
Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
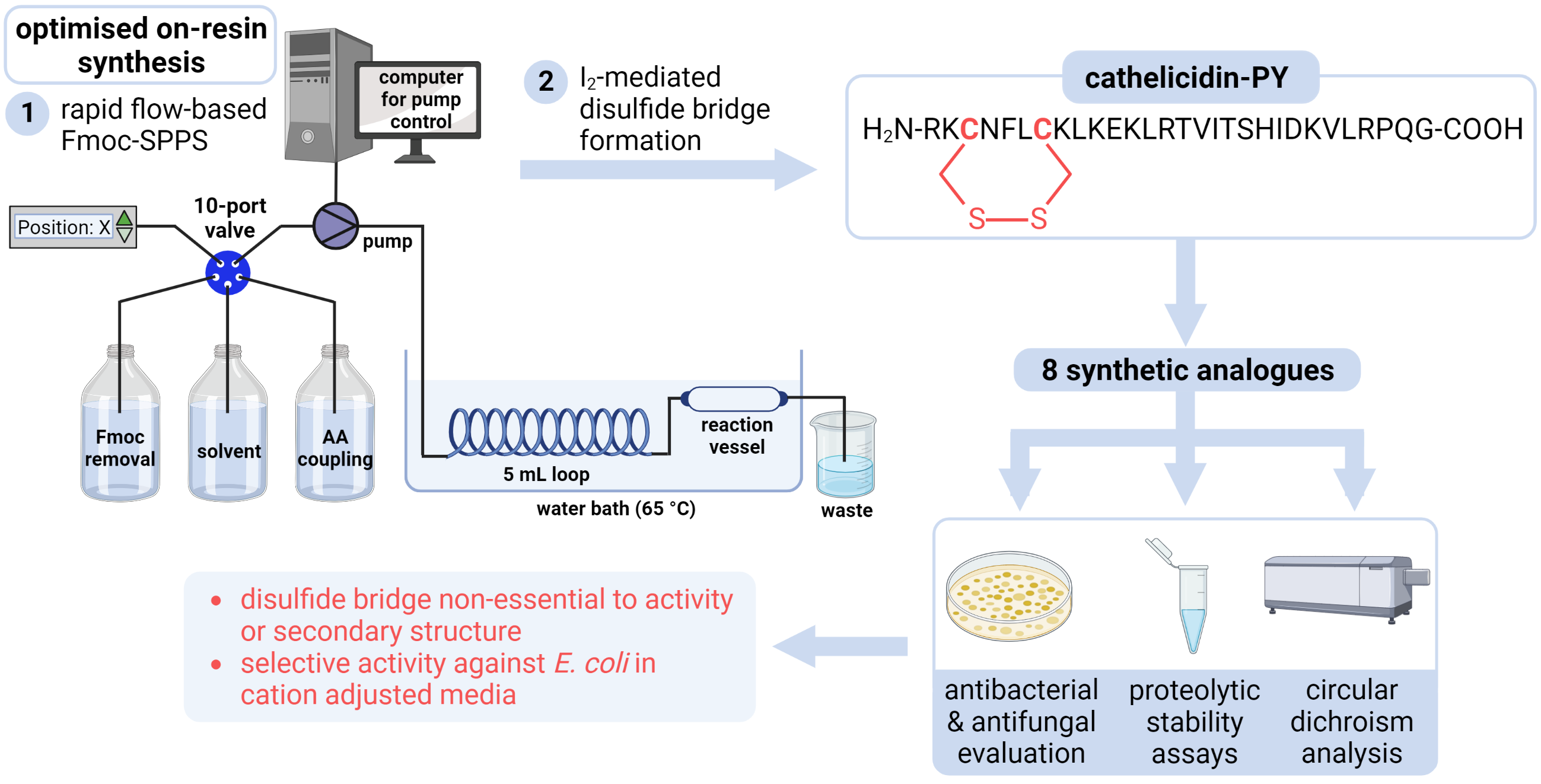
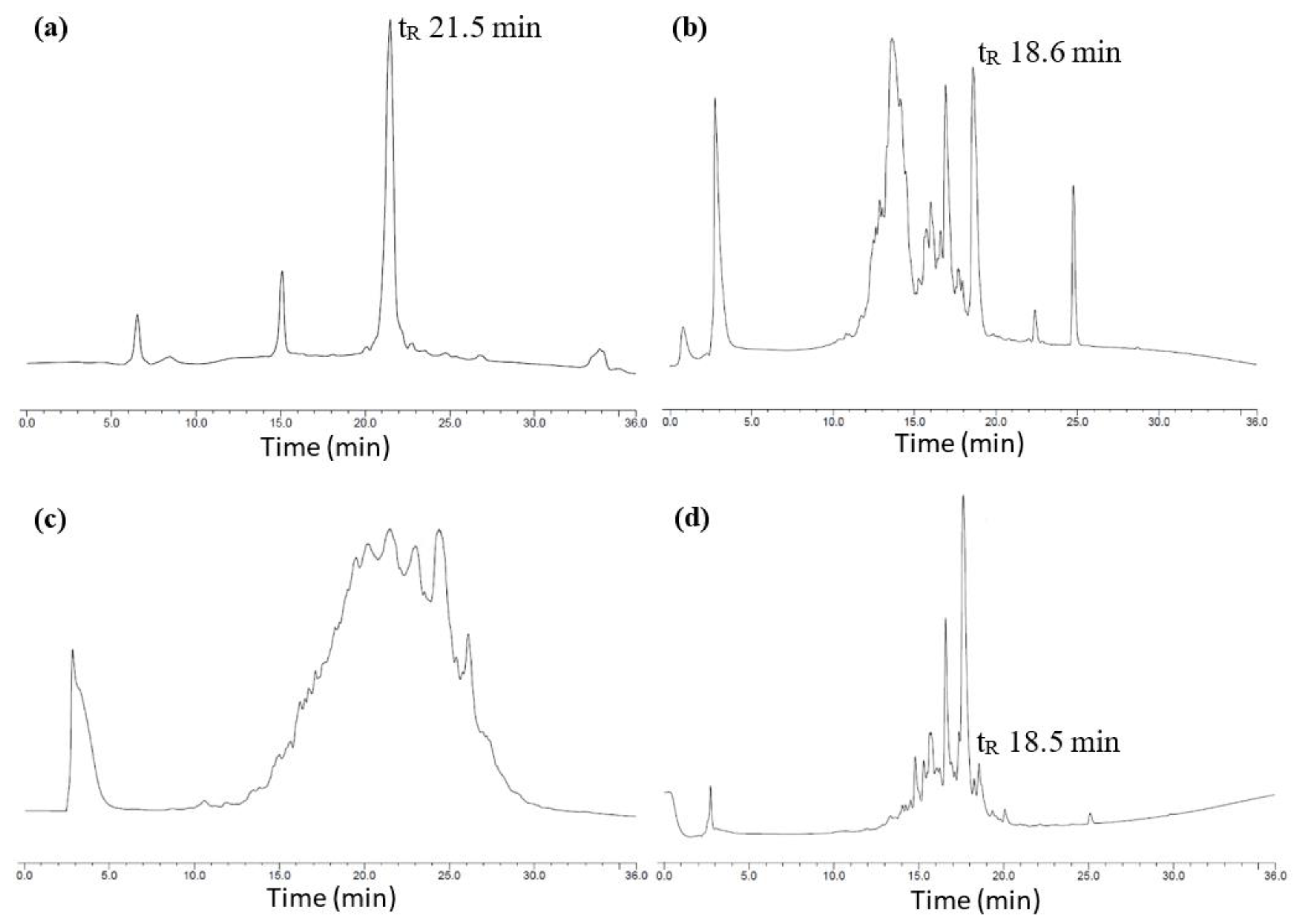
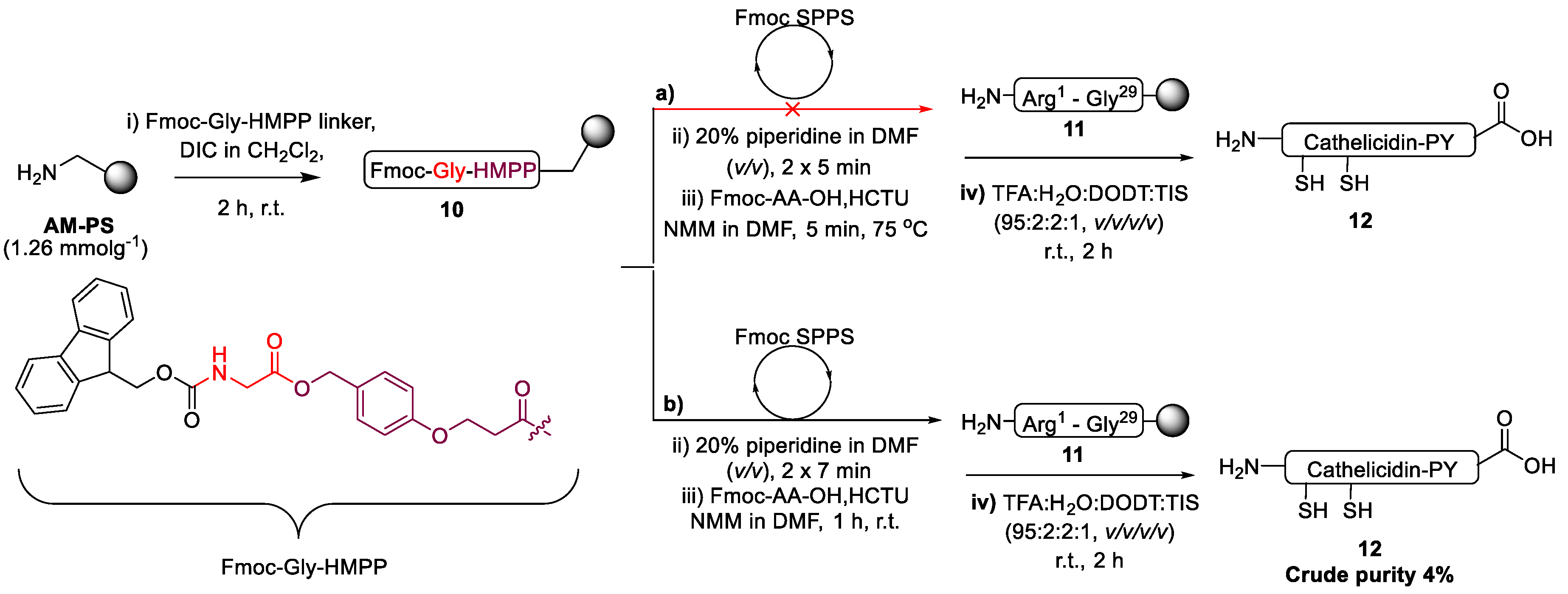
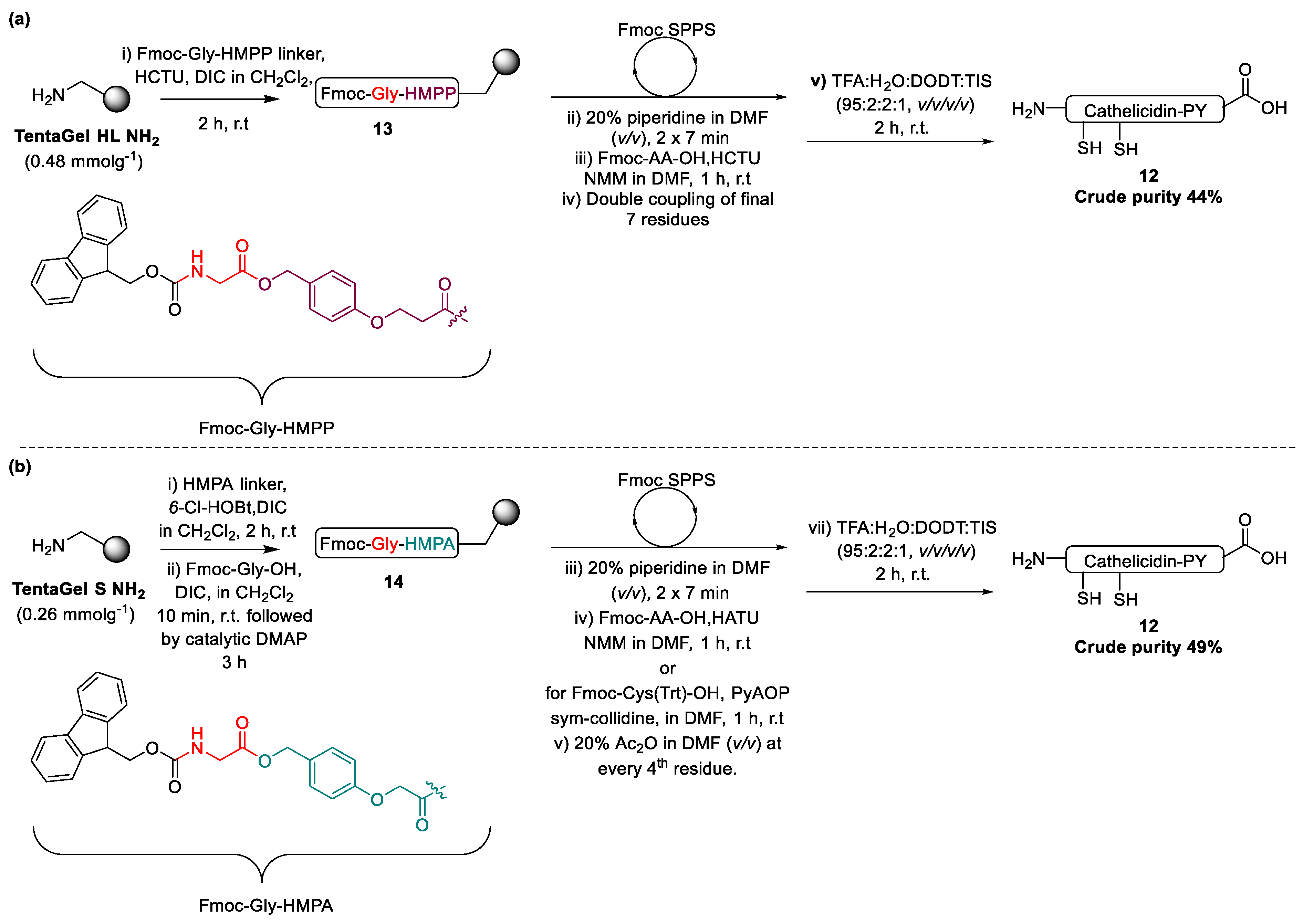
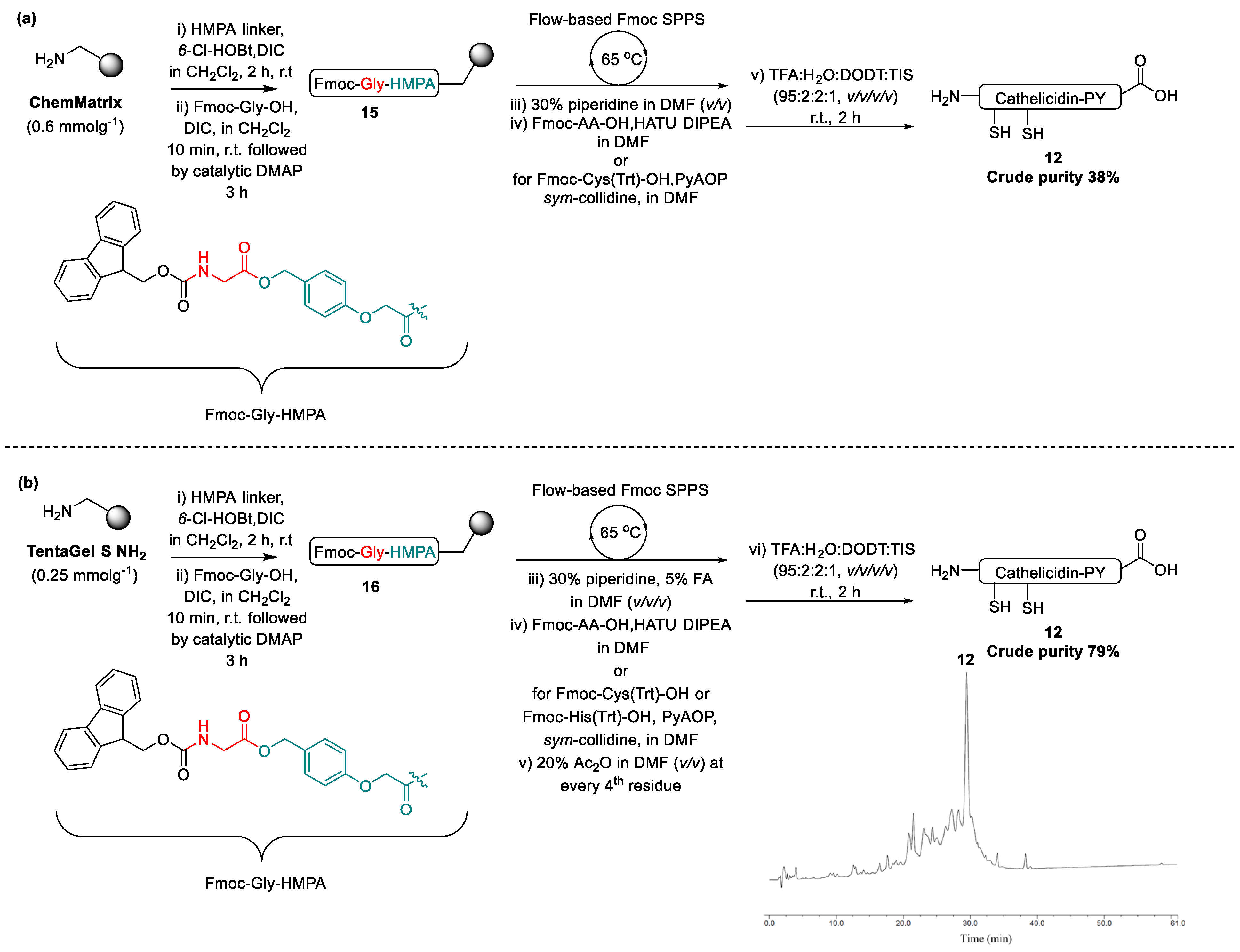
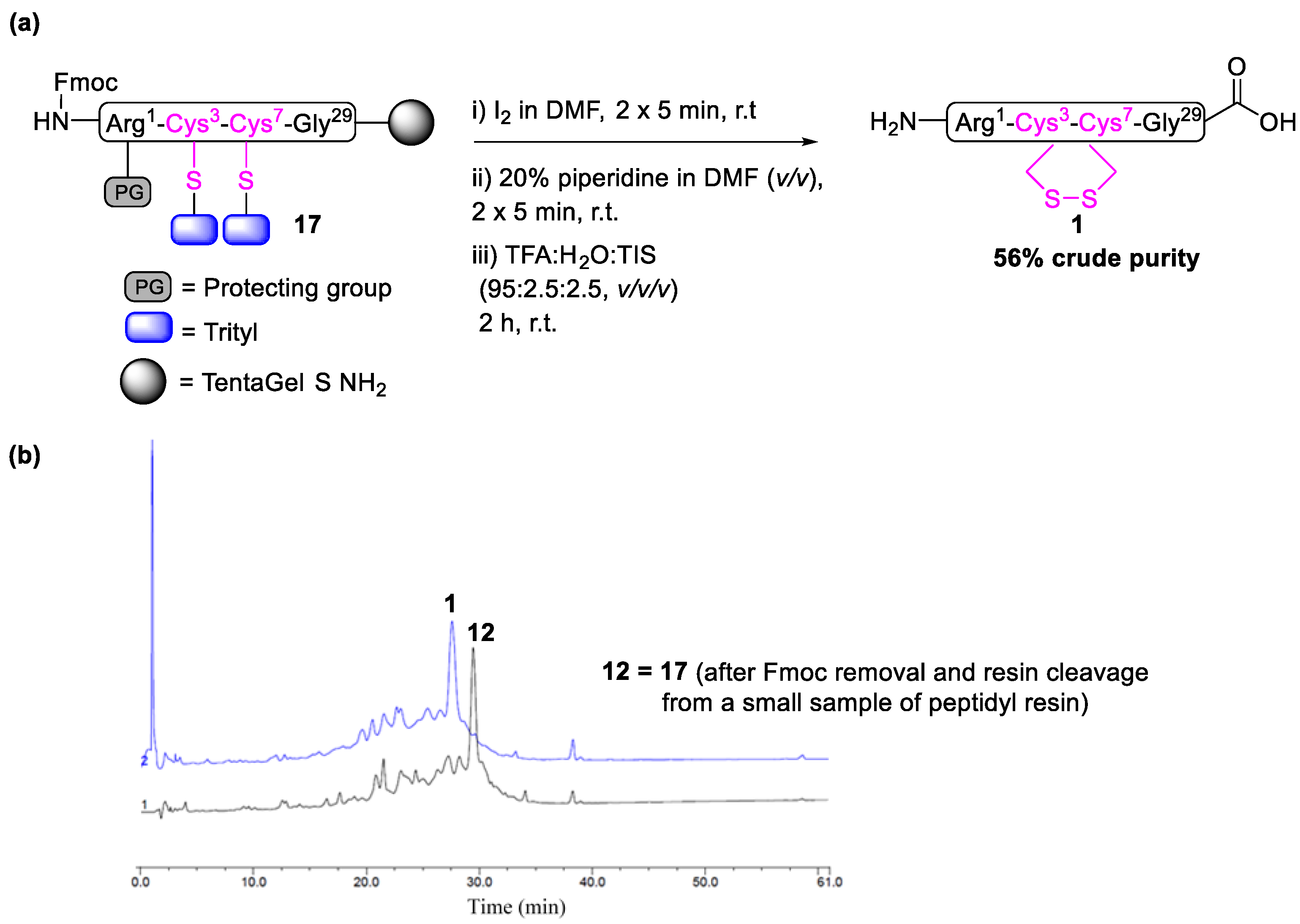
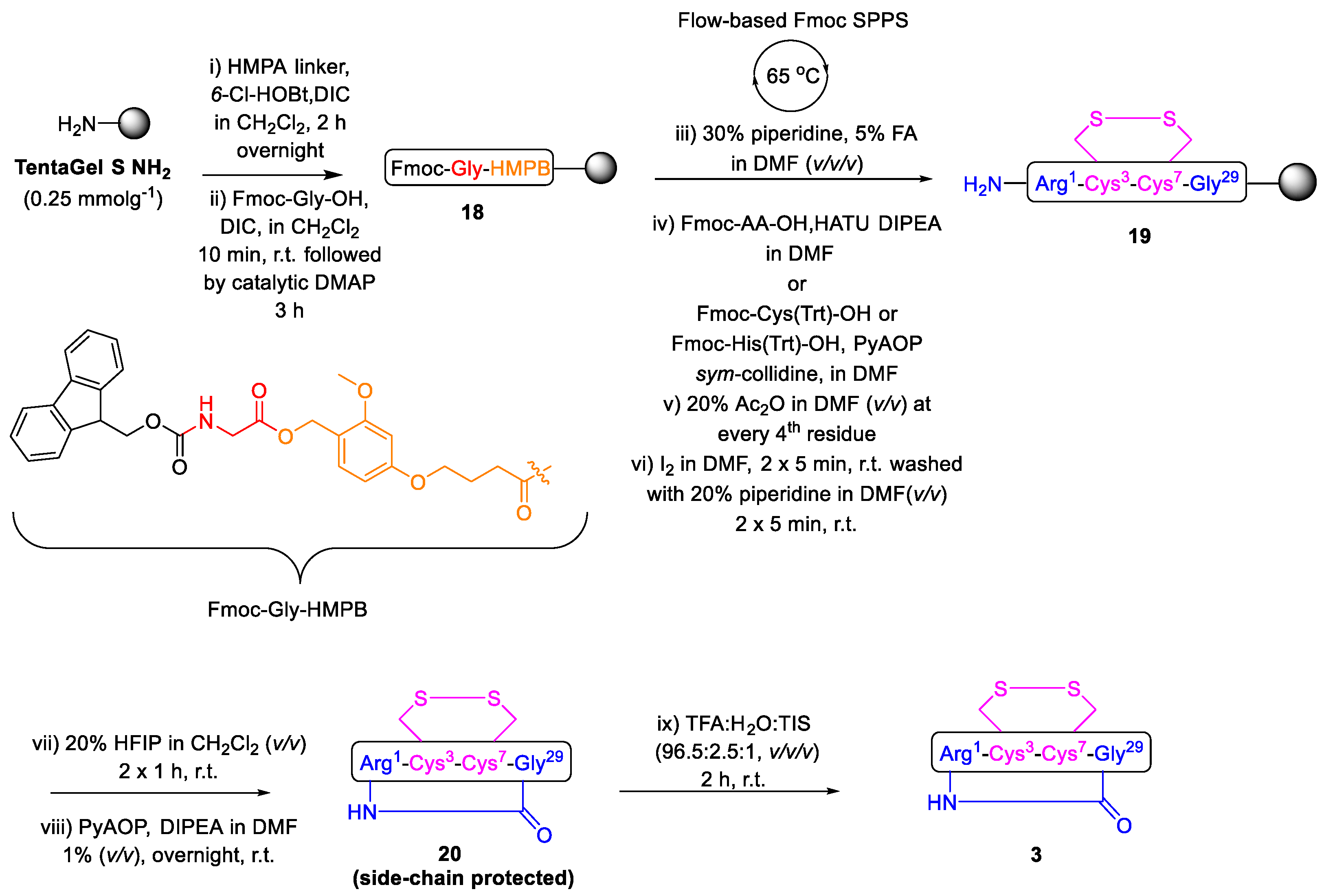
2.1. Total Synthesis of Cathelicidin-PY
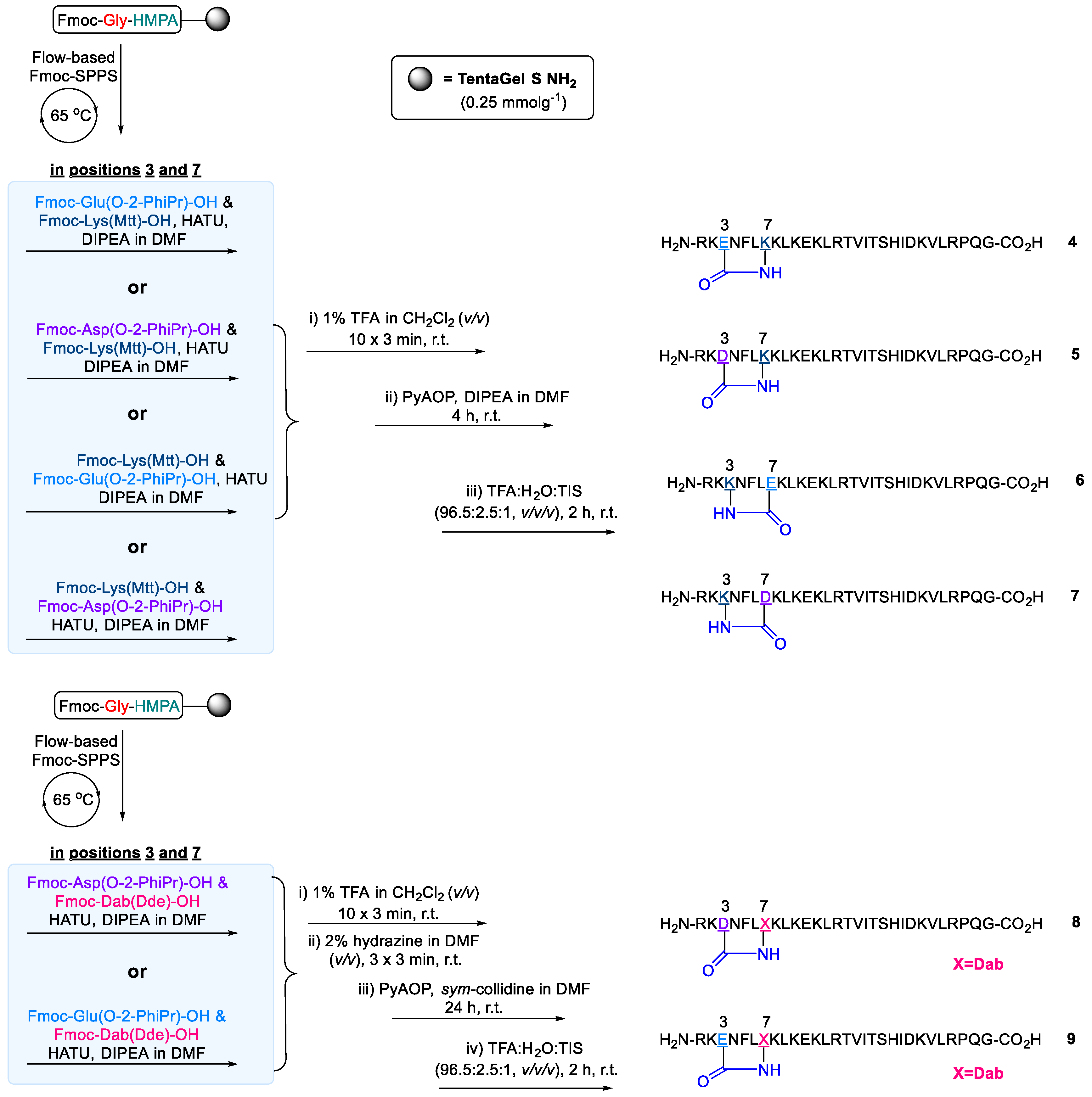
2.2. Design and Synthesis of Cathelicidin-PY Analogues
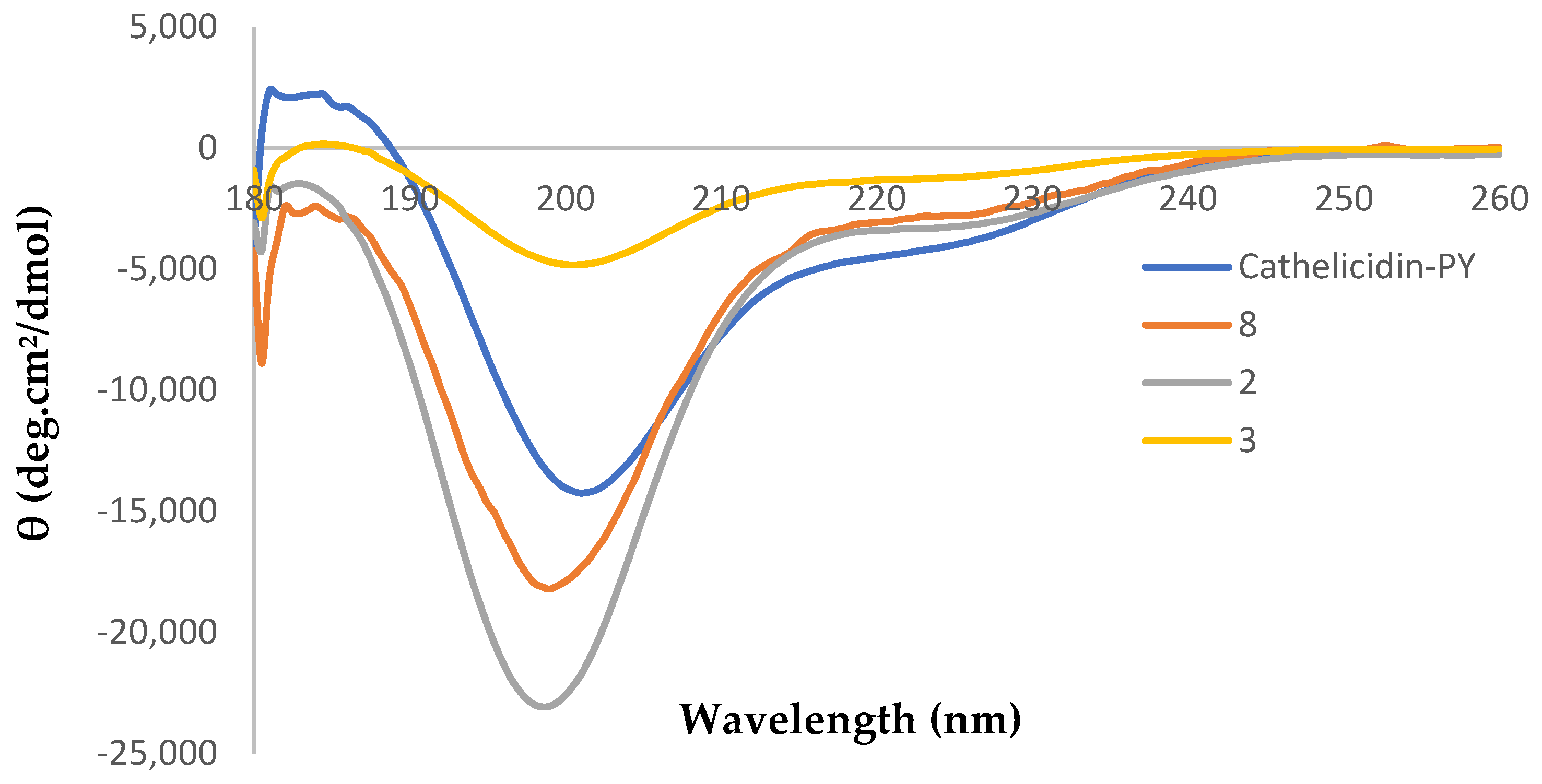
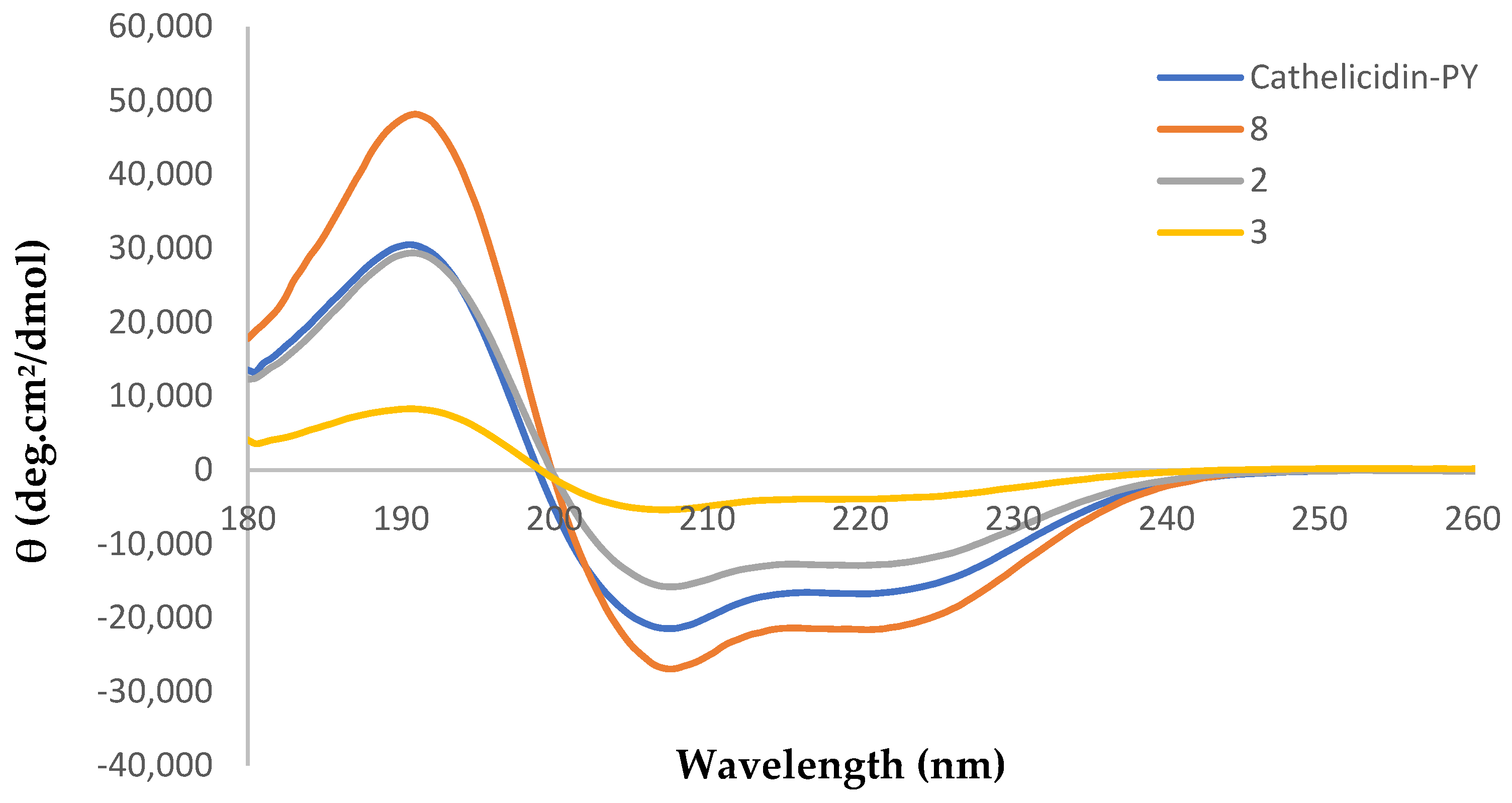
2.3. Circular Dichroism Analysis
2.4. Antimicrobial Activity
2.5. Peptide Stability
3. Conclusions
4. Materials and Methods
4.1. General Procedures and Reagents
4.2. Peptide Synthesis
4.3. Circular Dichroism (CD) Spectroscopy
4.4. Bacterial Minimum Inhibitory Concentration Assay
4.5. Fungal Minimum Inhibitory Concentration Assay
4.6. Human Serum Stability Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.F. The resistance tsunami, antimicrobial stewardship, and the golden age of microbiology. Vet. Microbiol. 2014, 171, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuji, T.; Gallo, R.L. Antimicrobial peptides: Old molecules with new ideas. J. Investig. Dermatol. 2012, 132 Pt 2, 887–895. [Google Scholar] [CrossRef] [Green Version]
- De Smet, K.; Contreras, R. Human antimicrobial peptides: Defensins, cathelicidins and histatins. Biotechnol. Lett. 2005, 27, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Bauer, F.; Schweimer, K.; Kluver, E.; Conejo-Garcia, J.R.; Forssmann, W.G.; Rosch, P.; Adermann, K.; Sticht, H. Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 2001, 10, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Peptide Antibiotics and Their Role in Innate Immunity. Annu. Rev. Immunol. 1995, 13, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, M.; Skerlavaj, B.; Romeo, D.; Gennaro, R. Proteolytic cleavage by neutrophil elastase converts inactive storage proforms to antibacterial bactenecins. Eur. J. Biochem. 1992, 209, 589–595. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Karimi, A.; Tohidpour, A.; Abbasi, N.; Fallah, F.; Akhavan, M.M. The antimicrobial potential of a new derivative of cathelicidin from Bungarus fasciatus against methicillin-resistant Staphylococcus aureus. J. Microbiol. 2018, 56, 128–137. [Google Scholar] [CrossRef]
- Scheb-Wetzel, M.; Rohde, M.; Bravo, A.; Goldmann, O. New insights into the antimicrobial effect of mast cells against enterococcus faecalis. Infect. Autoimmun. 2014, 82, 4496–4507. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Soundrarajan, N.; Lee, J.; Cho, H.S.; Choi, M.; Cha, S.Y.; Ahn, B.; Jeon, H.; Le, M.T.; Song, H.; et al. Genomewide analysis of the antimicrobial peptides in python bivittatus and characterization of cathelicidins with potent antimicrobial activity and low cytotoxicity. Antimicrob. Agents Chemother. 2017, 61, e00530–e00617. [Google Scholar] [CrossRef] [Green Version]
- Coorens, M.; Scheenstra, M.R.; Veldhuizen, E.J.A.; Haagsman, H.P. Interspecies cathelicidin comparison reveals divergence in antimicrobial activity, TLR modulation, chemokine induction and regulation of phagocytosis. Sci. Rep. 2017, 7, 40874. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, I.; Said, D.G.; Nubile, M.; Mastropasqua, L.; Dua, H.S. Cathelicidin-derived synthetic peptide improves therapeutic potential of vancomycin against Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bescucci, D.M.; Clarke, S.T.; Brown, C.L.J.; Boras, V.F.; Montina, T.; Uwiera, R.R.E.; Inglis, G.D. The absence of murine cathelicidin-related antimicrobial peptide impacts host responses enhancing Salmonella enterica serovar Typhimurium infection. Gut Pathog. 2020, 12, 53. [Google Scholar] [CrossRef]
- Benincasa, M.; Scocchi, M.; Pacor, S.; Tossi, A.; Nobili, D.; Basaglia, G.; Busetti, M.; Gennaro, R. Fungicidal activity of five cathelicidin peptides against clinically isolated yeasts. J. Antimicrob. Chemother. 2006, 58, 950–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wetering, S.; Sterk, P.J.; Rabe, K.F.; Hiemstra, P.S. Defensins: Key players or bystanders in infection, injury, and repair in the lung. J. Allergy Clin. Immunol. 1999, 104, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.; Walter-Jallow, L.; Broliden, K.; Agerberth, B.; Soderlund, J. The antimicrobial peptide LL-37 inhibits HIV-1 replication. Curr. HIV Res. 2007, 5, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Verma, A.; Kim, E.J.; White, M.R.; Hartshorn, K.L. LL-37 modulates human neutrophil responses to influenza A virus. J. Leukoc. Biol. 2014, 96, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harcourt, J.L.; McDonald, M.; Svoboda, P.; Pohl, J.; Tatti, K.; Haynes, L.M. Human cathelicidin, LL-37, inhibits respiratory syncytial virus infection in polarized airway epithelial cells. BMC Res. Notes 2016, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Sousa, F.H.; Casanova, V.; Findlay, F.; Stevens, C.; Svoboda, P.; Pohl, J.; Proudfoot, L.; Barlow, P.G. Cathelicidins display conserved direct antiviral activity towards rhinovirus. Peptides 2017, 95, 76–83. [Google Scholar] [CrossRef]
- Lee, C.J.; Buznyk, O.; Kuffova, L.; Rajendran, V.; Forrester, J.V.; Phopase, J.; Islam, M.M.; Skog, M.; Ahlqvist, J.; Griffith, M. Cathelicidin LL-37 and HSV-1 corneal infection: Peptide versus gene therapy. Transl. Vis. Sci. Technol. 2014, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, T.; Sugiyama, N.; Murayama, A.; Yamada, N.; Shiina, M.; Asabe, S.; Wakita, T.; Imawari, M.; Kato, T. Antimicrobial peptide LL-37 attenuates infection of hepatitis C virus. Hepatol. Res. 2016, 46, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Findlay, F.; Proudfoot, L.; Stevens, C.; Barlow, P.G. Cationic host defense peptides; novel antimicrobial therapeutics against category A pathogens and emerging infections. Pathog. Glob. Health 2016, 110, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieczorek, M.; Jenssen, H.; Kindrachuk, J.; Scott, W.R.; Elliott, M.; Hilpert, K.; Cheng, J.T.; Hancock, R.E.; Straus, S.K. Structural studies of a peptide with immune modulating and direct antimicrobial activity. Chem. Biol. 2010, 17, 970–980. [Google Scholar] [CrossRef] [Green Version]
- Schneider, V.A.F.; Coorens, M.; Ordonez, S.R.; Tjeerdsma-van Bokhoven, J.L.M.; Posthuma, G.; van Dijk, A.; Haagsman, H.P.; Veldhuizen, E.J.A. Imaging the antimicrobial mechanism(s) of cathelicidin-2. Sci. Rep. 2016, 6, 32948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Ouyang, J.; Wang, Y.; Zhang, M.; Fu, L.; Xiao, N.; Gao, L.; Zhang, P.; Zhou, J.; Wang, Y. A novel anionic cathelicidin lacking direct antimicrobial activity but with potent anti-inflammatory and wound healing activities from the salamander Tylototriton kweichowensis. Biochimie 2021, 191, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, R.; Skerlavaj, B.; Romeo, D. Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect. Immun. 1989, 57, 3142–3146. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Wang, Y.; Wu, C.; Li, X.; Fu, Z.; Yang, M.; Bian, W.; Wang, S.; Song, Y.; Tang, J.; et al. Cathelicidin-OA1, a novel antioxidant peptide identified from an amphibian, accelerates skin wound healing. Sci. Rep. 2018, 8, 15906. [Google Scholar] [CrossRef] [Green Version]
- Agier, J.; Efenberger, M.; Brzezinska-Blaszczyk, E. Cathelicidin impact on inflammatory cells. Cent. Eur. J. Immunol. 2015, 40, 225–235. [Google Scholar] [CrossRef]
- Hao, X.; Yang, H.; Wei, L.; Yang, S.; Zhu, W.; Ma, D.; Yu, H.; Lai, R. Amphibian cathelicidin fills the evolutionary gap of cathelicidin in vertebrate. Amino Acids 2012, 43, 677–685. [Google Scholar] [CrossRef]
- Yu, H.; Cai, S.; Gao, J.; Zhang, S.; Lu, Y.; Qiao, X.; Yang, H.; Wang, Y. Identification and polymorphism discovery of the cathelicidins, Lf-CATHs in ranid amphibian (Limnonectes fragilis). FEBS J. 2013, 280, 6022–6032. [Google Scholar] [CrossRef]
- Ling, G.; Gao, J.; Zhang, S.; Xie, Z.; Wei, L.; Yu, H.; Wang, Y. Cathelicidins from the bullfrog Rana catesbeiana provides novel template for peptide antibiotic design. PLoS ONE 2014, 9, e93216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, L.; Zhou, L.; Yang, J.; Zhuang, L.; Tang, J.; Liu, T.; Wu, J.; Yang, H. The first identified cathelicidin from tree frogs possesses anti-inflammatory and partial LPS neutralization activities. Amino Acids 2017, 49, 1571–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Yang, J.; He, X.; Mo, G.; Hong, J.; Yan, X.; Lin, D.; Lai, R. Structure and function of a potent lipopolysaccharide-binding antimicrobial and anti-inflammatory peptide. J. Med. Chem. 2013, 56, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Tang, J.; Liu, H.; Shen, C.; Rong, M.; Zhang, Z.; Lai, R. A potential wound-healing-promoting peptide from salamander skin. FASEB J. 2014, 28, 3919–3929. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.Y.; Zhan, B.; Gao, Y.Y. A novel cathelicidin from Bufo bufo gargarizans Cantor showed specific activity to its habitat bacteria. Gene 2015, 571, 172–177. [Google Scholar] [CrossRef]
- Gao, F.; Xu, W.F.; Tang, L.P.; Wang, M.M.; Wang, X.J.; Qian, Y.C. Characteristics of cathelicidin-Bg, a novel gene expressed in the ear-side gland of Bufo gargarizans. Genet. Mol. Res. 2016, 15, 15038481. [Google Scholar] [CrossRef]
- Rhodes, N.J.; Cruce, C.E.; O’Donnell, J.N.; Wunderink, R.G.; Hauser, A.R. Resistance trends and treatment options in Gram-negative ventilator-associated pneumonia. Curr. Infect. Dis. Rep. 2018, 20, 3. [Google Scholar] [CrossRef]
- Van Hoang, C.; Nguyen, T.T.; Phan, T.Q.; Pham, C.T.; Ninh, H.T.; Wang, B.; Jiang, J.; Ziegler, T.; Nguyen, T.Q. Distribution pattern of the Microhyla heymonsi group (Anura, Microhylidae) with descriptions of two new species from Vietnam. Eur. J. Taxon 2022, 846, 1–41. [Google Scholar] [CrossRef]
- Chai, J.; Chen, X.; Ye, T.; Zeng, B.; Zeng, Q.; Wu, J.; Kascakova, B.; Martins, L.A.; Prudnikova, T.; Smatanova, I.K.; et al. Characterization and functional analysis of cathelicidin-MH, a novel frog-derived peptide with anti-septicemic properties. eLife 2021, 10, e64411. [Google Scholar] [CrossRef]
- Skerlavaj, B.; Scocchi, M.; Tossi, A.; Romeo, D.; Gennaro, R. A Synthetic Approach for a SAR Study of the Pro-and Arg-rich Bactenecin bac7. In Proceedings of the 5th International Symposium on Innovation and Perspectives in Solid Phase Synthesis & Combinatorial Chemical Libraries, London, UK, 31 July 1999; Mayflower Scientific Ltd.: London, UK, 1999; pp. 395–398. [Google Scholar]
- Zanetti, M.; Gennaro, R.; Skerlavaj, B.; Tomasinsig, L.; Circo, R. Cathelicidin Peptides as Candidates for a Novel Class of Antimicrobials. Curr. Pharm. Des. 2002, 8, 779–793. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure–activity relationship studies. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, R.H.; Chen, Y.; Guo, Z.L.; Zhang, F.; Fang, Z.; Huang, K.; Yu, H.N.; Wang, Y.P. Identification and characterization of two novel cathelicidins from the frog Odorrana livida. Zool. Res. 2019, 40, 94–101. [Google Scholar]
- Murray, J.K.; Aral, J.; Miranda, L.P. Solid-phase peptide synthesis using microwave irradiation. Methods Mol. Biol. 2011, 716, 73–88. [Google Scholar]
- Varela, Y.F.; Vanegas Murcia, M.; Patarroyo, M.E. Synthetic Evaluation of Standard and Microwave-Assisted Solid Phase Peptide Synthesis of a Long Chimeric Peptide Derived from Four Plasmodium falciparum Proteins. Molecules 2018, 23, 2877. [Google Scholar] [CrossRef] [Green Version]
- Postma, T.M.; Albericio, F. Disulfide formation strategies in peptide synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Pedersen, S.L.; Tofteng, A.P.; Malik, L.; Jensen, K.J. Microwave heating in solid-phase peptide synthesis. Chem. Soc. Rev. 2012, 41, 1826–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadi-Kamalu, C.; Clarke, H.; McRobie, M.; Mortimer, J.; North, M.; Ran, Y.; Routledge, A.; Sibbald, D.; Tickias, M.; Tse, K.; et al. Investigation of Parameters that Affect Resin Swelling in Green Solvents. ChemistryOpen 2020, 9, 431–441. [Google Scholar] [CrossRef]
- Shepperson, O.A.; Hanna, C.C.; Brimble, M.A.; Harris, P.W.R.; Cameron, A.J. Total synthesis of novel antimicrobial β-Hairpin capitellacin via rapid flow-based SPPS assembly and regioselective on-resin disulfide syclisation. Int. J. Pept. Res. Ther. 2021, 28, 32. [Google Scholar] [CrossRef]
- Han, Y.; Albericio, F.; Barany, G. Occurrence and Minimization of Cysteine Racemization during Stepwise Solid-Phase Peptide Synthesis(1)(,)(2). J. Org. Chem. 1997, 62, 4307–4312. [Google Scholar] [CrossRef]
- Gates, Z.P.; Hartrampf, N. Flow-based SPPS for protein synthesis: A perspective. J. Pept. Sci. 2020, 112, e24198. [Google Scholar] [CrossRef]
- Simon, M.D.; Heider, P.L.; Adamo, A.; Vinogradov, A.A.; Mong, S.K.; Li, X.; Berger, T.; Policarpo, R.L.; Zhang, C.; Zou, Y.; et al. Rapid flow-based peptide synthesis. Chembiochem. Eur. J. Chem. Biol. 2014, 15, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohapatra, S.; Hartrampf, N.; Poskus, M.; Loas, A.; Gómez-Bombarelli, R.; Pentelute, B.L. Deep learning for prediction and pptimization of fast-flow peptide synthesis. ACS Cent. Sci. 2020, 6, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Mijalis, A.J.; Thomas, D.A., 3rd; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017, 13, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Michels, T.; Dolling, R.; Haberkorn, U.; Mier, W. Acid-mediated prevention of aspartimide formation in solid phase peptide synthesis. Org. Lett. 2012, 14, 5218–5221. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Xie, H.B.; Tian, G.L.; Ye, Y.H. Synthesis of cyclopentapeptides and cycloheptapeptides by DEPBT and the influence of some factors on cyclization. Pept. Res. 2002, 60, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Bjornstedt, M. Thioredoxin and thioredoxin reductase. Meth. Enzymol. 1995, 252, 199–208. [Google Scholar]
- Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Cyclization increases the antimicrobial activity and selectivity of arginine- and tryptophan-containing hexapeptides. Biochemistry 2004, 43, 9140–9150. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.; Muhammad, T.; Strömstedt, A.A.; Rosengren, K.J.; Göransson, U. Backbone cyclization and dimerization of LL-37-derived peptides enhance antimicrobial activity and proteolytic stability. Front. Microbiol. 2020, 11, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Mochon, J.J.; Bialy, L.; Bradley, M. Full orthogonality between Dde and Fmoc: The direct synthesis of PNA-peptide conjugates. Org. Lett. 2004, 6, 1127–1129. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; Sixteenth Informational Supplement; CLSI Document M100-S16; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2006. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, 2nd ed.; M27; Clinical and Laboratory Standards Institute: Malvern, PA, USA, 2002. [Google Scholar]
- Konstantinidis, T.; Tsigalou, C.; Karvelas, A.; Stavropoulou, E.; Voidarou, C.; Bezirtzoglou, E. Effects of antibiotics upon the gut microbiome: A review of the literature. Biomedicines. 2020, 8, 502. [Google Scholar] [CrossRef]
- Patangia, D.V.; Anthony Ryan, C.; Dempsey, E.; Paul Ross, R.; Stanton, C. Impact of antibiotics on the human microbiome and consequences for host health. MicrobiologyOpen 2022, 11, e1260. [Google Scholar] [CrossRef]
- Alm, R.A.; Lahiri, S.D. Narrow-spectrum antibacterial agents—Benefits and challenges. Antibiotics 2020, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Avis, T.; Wilson, F.X.; Khan, N.; Mason, C.S.; Powell, D.J. Targeted microbiome-sparing antibiotics. Drug Discov. 2021, 26, 2198–2203. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bajinka, O.; Jarju, P.O.; Tan, Y.; Taal, A.M.; Ozdemir, G. The varying effects of antibiotics on gut microbiota. AMB Express 2021, 11, 116. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
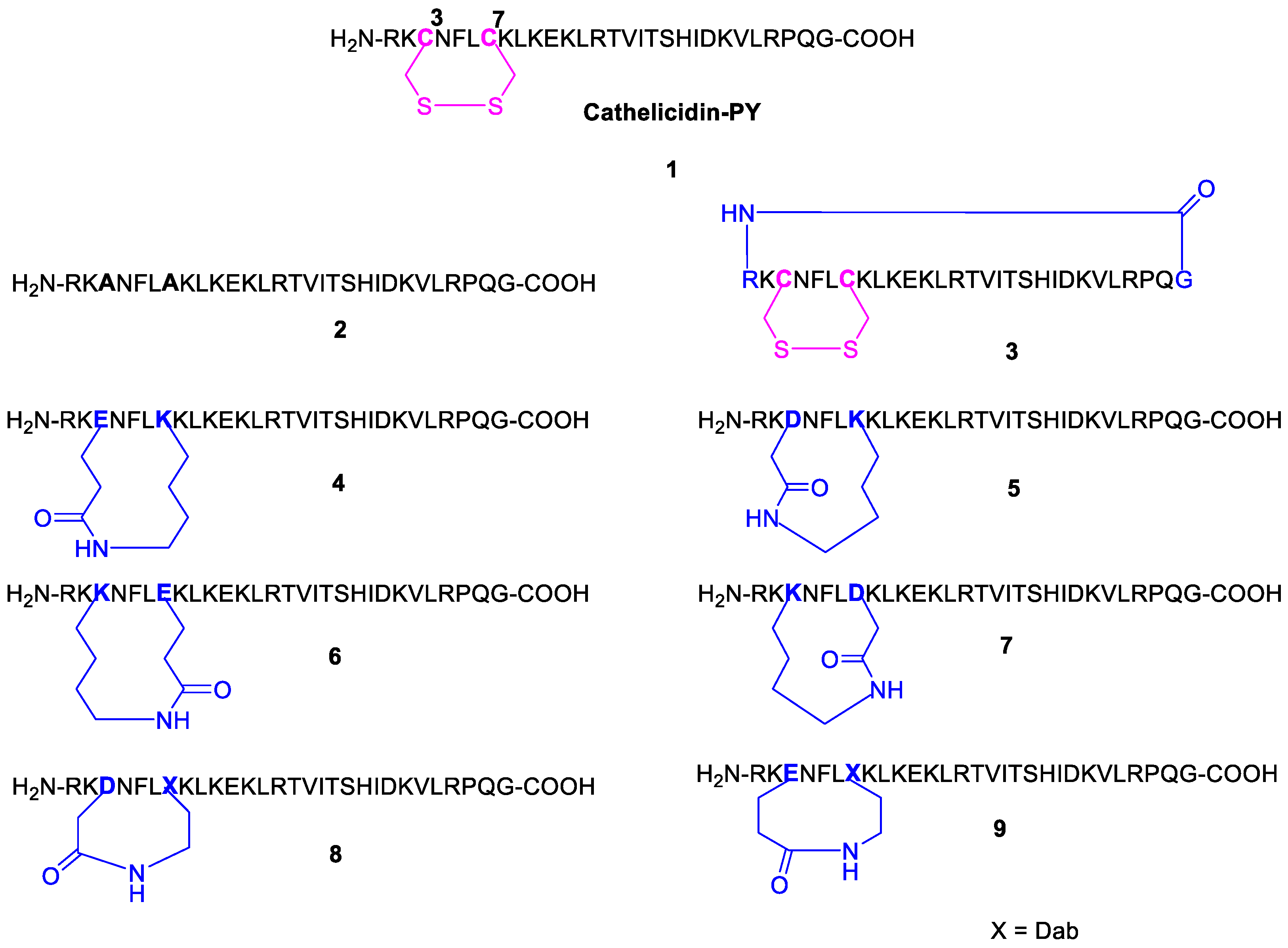{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microorganism | Minimum Inhibitory Concentration (μM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cation Adjusted Media | Compound | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | P * | A * | ||
| Gram-negative | ||||||||||||
| E. coli (ATCC 25922) | Y | 2 | 4 | 16 | 4 | 8 | 4 | 8 | 4 | 8 | 1 | - |
| N | 1 | 1 | 8 | 2 | 2 | 2 | 2 | 1 | 2 | 1 | - | |
| P. aeruginosa (SVB-B9) T | Y | 16 | 32 | >64 | >64 | >64 | 64 | >64 | 32 | 64 | 2 | |
| N | 2 | 4 | 32 | 16 | 16 | 8 | 16 | 8 | 16 | 2 | - | |
| Gram-positive | ||||||||||||
| S. aureus (ATCC 29213) | Y | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | - | >64 |
| N | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | - | >64 | |
| Fungi | ||||||||||||
| C. albicans (SC5314) T | N/A | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | >64 | 1 | |
| Time Point and % Sample Remaining | ||||
|---|---|---|---|---|
| Test Compound | t10 min | t1 h | t3 h | t6 h |
| Cathelicidin-PY | 84.7% | 20.8% | 8.8% | 2.4% |
| 2 | 92.9% | 61.6% | 8.1% | 7.4% |
| 8 | 93.9% | 64.6% | 13.4% | 10.0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dissanayake, S.; He, J.; Yang, S.H.; Brimble, M.A.; Harris, P.W.R.; Cameron, A.J. Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity. Molecules 2023, 28, 1993. https://doi.org/10.3390/molecules28041993
Dissanayake S, He J, Yang SH, Brimble MA, Harris PWR, Cameron AJ. Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity. Molecules. 2023; 28(4):1993. https://doi.org/10.3390/molecules28041993
Chicago/Turabian StyleDissanayake, Shama, Junming He, Sung H. Yang, Margaret A. Brimble, Paul W. R. Harris, and Alan J. Cameron. 2023. "Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity" Molecules 28, no. 4: 1993. https://doi.org/10.3390/molecules28041993
APA StyleDissanayake, S., He, J., Yang, S. H., Brimble, M. A., Harris, P. W. R., & Cameron, A. J. (2023). Flow-Based Fmoc-SPPS Preparation and SAR Study of Cathelicidin-PY Reveals Selective Antimicrobial Activity. Molecules, 28(4), 1993. https://doi.org/10.3390/molecules28041993