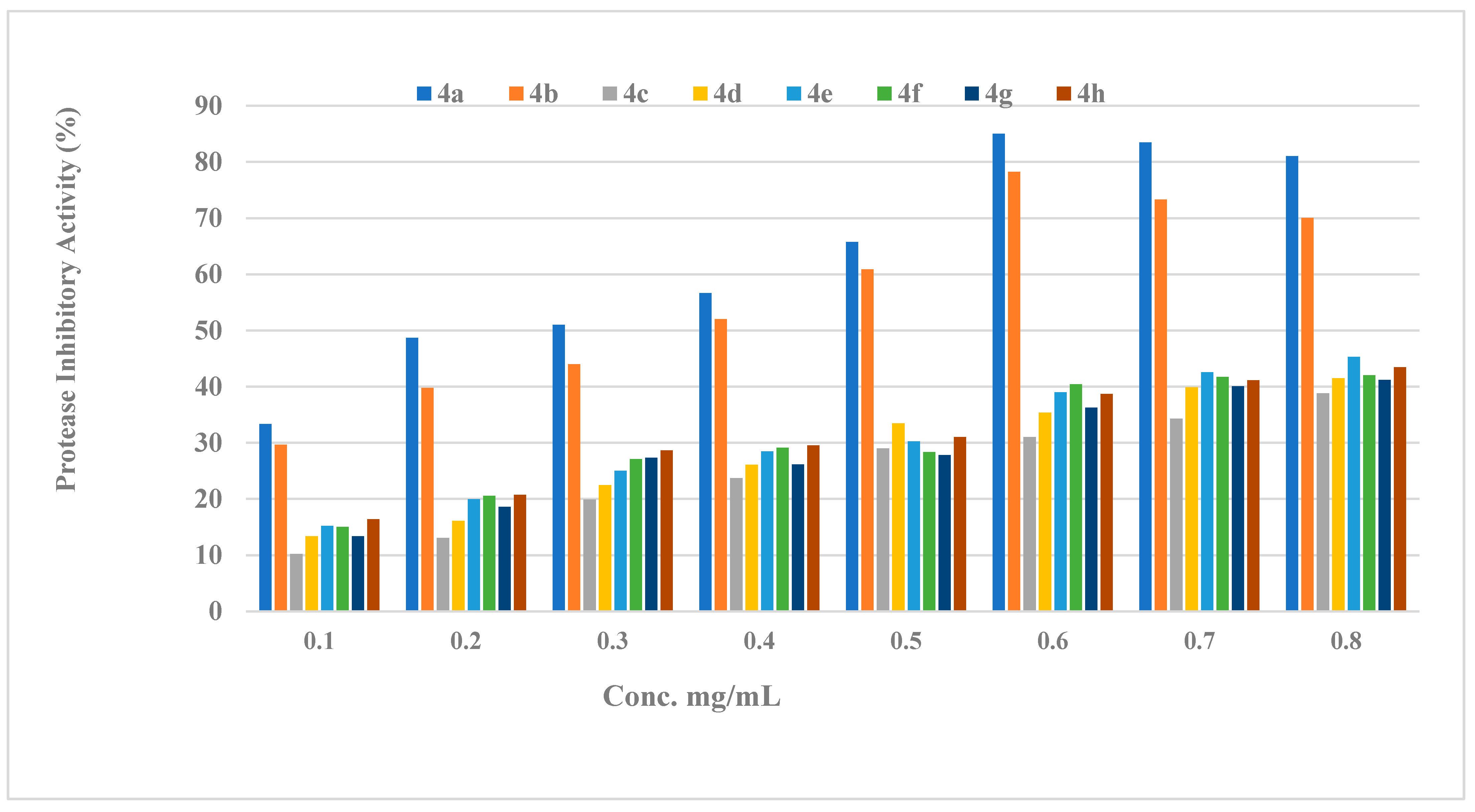
2.2.2. Protease Activity
2.2.3. Structure Activity Relationship (SAR) of Compounds (4a–h)
2.3. In Silico Studies
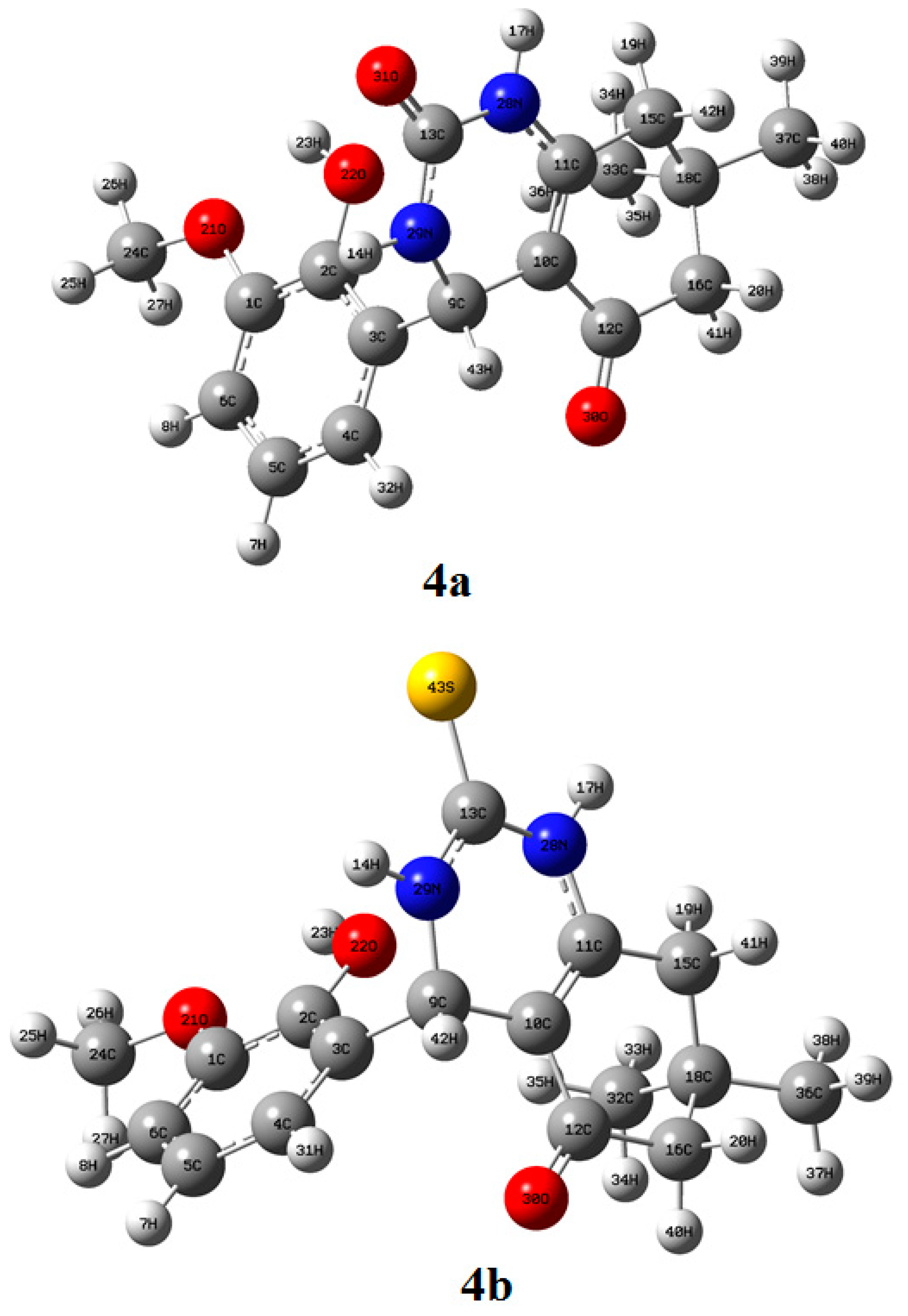
2.3.1. Structure Elucidation of Compounds 4a and 4b
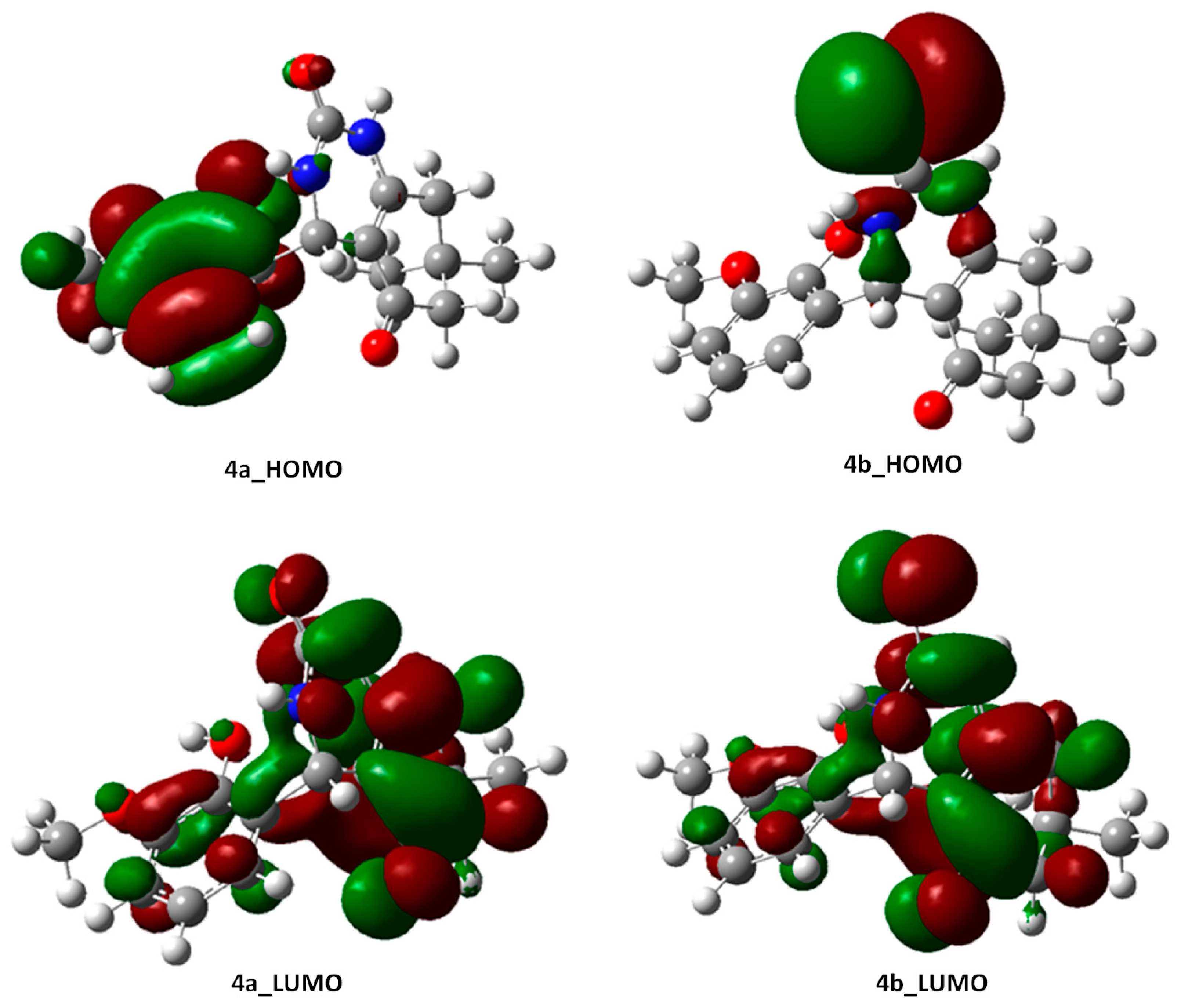
2.3.2. Frontier Molecular Orbital (FMO) Analysis of Compounds 4a and 4b
2.3.3. Nature Bonding Orbital (NBO) Analysis of Molecules 4a and 4b
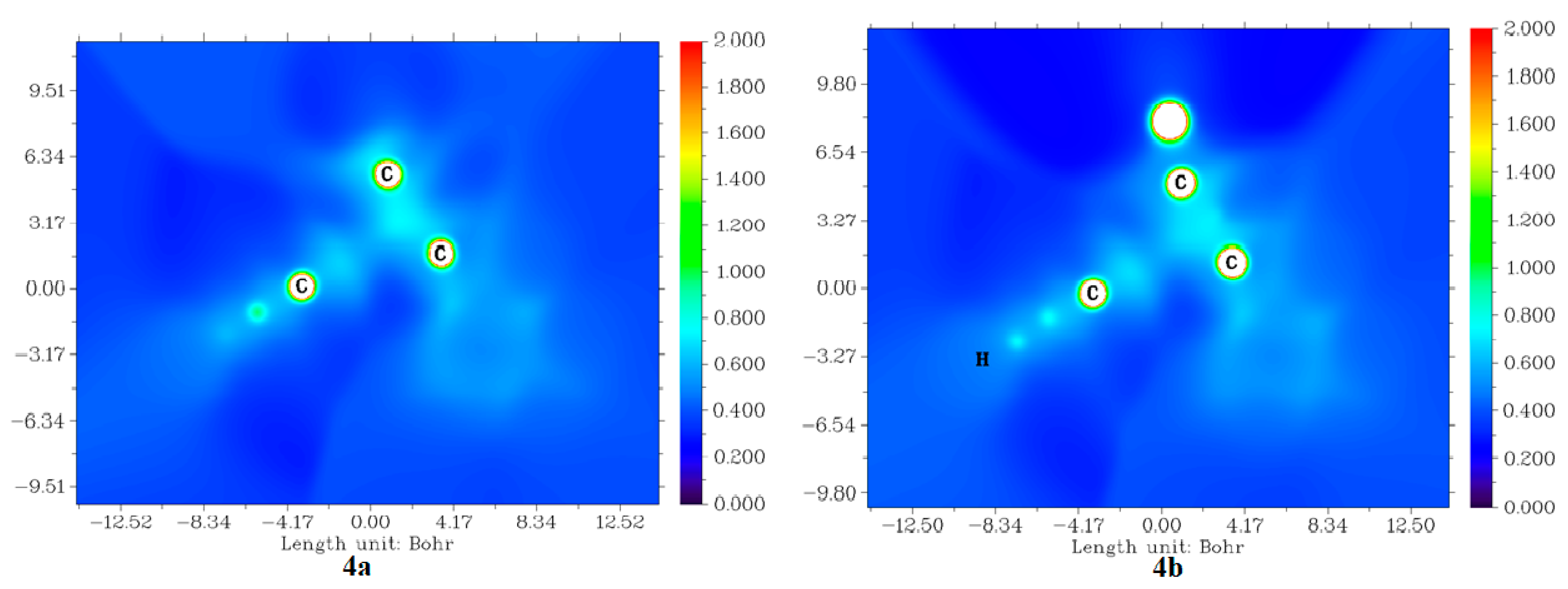
2.3.4. Average Localized Ionization Energy (ALIE) of Molecules 4a and 4b
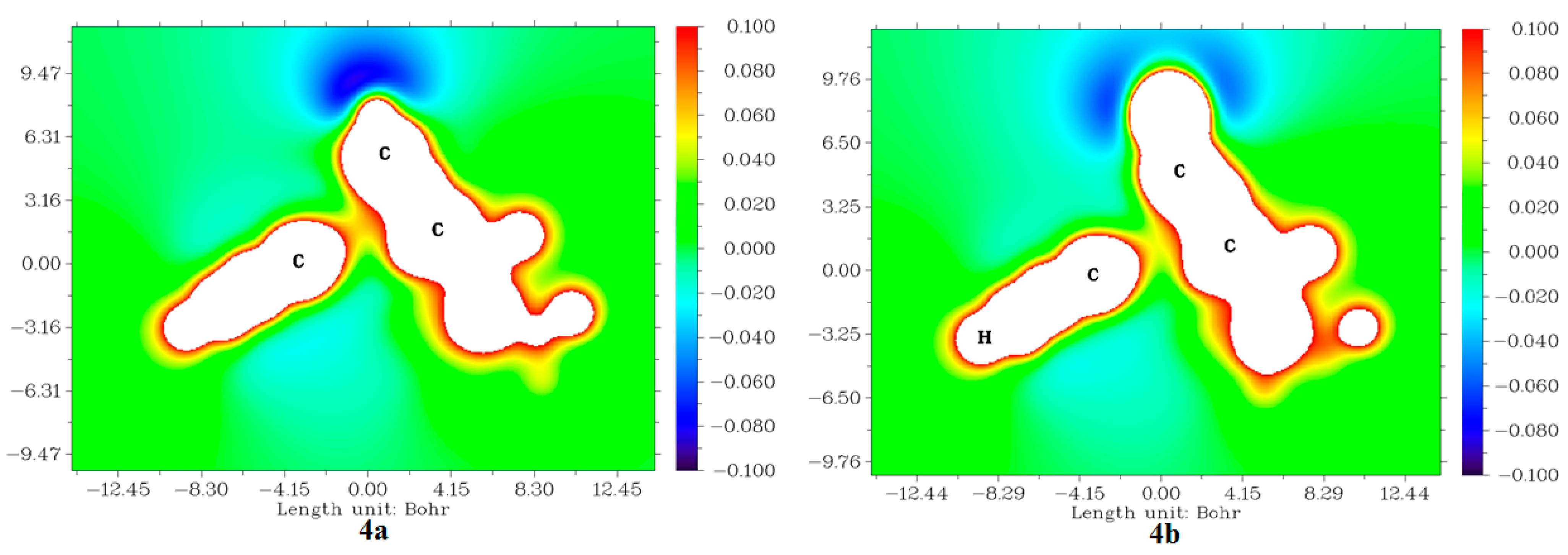
2.3.5. Molecular Electrostatic Potentials (MESP) from Electronic Charges and Nuclear Charges of Compounds 4a and 4b
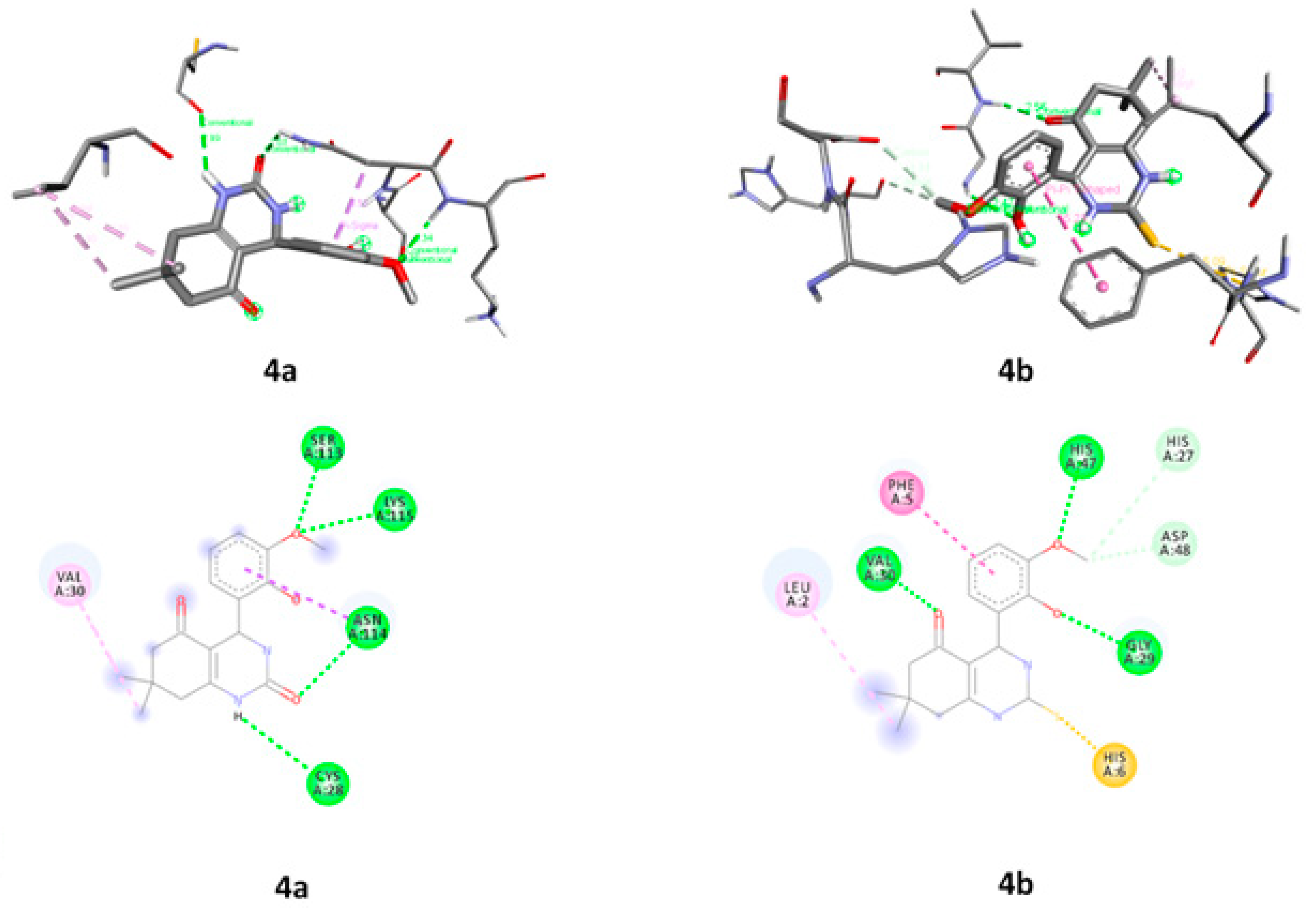
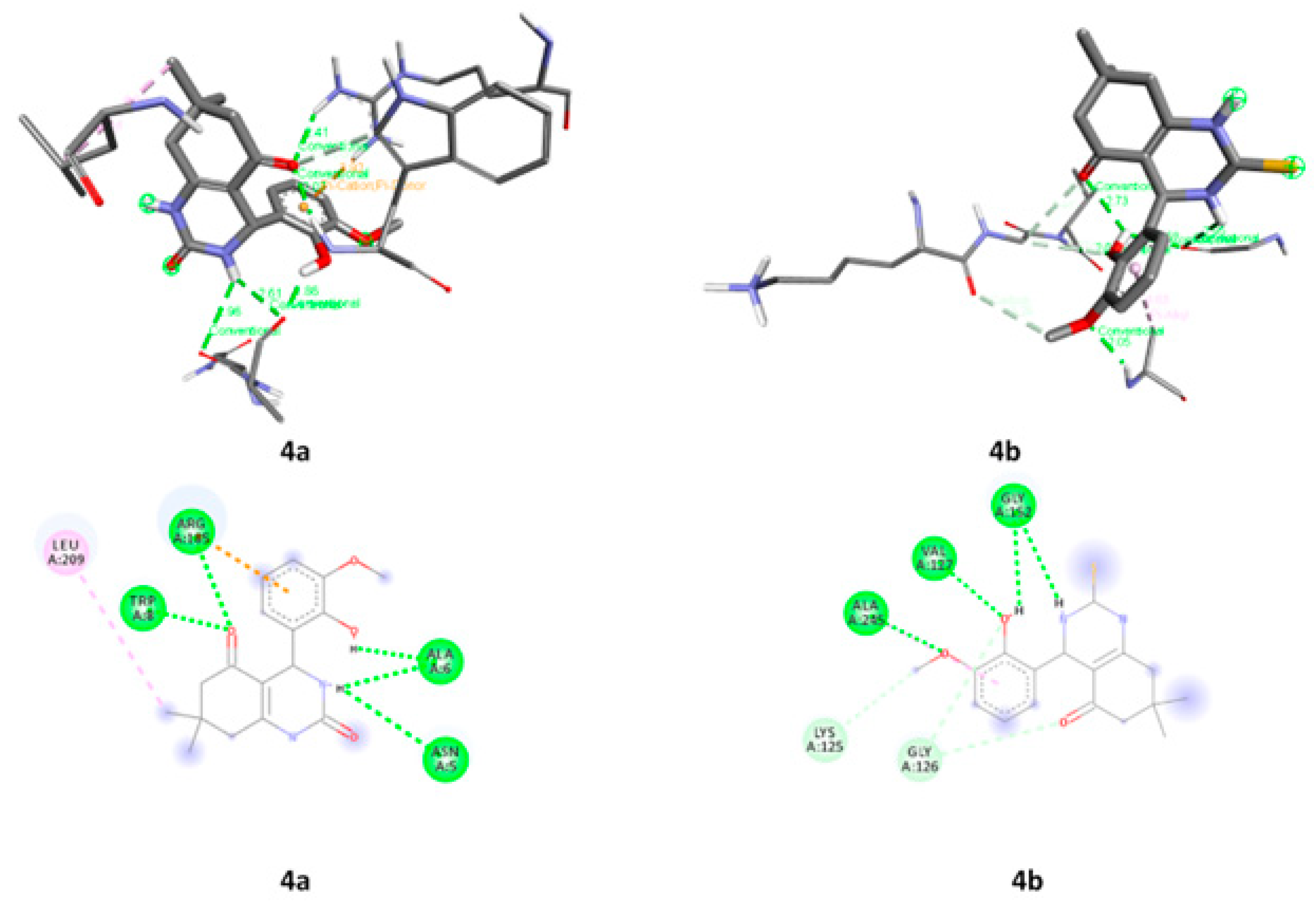
2.3.6. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. Preparation of Pd-HPW/SiO2 Catalyst
3.1.2. General Procedure for the Synthesis of Octahydrquinazoline Derivatives (4a–b)
3.2. In Silico Studies
3.2.1. DFT Studies
3.2.2. Molecular Docking
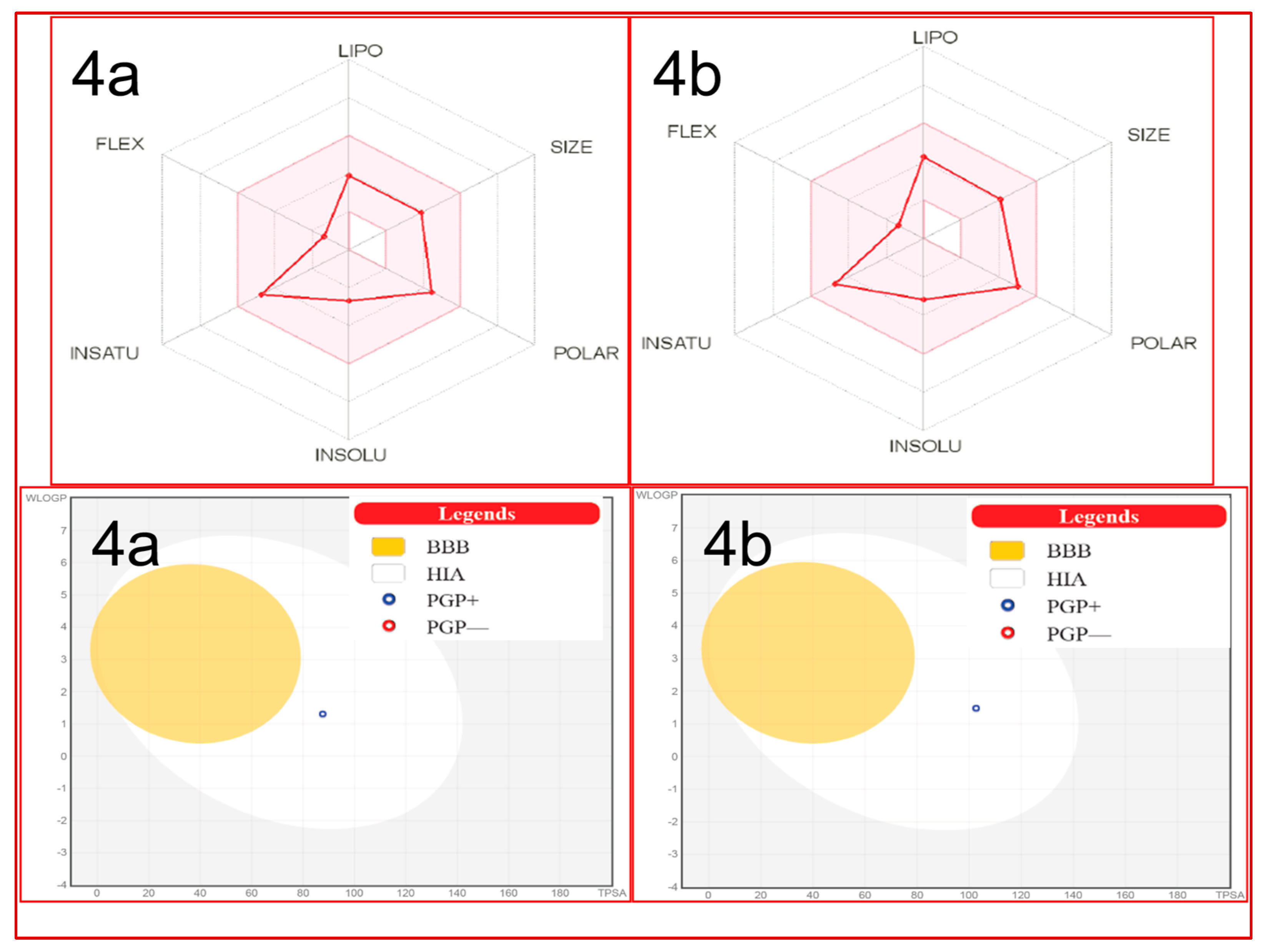
ADME Studies
3.3. Biological Evaluation
3.3.1. Inhibitory Activity of Phospholipase (PLA2) Enzyme
3.3.2. Protease Inhibitory Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sabir, S.; Alhazza, M.I.; Ibrahim, A.A. A review on heterocyclic moieties and their applications. Catal. Sustain. Energy 2016, 2, 99–115. [Google Scholar] [CrossRef]
- Riadi, Y.; Alamri, M.A.; Geesi, M.H.; Anouar, E.H.; Ouerghi, O.; Alabbas, A.B.; Alossaimi, M.A.; Altharawi, A.; Dehbi, O.; Alqahtani, S.M. Synthesis, characterization, biological evaluation and molecular docking of a new quinazolinone-based derivative as a potent dual inhibitor for VEGFR-2 and EGFR tyrosine kinases. J. Biomol. Struct. Dyn. 2021, 40, 6810–6816. [Google Scholar] [CrossRef] [PubMed]
- Allah, M.A.A.H.; Balakit, A.A.; Salman, H.I.; Abdulridha, A.A.; Sert, Y. New Heterocyclic Compound as Carbon Steel Corrosion Inhibitor in 1 M H2SO4, High Efficiency at Low Concentration: Experimental and Theoretical Studies. J. Adhes. Sci. Technol. 2022, 37, 525–547. [Google Scholar] [CrossRef]
- Abdulridha, A.A.; Allah, M.A.A.H.; Makki, S.Q.; Sert, Y.; Salman, H.E.; Balakit, A.A. Corrosion inhibition of carbon steel in 1 M H2SO4 using new Azo Schiff compound: Electrochemical, gravimetric, adsorption, surface and DFT studies. J. Mol. Liq. 2020, 315, 113690. [Google Scholar] [CrossRef]
- Balakit, A.A.; Makki, S.Q.; Sert, Y.; Ucun, F.; Alshammari, M.B.; Thordarson, P.; El-Hiti, G.A. Synthesis, spectrophotometric and DFT studies of new Triazole Schiff bases as selective naked-eye sensors for acetate anion. Supramol. Chem. 2020, 32, 519–526. [Google Scholar] [CrossRef]
- Gümüş, M.; Babacan, N.; Demir, Y.; Sert, Y.; Koca, I.; Gülçin, I. Discovery of sulfadrug–pyrrole conjugates as carbonic anhydrase and acetylcholinesterase inhibitors. Arch. Pharm. 2021, 355, e2100242. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, S.; Zheng, S.; Liang, E.; Liu, S.; Yao, X.; Zhu, Q. A series of octahydroquinazoline-5-ones as novel inhibitors against dengue virus. Eur. J. Med. Chem. 2020, 200, 112318. [Google Scholar] [CrossRef] [PubMed]
- Kidwai, M.; Bhatnagar, D.; Kumar, R.; Luthra, P.M. Synthesis of 2-Oxo/Thioxooctahydroquinazolin-5-one Derivatives and Their Evaluation as Anticancer Agents. Chem. Pharm. Bull. 2010, 58, 1320–1323. [Google Scholar] [CrossRef] [Green Version]
- Chandrika, P.M.; Yakaiah, T.; Rao, A.R.R.; Narsaiah, B.; Reddy, N.C.; Sridhar, V.; Rao, J.V. Synthesis of novel 4,6-disubstituted quinazoline derivatives, their anti-inflammatory and anti-cancer activity (cytotoxic) against U937 leukemia cell lines. Eur. J. Med. Chem. 2008, 43, 846–852. [Google Scholar] [CrossRef]
- Saleh, E.A.M.; Al Dawsari, A.M.; Husain, K.; Kutty, I.H.; Rai, K. Synthesis, Antibacterial, and Antioxidant Evaluation of Novel Series of Condensed Thiazoloquinazoline with Pyrido, Pyrano, and Benzol Moieties as Five- and Six-Membered Heterocycle Derivatives. Molecules 2021, 26, 357. [Google Scholar] [CrossRef]
- Hassani, Z.; Islami, M.R.; Kalantari, M. An efficient one-pot synthesis of octahydroquinazolinone derivatives using catalytic amount of H2SO4 in water. Bioorganic Med. Chem. Lett. 2006, 16, 4479–4482. [Google Scholar] [CrossRef] [PubMed]
- Kuraitheerthakumaran, A.; Pazhamalai, S.; Manikandan, H.; Gopalakrishnan, M. Rapid and efficient one-pot synthesis of octahydroquinazolinone derivatives using lanthanum oxide under solvent-free condition. J. Saudi Chem. Soc. 2014, 18, 920–924. [Google Scholar] [CrossRef] [Green Version]
- Misono, M. Unique acid catalysis of heteropoly compounds (heteropolyoxometalates) in the solid state. Chem. Commun. 2001, 13, 1141–1152. [Google Scholar] [CrossRef]
- Furimsky, E.; Massoth, F.E. Introduction. Catal. Today 1993, 17, 537–659. [Google Scholar] [CrossRef]
- Krayem, N.; Gargouri, Y. Scorpion venom phospholipases A2: A minireview. Toxicon 2020, 184, 48–54. [Google Scholar] [CrossRef]
- Valentin, E.; Lambeau, G. Increasing molecular diversity of secreted phospholipases A2 and their receptors and binding proteins. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2000, 1488, 59–70. [Google Scholar] [CrossRef]
- Murakami, M.; Kudo, I. Diversity and regulatory functions of mammalian secretory phospholipase A2s. Adv. Immunol. 2001, 77, 163–194. [Google Scholar] [CrossRef]
- El-Sayed, N.N.E.; Alafeefy, A.M.; Bakht, M.A.; Masand, V.H.; Aldalbahi, A.; Chen, N.; Fan, C.; Ben Bacha, A. Synthesis, Antiphospholipase A2, Antiprotease, Antibacterial Evaluation and Molecular Docking Analysis of Certain Novel Hydrazones. Molecules 2016, 21, 1664. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [Green Version]
- Rae, D.; Beechey-Newman, N.; Sumar, N.; Hermon-Taylor, J.; Porter, J.; Bennett, D. Type 1 prophospholipase A2 propeptide in acute lung injury. Lancet 1994, 344, 1472–1473. [Google Scholar] [CrossRef]
- Peuravuori, H.J.; Funatomi, H.; Nevalainen, T.J. Group I and Group II Phospholipases A2 in Serum in Uraemia. Clin. Chem. Lab. Med. 1993, 31, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carreno, F.; Navarrete, M.; Toro, D. Classification of Proteases without Tears. Biochem. Educ. 1997, 25, 161–167. [Google Scholar] [CrossRef]
- El-Sayed, N.; Almaneai, N.; Bacha, A.; El-Ashrey, M.; Al-Zaben, M.; Almarhoon, Z. Biological Evaluation, Molecular Docking Analyses, and ADME Profiling of Certain New Quinazolinones as Anti-colorectal Agents. ACS Omega 2022, 7, 18443–18458. [Google Scholar] [CrossRef] [PubMed]
- Kirincich, S.J.; Xiang, J.; Green, N.; Tam, S.; Yang, H.; Shim, J.; Shen, M.; Clark, J.; McKew, J. Benzhy-drylquinazolinediones: Novel cytosolic phospholipase A2α inhibitors with improved physicochemical properties. Bioorg. Med. Chem. 2009, 17, 4383. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.N.E.; Almaneai, N.M.; Ben Bacha, A.; Al-Obeed, O.; Ahmad, R.; Abdulla, M.; Alafeefy, A.M. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3H-quinazolin-4-one derivatives. J. Enzym. Inhib. Med. Chem. 2019, 34, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakht, A.; Alotaibi, M.; Alharthi, A.I.; Din, I.U.; Ali, A.; Ali, A.; Ahsan, M.J. Pd-HPW/SiO2 Bi-Functional Catalyst: Sonochemical Synthesis, Characterization, and Effect on Octahydroquinazolinone Synthesis. Catalysts 2021, 11, 1273. [Google Scholar] [CrossRef]
- Yarım, M.; Saraç, S.; Kılıç, F.; Erol, K. Synthesis and in vitro calcium antagonist activity of 4-aryl-7,7-dimethyl/1,7,7-trimethyl-1,2,3,4,5,6,7,8-octahydroquinazoline-2,5-dione derivatives. II Farm. 2003, 58, 17–24. [Google Scholar] [CrossRef]
- Le Parc, R.; Freitas, V.T.; Hermet, P.; Cojocariu, A.M.; Cattoën, X.; Wadepohl, H.; Maurin, D.; Tse, C.H.; Bartlett, J.R.; Ferreira, R.A.S.; et al. Infrared and Raman spectroscopy of non-conventional hydrogen bonding between N,N′-disubstituted urea and thiourea groups: A combined experimental and theoretical investigation. Phys. Chem. Chem. Phys. 2019, 21, 3310–3317. [Google Scholar] [CrossRef]
- Tiwari, D.; Fermin, D.J.; Chaudhuri, T.K.; Ray, A. Solution Processed Bismuth Ferrite Thin Films for All-Oxide Solar Photovoltaics. J. Phys. Chem. C 2015, 119, 5872–5877. [Google Scholar] [CrossRef]
- Panicker, C.Y.; Varghese, H.T.; Ambujakshan, K.; Mathew, S.; Ganguli, S.; Nanda, A.K.; Van Alsenoy, C. FT-IR and FT-Raman spectra and ab initio calculations of 3-{[(2-hydroxyphenyl) methylene]amino}-2-phenylquinazolin-4(3H)-one. J. Raman Spectrosc. 2009, 40, 1262–1273. [Google Scholar] [CrossRef]
- Colthup, N.; Daly, L.; Wiberly, S. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: Boston, MA, USA, 1990. [Google Scholar]
- Khairul, W.M.; Isa, M.I.N.; Samsudin, A.S.; Adli, H.K.; Ghazali, S.R. Conductive biodegradable film of N-octyloxyphenyl-N′-(4-methylbenzoyl)thiourea. Bull. Mater. Sci. 2014, 37, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Alafeefy, A.M.; Awaad, A.; Abdel-Aziz, H.; El-Meligy, R.M.; Zain, M.E.; Al-Outhman, M.R.; Ben Bacha, A. Synthesis and biological evaluation of certain 3-substituted benzylideneamino-2-(4-nitrophenyl)quinazolin-4(3H)-one derivatives. J. Enzym. Inhib. Med. Chem. 2014, 30, 270–276. [Google Scholar] [CrossRef]
- Thomas, R.; Pooventhiran, T. Study of the dynamics of the interaction of glycine and GABA with water and ethanol using theoretical tools. J. Mol. Liq. 2022, 368, 120721. [Google Scholar] [CrossRef]
- Thomas, R.; Pooventhiran, T.; Bakht, A.; Alzahrani, A.Y.; Salem, M.A. Study of interaction between different solvents and neurotransmitters dopamine, l-adrenaline, and l-noradrenaline using LED, QTAIM and AIMD. J. Mol. Liq. 2022, 368, 120708. [Google Scholar] [CrossRef]
- Thomas, R.; Pooventhiran, T. Comprehensive Quantum Mechanical Study of Structural Features, Reactivity, Molecular Properties and Wave Function-Based Characteristics of Capmatinib. In Advanced Materials and Nano Systems: Theory and Experiment—Part 2; Rai, D.P., Ed.; Bentham Science Publisher: Sharjah, United Arab Emirates, 2022; pp. 252–270. [Google Scholar] [CrossRef]
- Pooventhiran, T.; Rajashekaraiah, K.; Murthy, S.; Joseph, R.; Thomas, R. Study of the Electronic Properties of a Fluoropyrazolecarbonitrile Derivative and Enhancement of Spectral Properties on Adsorption with Fullerene. Biointerface Res. Appl. Chem. 2023, 13, 1–28. [Google Scholar] [CrossRef]
- Anaikutti, P.; Selvaraj, M.; Prabhakaran, J.; Pooventhiran, T.; Jeyakumar, T.C.; Thomas, R.; Makam, P. Indolyl-4H-chromenes: Multicomponent one-pot green synthesis, in vitro and in silico, anticancer and antioxidant studies. J. Mol. Struct. 2022, 1266, 133464. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, UK, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Schmider, H.; Becke, A. Chemical content of the kinetic energy density. J. Mol. Struct. THEOCHEM 2000, 527, 51–61. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D.; Johnson, E.R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Longuet-Higgins, H.C.; Pople, J.A. Electronic Spectral Shifts of Nonpolar Molecules in Nonpolar Solvents. J. Chem. Phys. 1957, 27, 192–194. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [Green Version]
- Filimonov, D.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Mary, Y.S.; Resmi, K.; Kumar, V.S.; Thomas, R.; Sureshkumar, B. Detailed quantum mechanical, molecular docking, QSAR prediction, photovoltaic light harvesting efficiency analysis of benzil and its halogenated analogues. Heliyon 2019, 5, e02825. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2018, 47, D464–D474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Discovery Studio BIOVA. Discovery Studio Vizualizer Client, V17; Dassault Systems: San Diego, CA, USA, 2017. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, A.L.; Radvanyi, F. Determination of phospholipase A2 activity by a colorimetric assay using a pH indicator. Toxicon 1987, 25, 1181–1188. [Google Scholar] [CrossRef]
- Kunitz, M. Crystalline soybean trypsin inhibitor. J. Gen. Physiol. 1947, 30, 291–310. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 |
|---|---|
| 4a | 0.029 g/L (0.027–0.031, n = 3) |
| 4b | 0.030 g/L (0.028–0.031, n = 3) |
| 4c | 0.037 g/L (0.030–0.044, n = 3) |
| 4c | 0.041 g/L (0.037–0.046, n = 3) |
| 4e | 0.035 g/L (0.029–0.042, n = 3) |
| 4f | 0.028 g/L (0.011–0.044, n = 3) |
| 4g | 0.049 g/L (0.046–0.052, n = 3) |
| 4h | 0.048 g/L (0.044–0.053, n = 3) |
| Compound | IC50 |
|---|---|
| 4a | 0.039 mg/L (0.031–0.046, n = 3) |
| 4b | 0.037 mg/L (0.032–0.042, n = 3) |
| 4c | 0.039 mg/L (0.023–0.042, n = 3) |
| 4c | 0.037 mg/L (0.033–0.040, n = 3) |
| 4e | 0.040 mg/L (0.032–0.049, n = 3) |
| 4f | 0.047 mg/L (0.011–0.067, n = 3) |
| 4g | 0.963 mg/L (0.315–2.94, n = 3) |
| 4h | 0.826 mg/L (0.382–1.78, n = 3) |
| Parameters | 4a | 4b | ||
|---|---|---|---|---|
| eV | kcal/mol | eV | kcal/mol | |
| HOMO | −8.6269 | −198.42 | −7.5015 | −172.54 |
| LUMO | −5.2689 | −121.19 | −5.3187 | −122.33 |
| Energy gap (∆E) | −3.3580 | −77.234 | −2.1828 | −50.204 |
| Ionization energy (I = ɛHOMO = −HOMO) | 8.62693 | 198.419 | 7.50153 | 172.535 |
| Electron affinity (A= ɛLUMO = −LUMO) | 5.26894 | 121.186 | 5.31874 | 122.331 |
| Global hardness (η = (I − A)/2) | 1.67899 | 38.6168 | 1.09139 | 25.102 |
| Global softness (S = 1/2η) | 0.8395 | 19.3084 | 0.5457 | 12.551 |
| Chemical potential (μ = −(I + A)/2) | −6.9479 | −159.8 | −6.4101 | −147.43 |
| Electronegativity (χ = −μ) | 6.94794 | 159.803 | 6.41013 | 147.433 |
| Electrophilicity index (ω = μ2/2η | 14.3758 | 330.644 | 18.8245 | 432.963 |
| Nucleophilicity index (N = 1/ω) | 0.06956 | 1.59991 | 0.05312 | 1.22181 |
| Electron accepting power (ω+ = A2/2(I − A)) | 4.1337 | 95.0751 | 6.48002 | 149.04 |
| Electron donating power (ω− = I2/2(I − A)) | 11.0816 | 254.8772 | 12.8901 | 296.473 |
| NBOs | Donor NBO (i) | NBOs | Acceptor NBO (j) | E(2) kcal/mol | E(j)-E(i) a.u. | F(i,j) a.u. |
|---|---|---|---|---|---|---|
| 4a | ||||||
| 3 | BD (2) C1–C6 | 377 | BD*(2) C2–C3 | 18.45 | 0.31 | 0.069 |
| 3 | BD (2) C1–C6 | 382 | BD*(2) C4–C5 | 17.84 | 0.31 | 0.067 |
| 6 | BD (2) C2–C3 | 374 | BD*(2) C1–C6 | 20.45 | 0.28 | 0.069 |
| 6 | BD (2) C2–C3 | 382 | BD*(2) C4–C5 | 20.36 | 0.30 | 0.07 |
| 11 | BD (2) C4–C5 | 374 | BD*(2) C1–C6 | 19.94 | 0.27 | 0.067 |
| 11 | BD (2) C4–C5 | 377 | BD*(2) C2–C3 | 19.06 | 0.28 | 0.067 |
| 20 | BD (2) C10–C11 | 397 | BD*(2) C12–O30 | 25.42 | 0.31 | 0.079 |
| 78 | LP (2) O22 | 377 | BD*(2) C2–C3 | 27.61 | 0.36 | 0.095 |
| 80 | LP (1) N29 | 401 | BD*(2) C13–O31 | 48.11 | 0.32 | 0.112 |
| 81 | LP (1) O30 | 174 | RY*(1) C12 | 15.00 | 1.30 | 0.125 |
| 82 | LP (2) O30 | 392 | BD*(1) C10–C12 | 17.29 | 0.75 | 0.103 |
| 82 | LP (2) O30 | 395 | BD*(1) C12–C16 | 19.62 | 0.65 | 0.102 |
| 83 | LP (1) O31 | 183 | RY*(1) C13 | 16.11 | 1.21 | 0.125 |
| 84 | LP (2) O31 | 398 | BD*(1) C13–N28 | 26.39 | 0.65 | 0.119 |
| 84 | LP (2) O31 | 399 | BD*(1) C13–N29 | 23.39 | 0.71 | 0.117 |
| 374 | BD*(2) C1–C6 | 377 | BD*(2) C2–C3 | 301.87 | 0.01 | 0.085 |
| 374 | BD*(2) C1–C6 | 382 | BD*(2) C4–C5 | 171.54 | 0.02 | 0.082 |
| 397 | BD*(2) C12–O30 | 391 | BD*(2) C10–C11 | 153.69 | 0.01 | 0.075 |
| 4b | ||||||
| 3 | BD (2) C1–C6 | 381 | BD*(2) C2–C3 | 18.65 | 0.30 | 0.069 |
| 3 | BD (2) C1–C6 | 386 | BD*(2) C4–C5 | 17.90 | 0.31 | 0.067 |
| 6 | BD (2) C2–C3 | 378 | BD*(2) C1–C6 | 20.08 | 0.28 | 0.068 |
| 6 | BD (2) C2–C3 | 386 | BD*(2) C4–C5 | 20.38 | 0.30 | 0.07 |
| 11 | BD (2) C4–C5 | 378 | BD*(2) C1–C6 | 19.89 | 0.27 | 0.067 |
| 11 | BD (2) C4–C5 | 381 | BD*(2) C2–C3 | 18.99 | 0.28 | 0.067 |
| 20 | BD (2) C10–C11 | 401 | BD*(2) C12–O30 | 24.62 | 0.31 | 0.078 |
| 80 | LP (2) O21 | 378 | BD*(2) C1–C6 | 26.26 | 0.36 | 0.093 |
| 82 | LP (2) O22 | 381 | BD*(2) C2–C3 | 28.02 | 0.36 | 0.096 |
| 83 | LP (1) N28 | 395 | BD*(2) C10–C11 | 42.27 | 0.31 | 0.106 |
| 83 | LP (1) N28 | 405 | BD*(2) C13–S43 | 22.72 | 0.35 | 0.08 |
| 84 | LP (1) N29 | 404 | BD*(1) C13–S43 | 14.16 | 0.43 | 0.071 |
| 84 | LP (1) N29 | 405 | BD*(2) C13–S43 | 24.23 | 0.34 | 0.081 |
| 85 | LP (1) O30 | 178 | RY*(1) C12 | 14.98 | 1.30 | 0.125 |
| 86 | LP (2) O30 | 396 | BD*(1) C10–C12 | 17.49 | 0.75 | 0.104 |
| 86 | LP (2) O30 | 399 | BD*(1) C12–C16 | 19.59 | 0.66 | 0.103 |
| 378 | BD*(2) C1–C6 | 381 | BD*(2) C2–C3 | 351.67 | 0.01 | 0.085 |
| 378 | BD*(2) C1–C6 | 386 | BD*(2) C4–C5 | 177.80 | 0.02 | 0.082 |
| 405 | BD*(2) C13–S43 | 404 | BD*(1) C13–S43 | 110.50 | 0.09 | 0.166 |
| Mode | Affinity kcal/mol | Distribution from Best Mode | Mode | Affinity kcal/mol | Distribution from Best Mode | ||
|---|---|---|---|---|---|---|---|
| rmsdl.b | rmsdu.b. | rmsdl.b | rmsdu.b. | ||||
| 1DB4 vs. 4a | 1DB4 vs. 4b | ||||||
| 1 | −7.80 | 0 | 0 | 1 | −7.30 | 0 | 0 |
| 2 | −6.80 | 1.806 | 2.167 | 2 | −7.10 | 3.214 | 4.218 |
| 3 | −6.70 | 9.86 | 11.519 | 3 | −6.90 | 2.703 | 3.421 |
| 4 | −6.60 | 2.494 | 5.878 | 4 | −6.60 | 3.382 | 4.898 |
| 5 | −6.50 | 1.87 | 2.322 | 5 | −6.50 | 3.116 | 6.516 |
| 6 | −6.20 | 2.225 | 5.731 | 6 | −6.30 | 2.657 | 3.351 |
| 7 | −6.00 | 9.6 | 11.567 | 7 | −6.30 | 2.416 | 5.575 |
| 8 | −5.90 | 3.292 | 4.99 | 8 | −6.30 | 4.985 | 6.505 |
| 9 | −5.90 | 2.104 | 2.917 | 9 | −6.20 | 3.059 | 5.453 |
| Mode | Affinity kcal/mol | Distribution from Best Mode | Mode | Affinity kcal/mol | Distribution from Best Mode | ||
| rmsdl.b | rmsdu.b. | rmsdl.b | rmsdu.b. | ||||
| 2PWB vs. 4a | 2PWB vs. 4b | ||||||
| 1 | −7.00 | 0 | 0 | 1 | −6.60 | 0 | 0 |
| 2 | −6.60 | 2.572 | 4.249 | 2 | −6.50 | 3.067 | 4.547 |
| 3 | −6.40 | 16.378 | 18.226 | 3 | −6.30 | 4.012 | 7.627 |
| 4 | −6.40 | 2.717 | 5.942 | 4 | −6.30 | 2.764 | 6.511 |
| 5 | −6.30 | 2.172 | 5.757 | 5 | −6.20 | 17.163 | 19.589 |
| 6 | −6.10 | 2.813 | 5.743 | 6 | −6.00 | 23.322 | 25.106 |
| 7 | −6.00 | 20.991 | 22.976 | 7 | −5.90 | 2.767 | 4.068 |
| 8 | −5.90 | 23.184 | 24.946 | 8 | −5.80 | 27.71 | 29.173 |
| 9 | −5.80 | 27.267 | 29.093 | 9 | −5.70 | 29.691 | 30.831 |
| Compounds | Proteins | Protein Residues |
|---|---|---|
| 4a | 1DB4 | CYS A:28, VAL A:30, SER A:113, ASN A:114 and LYS A:115 |
| 4b | LEU A:2, PHE A:5, HIS A6, HIS A:27, GLY A:29, VAL A:30, HIS A:47 and ASP A:48 | |
| 4a | 2PWB | ASN A:5, ALA A:6, TRP A:8, ARG A:185 and LEU A:209 |
| 4b | LYS A:125, GLY A:126, VAL A:127, GLY A:152 AND ALA A:245 |
| Parameters | Compounds | |
|---|---|---|
| 4a | 4b | |
| Molecular weight | 316.35 | 332.42 |
| No. H-bond acceptors | 4 | 3 |
| No. H-bond donors | 3 | 3 |
| LogPO/W(iLOGP) | 2.36 | 2.76 |
| No. rotatable bonds | 2 | 2 |
| TPSA | 87.66 | 102.68 |
| Log KP (skin permeation) | −7.28 | −6.95 |
| Lipinski’s rule violation | No | No |
| Bioavailability score | 0.55 | 0.55 |
| GI absorption | High | High |
| BBB permeation | No | No |
| Hepatotoxicity | − | − |
| Immunotoxicity | − | − |
| Mutagenicity | − | − |
| Cytotoxicity | − | − |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakht, M.A.; Pooventhiran, T.; Thomas, R.; Kamal, M.; Din, I.U.; Rehman, N.U.; Ali, I.; Ajmal, N.; Ahsan, M.J. Synthesis and Biological Evaluation of Octahydroquinazolinones as Phospholipase A2, and Protease Inhibitors: Experimental and Theoretical Exploration. Molecules 2023, 28, 1944. https://doi.org/10.3390/molecules28041944
Bakht MA, Pooventhiran T, Thomas R, Kamal M, Din IU, Rehman NU, Ali I, Ajmal N, Ahsan MJ. Synthesis and Biological Evaluation of Octahydroquinazolinones as Phospholipase A2, and Protease Inhibitors: Experimental and Theoretical Exploration. Molecules. 2023; 28(4):1944. https://doi.org/10.3390/molecules28041944
Chicago/Turabian StyleBakht, Md. Afroz, Thangaiyan Pooventhiran, Renjith Thomas, Mehnaz Kamal, Israf Ud Din, Najeeb Ur Rehman, Imtiaz Ali, Noushin Ajmal, and Mohamed Jawed Ahsan. 2023. "Synthesis and Biological Evaluation of Octahydroquinazolinones as Phospholipase A2, and Protease Inhibitors: Experimental and Theoretical Exploration" Molecules 28, no. 4: 1944. https://doi.org/10.3390/molecules28041944
APA StyleBakht, M. A., Pooventhiran, T., Thomas, R., Kamal, M., Din, I. U., Rehman, N. U., Ali, I., Ajmal, N., & Ahsan, M. J. (2023). Synthesis and Biological Evaluation of Octahydroquinazolinones as Phospholipase A2, and Protease Inhibitors: Experimental and Theoretical Exploration. Molecules, 28(4), 1944. https://doi.org/10.3390/molecules28041944