
Influence of Mutations of Conserved Arginines on Neuropeptide Binding in the DPP III Active Site


Abstract
:1. Introduction
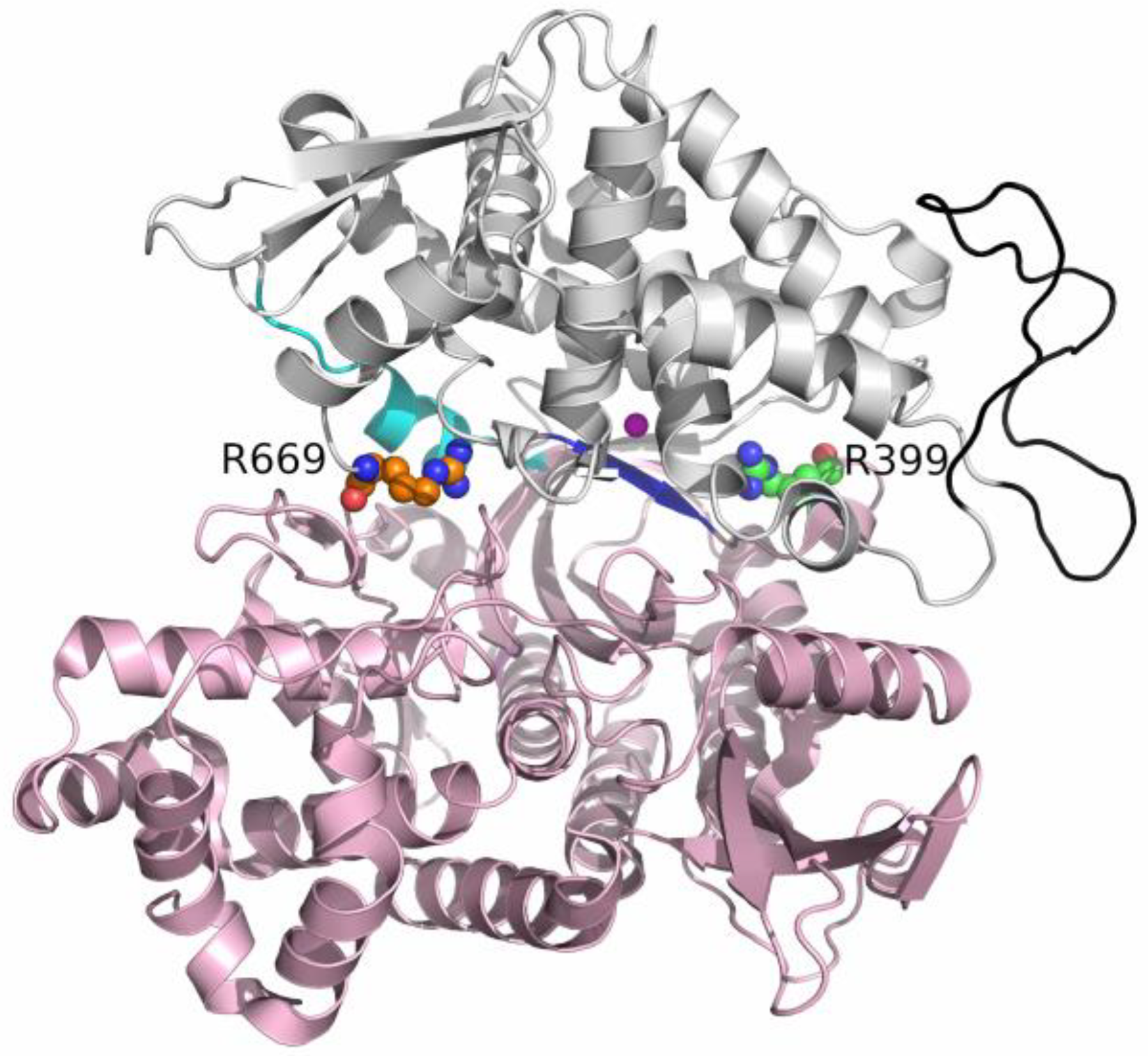
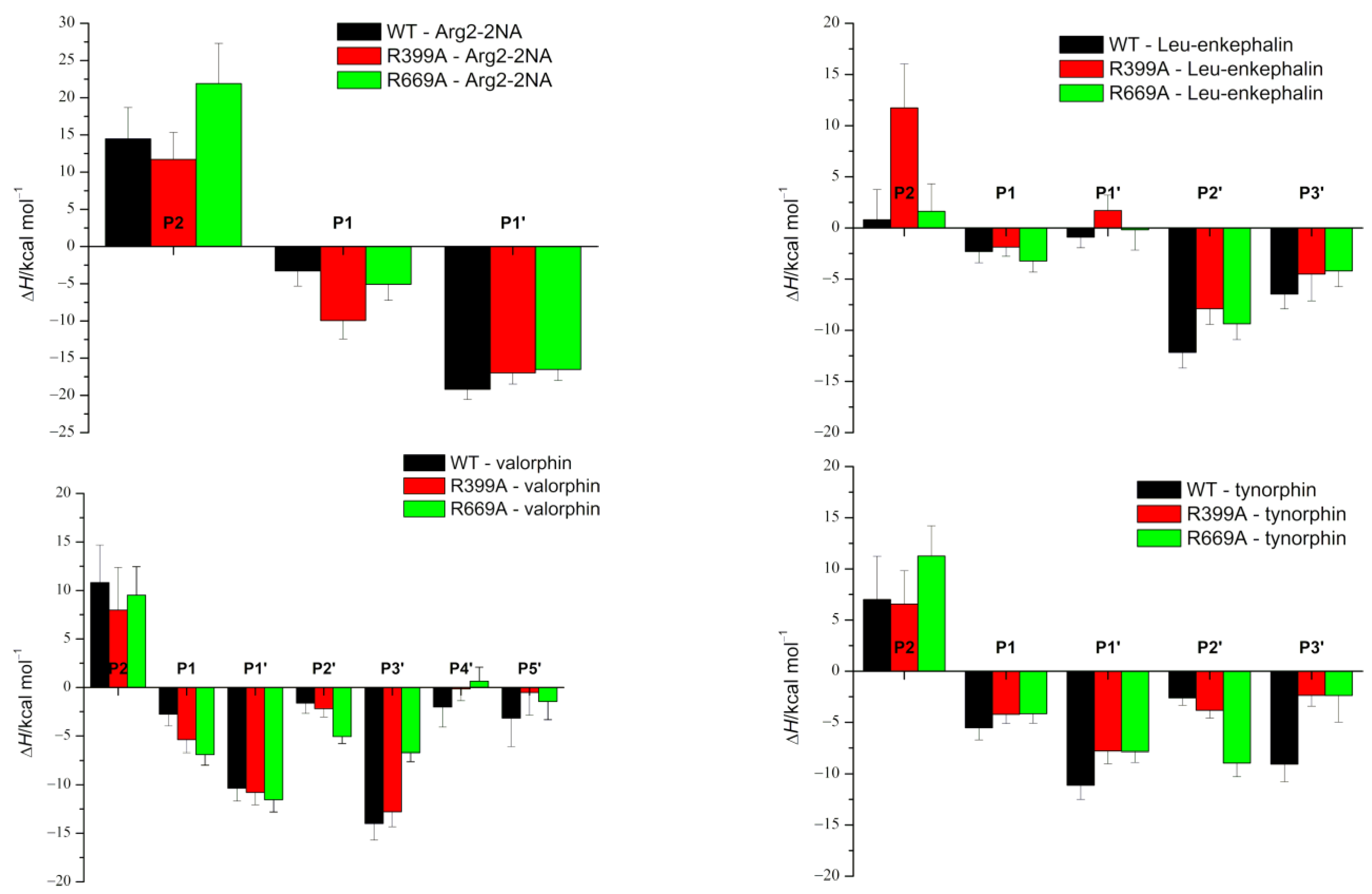
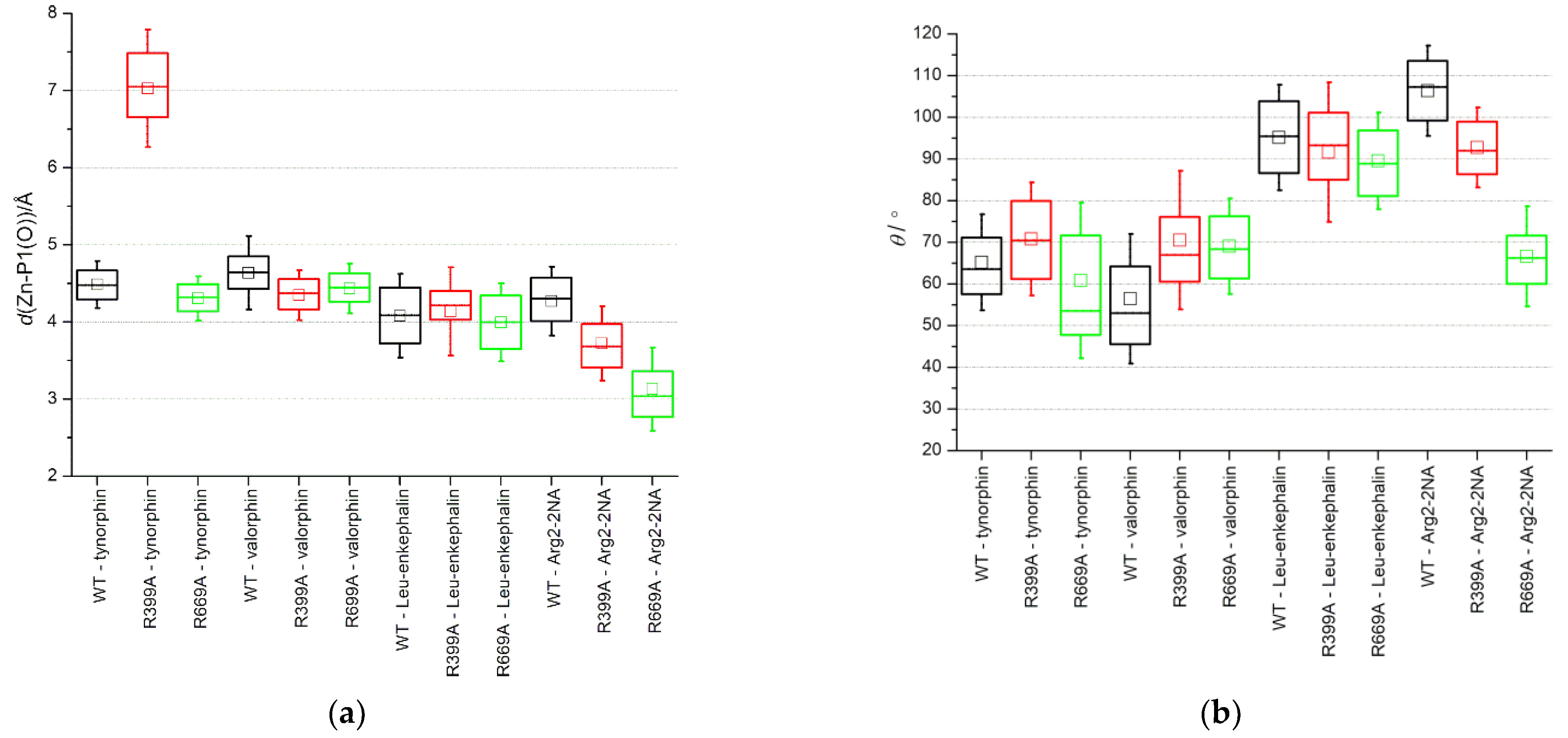
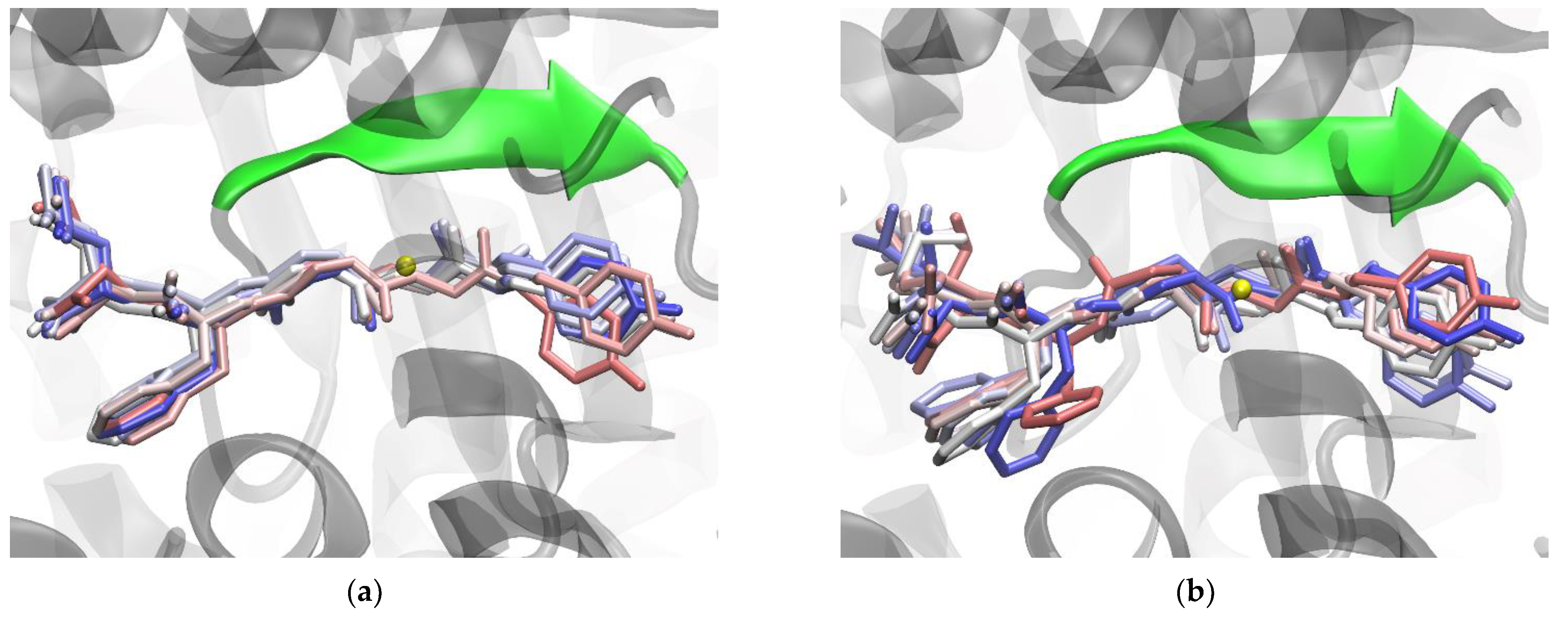
2. Results and Discussion
2.1. Impact of R669A Mutation on Ligand Binding
2.2. Impact of R399A Mutation on Ligand Binding
3. Materials and Methods
3.1. Experimental Methods
3.1.1. Protein Expression and Purification
3.1.2. Enzyme Kinetics and Inhibition
3.2. Computational Methods
3.2.1. System Preparation
3.2.2. MD Simulations
3.2.3. MM/PBSA and Per-Residue MM/GBSA Calculations
3.2.4. Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chen, J.M.; Barrett, A.J. Dipeptidyle-Peptidase III. In Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Elsevier: Amsterdam, The Netherlands; Academic Press: London, UK, 2004; pp. 809–812. [Google Scholar]
- Cruz-Diaz, N.; Wilson, B.A.; Pirro, N.T.; Brosnihan, K.B.; Marshall, A.C.; Chappell, M.C. Identification of Dipeptidyl Peptidase 3 as the Angiotensin-(1–7) Degrading Peptidase in Human HK-2 Renal Epithelial Cells. Peptides 2016, 83, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Shimizu, A.; Kurita, S.; Zankov, D.P.; Takeuchi, K.; Yasuda-Yamahara, M.; Kume, S.; Ishida, T.; Ogita, H. Novel Therapeutic Role for Dipeptidyl Peptidase III in the Treatment of Hypertension. Hypertension 2016, 68, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Tomić, A.; Kovačević, B.; Tomić, S. Concerted Nitrogen Inversion and Hydrogen Bonding to Glu451 Are Responsible for Protein-Controlled Suppression of the Reverse Reaction in Human DPP III. Phys. Chem. Chem. Phys. 2016, 18, 27245–27256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, T.; Li, Y.-H.; Yamane, T.; Ogikubo, O.; Fukuoka, M.; Arai, R.; Takahashi, S.; Ohtsuka, T.; Ohkubo, I.; Matsui, N. Inhibition of Recombinant Dipeptidyl Peptidase III by Synthetic Hemorphin-like Peptides. Peptides 2003, 24, 773–778. [Google Scholar] [CrossRef]
- Barsun, M.; Jajcanin, N.; Vukelić, B.; Spoljarić, J.; Abramić, M. Human Dipeptidyl Peptidase III Acts as a Post-Proline-Cleaving Enzyme on Endomorphins. Biol. Chem. 2007, 388, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic Analysis of Ubiquitin Ligase KEAP1 Reveals Associated Proteins That Inhibit NRF2 Ubiquitination. Cancer Res. 2013, 73, 2199–2210. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress–Induced DPP3–KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The Emerging Role of the Nrf2–Keap1 Signaling Pathway in Cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menale, C.; Robinson, L.J.; Palagano, E.; Rigoni, R.; Erreni, M.; Almarza, A.J.; Strina, D.; Mantero, S.; Lizier, M.; Forlino, A.; et al. Absence of Dipeptidyl Peptidase 3 Increases Oxidative Stress and Causes Bone Loss. J. Bone Miner. Res. 2019, 34, 2133–2148. [Google Scholar] [CrossRef] [PubMed]
- Matić, S.; Tomašić Paić, A.; Sobočanec, S.; Pinterić, M.; Pipalović, G.; Martinčić, M.; Matovina, M.; Tomić, S. Interdisciplinary Study of the Effects of Dipeptidyl-Peptidase III Cancer Mutations on the KEAP1-NRF2 Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 1994. [Google Scholar] [CrossRef]
- Bezerra, G.A.; Dobrovetsky, E.; Viertlmayr, R.; Dong, A.; Binter, A.; Abramic, M.; Macheroux, P.; Dhe-Paganon, S.; Gruber, K. Entropy-Driven Binding of Opioid Peptides Induces a Large Domain Motion in Human Dipeptidyl Peptidase III. Proc. Natl. Acad. Sci. USA. 2012, 109, 6525–6530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Reithofer, V.; Reisinger, M.; Wallner, S.; Pavkov-Keller, T.; Macheroux, P.; Gruber, K. Substrate Complexes of Human Dipeptidyl Peptidase III Reveal the Mechanism of Enzyme Inhibition. Sci. Rep. 2016, 6, 23787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomić, A.; Tomić, S. Demystifying DPP III Catalyzed Peptide Hydrolysis—Computational Study of the Complete Catalytic Cycle of Human DPP III Catalyzed Tynorphin Hydrolysis. Int. J. Mol. Sci. 2022, 23, 1858. [Google Scholar] [CrossRef] [PubMed]
- Karačić, Z.; Šupljika, F.; Tomić, A.; Brkljačić, L.; Paić, A.T.; Ćehić, M.; Tomić, S. Neuropeptides, Substrates and Inhibitors of Human Dipeptidyl Peptidase III, Experimental and Computational Study—A New Substrate Identified. Int. J. Biol. Macromol. 2022, 220, 1390–1401. [Google Scholar] [CrossRef]
- Tomić, A.; González, M.; Tomić, S. The Large Scale Conformational Change of the Human DPP III-Substrate Prefers the “Closed” Form. J. Chem. Inf. Model. 2012, 52, 1583–1594. [Google Scholar] [CrossRef]
- Chen, S.-B.; Zhang, H.; Chen, S.; Ye, X.-F.; Li, Z.-K.; Liu, W.-D.; Cui, Z.-L.; Huang, Y. Structural and Functional Characterization of a New Bacterial Dipeptidyl Peptidase III Involved in Fruiting Body Formation in Myxobacteria. Int. J. Mol. Sci. 2022, 24, 631. [Google Scholar] [CrossRef]
- Salopek-Sondi, B.; Vukelić, B.; Spoljarić, J.; Simaga, S.; Vujaklija, D.; Makarević, J.; Jajcanin, N.; Abramić, M. Functional Tyrosine Residue in the Active Center of Human Dipeptidyl Peptidase III. Biol. Chem. 2008, 389, 163–167. [Google Scholar] [CrossRef]
- Tomić, A.; Abramić, M.; Spoljarić, J.; Agić, D.; Smith, D.M.; Tomić, S. Human Dipeptidyl Peptidase III: Insights into Ligand Binding from a Combined Experimental and Computational Approach. J. Mol. Recognit. 2011, 24, 804–814. [Google Scholar] [CrossRef]
- B:urgi, H.B.; Dunitz, J.D.; Lehn, J.M.; Wipff, G. Stereochemistry of Reaction Paths at Carbonyl Centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar] [CrossRef]
- Abramić, M.; Špoljarić, J.; Šimaga, Š. Prokaryotic Homologs Help to Define Consensus Sequences in Peptidase Family M49. Period. Biol. 2004, 106, 161–168. [Google Scholar]
- Špoljarić, J.; Salopek-Sondi, B.; Makarević, J.; Vukelić, B.; Agić, D.; Simaga, S.; Jajcanin-Jozić, N.; Abramić, M. Absolutely Conserved Tryptophan in M49 Family of Peptidases Contributes to Catalysis and Binding of Competitive Inhibitors. Bioorg. Chem. 2009, 37, 70–76. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Tomić, A.; Horvat, G.; Ramek, M.; Agić, D.; Brkić, H.; Tomić, S. New Zinc Ion Parameters Suitable for Classical MD Simulations of Zinc Metallopeptidases. J. Chem. Inf. Model. 2019, 59, 3437–3453. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Loncharich, R.J.; Brooks, B.R.; Pastor, R.W. Langevin Dynamics of Peptides: The Frictional Dependence of Isomerization Rates OfN-Acetylalanyl-N?-Methylamide. Biopolymers 1992, 32, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Swanson, J.M.J.; Henchman, R.H.; McCammon, J.A. Revisiting Free Energy Calculations: A Theoretical Connection to MM/PBSA and Direct Calculation of the Association Free Energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild-Type | R669A | R669M | |
|---|---|---|---|
| Ki (tynorphin)/nM | 11.2 ± 0.8 | 328 ± 25 | 102 ± 16 |
| Ki (valorphin)/nM | 36.5 ± 2.9 | 1207 ± 224 | 406 ± 33 |
| Ki (Leu-enkephalin)/μM | 10.4 ± 1.4 | 52 ± 5.3 | 58 ± 7.0 |
| (ΔHMM/PBSA ± SD)/kcal mol−1 | ||||
|---|---|---|---|---|
| DPP III | Arg2-2NA | Leu-Enkephalin [YGGFL] | Valorphin [VVYPWTQ] | Tynorphin [VVYPW] |
| WT | −32.13 ± 7.36 | −20.04 ± 4.52 | −9.15 ± 7.01 | −21.58 ± 4.62 |
| R399A | −35.09 ± 7.96 | 0.00 ± 7.91 | −15.33 ± 7.11 | −18.82 ± 4.59 |
| R669A | −22.74 ± 6.25 | −13.67 ± 4.98 | −16.30 ± 6.05 | −7.32 ± 4.39 |
| Wild-Type | R669A | R669M | |
|---|---|---|---|
| KM/μM | 7.8 ± 0.6 | 3.4 ± 0.3 | 2.8 ± 0.2 |
| kcat/s−1 | 3.74 ± 0.07 | 2.24 ± 0.05 | 2.26 ± 0.03 |
| kcat/KM (M∙s)−1 | 4.8 × 104 | 6.6 × 104 | 8.07 × 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomić, A.; Karačić, Z.; Tomić, S. Influence of Mutations of Conserved Arginines on Neuropeptide Binding in the DPP III Active Site. Molecules 2023, 28, 1976. https://doi.org/10.3390/molecules28041976
Tomić A, Karačić Z, Tomić S. Influence of Mutations of Conserved Arginines on Neuropeptide Binding in the DPP III Active Site. Molecules. 2023; 28(4):1976. https://doi.org/10.3390/molecules28041976
Chicago/Turabian StyleTomić, Antonija, Zrinka Karačić, and Sanja Tomić. 2023. "Influence of Mutations of Conserved Arginines on Neuropeptide Binding in the DPP III Active Site" Molecules 28, no. 4: 1976. https://doi.org/10.3390/molecules28041976
APA StyleTomić, A., Karačić, Z., & Tomić, S. (2023). Influence of Mutations of Conserved Arginines on Neuropeptide Binding in the DPP III Active Site. Molecules, 28(4), 1976. https://doi.org/10.3390/molecules28041976