


Development of an LC-MS/MS Method for Quantification of Sapitinib in Human Liver Microsomes: In Silico and In Vitro Metabolic Stability Evaluation


Abstract
:1. Introduction
2. Results and Discussion
2.1. In Silico SPT Metabolic Stability
2.2. LC-MS/MS Method Development
2.3. Validation of UPLC-TQD MS Method
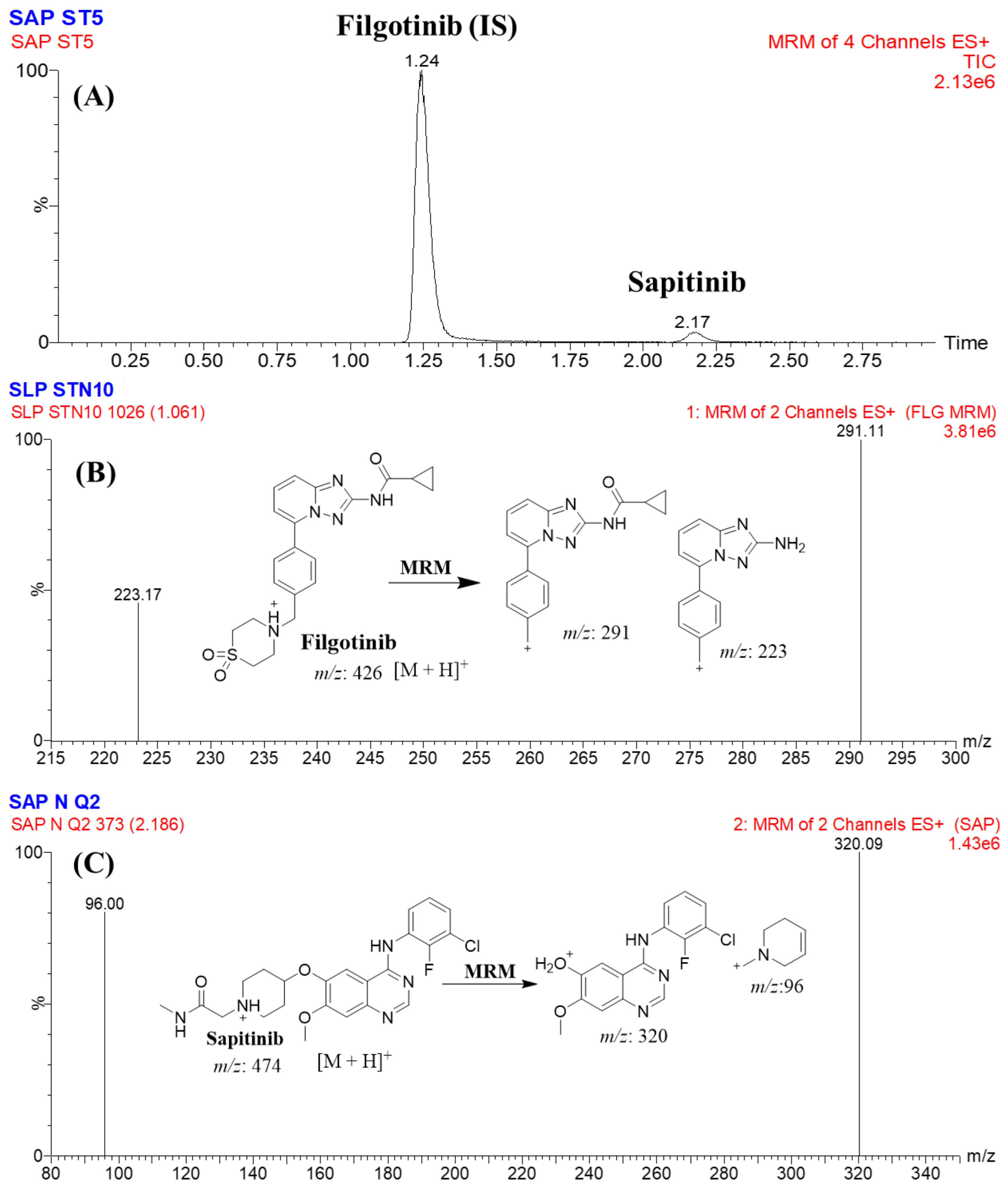
2.3.1. Specificity
2.3.2. Sensitivity and Linearity
2.3.3. Precision and Accuracy
2.3.4. SPT Extraction Recovery and Matrix Effects of HLMs
2.3.5. Stability of SPT in Stock Solution and HLM Matrix
2.4. In Vitro Metabolic Stability of STP
3. Material and Methods
3.1. Materials and Instruments
3.2. In Silico Evaluation of SPT Metabolic Stability
3.3. UPLC-TQD MS Optimized Parameters
3.3.1. UPLC Parameters
3.3.2. TQD MS Parameters
3.4. Preparation of SPT Working Solutions
3.5. Construction of Calibration Curve of SPT
3.6. Extraction of SPT and FGT from HLM Matrix
3.7. Validation of UPLC-TQD MS Method
3.7.1. Specificity
3.7.2. Linearity and Sensitivity
3.7.3. Accuracy and Precision
3.7.4. Matrix Effect and Extraction Recovery
3.8. Stability
3.9. In Vitro Evaluation of SPT Metabolic Stability
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| SPT | sapitinib |
| FGT | filgotinib |
| IS | internal standard |
| ESI | electrospray ionization |
| HLMs | human liver microsomes |
| CLint | intrinsic clearance |
| TKIs | tyrosine kinase inhibitors |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| EGFR | epidermal growth factor receptor |
| NSCLC | non-small-cell lung cancer |
| t1/2 | half-life |
| MRM | multiple reaction monitoring |
| DMSO | dimethyl sulfoxide |
| LLOQ | lower limit of quantification |
| QC | quality control |
| RE | relative error |
| RSD | relative standard deviation |
| SD | standard deviation |
| S/N | signal-to-noise ratio |
| AUC | area under the curve |
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barinaga, M. From Bench Top to Bedside. Science 1997, 278, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, Z.; Paul, B.T.; Craft, B.; ElShamy, W.M. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast Cancer Res. 2015, 17, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlaam, B.; Anderton, J.; Ballard, P.; Bradbury, R.H.; Hennequin, L.F.; Hickinson, D.M.; Kettle, J.G.; Kirk, G.; Klinowska, T.; Lambert-van der Brempt, C.; et al. Discovery of AZD8931, an Equipotent, Reversible Inhibitor of Signaling by EGFR, HER2, and HER3 Receptors. ACS Med. Chem. Lett. 2013, 4, 742–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharm. 1994, 47, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharm. Exp. 1997, 283, 46–58. [Google Scholar]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Alrabiah, H. A reliable and stable method for the determination of foretinib in human plasma by LC-MS/MS: Application to metabolic stability investigation and excretion rate. Eur. J. Mass Spectrom. 2018, 24, 344–351. [Google Scholar] [CrossRef]
- Darwish, H.W.; Kadi, A.A.; Attwa, M.W.; Almutairi, H.S. Investigation of metabolic stability of the novel ALK inhibitor brigatinib by liquid chromatography tandem mass spectrometry. Clin. Chim. Acta 2018, 480, 180–185. [Google Scholar] [CrossRef]
- Attwa, M.W.; Darwish, H.W.; Alhazmi, H.A.; Kadi, A.A. Investigation of metabolic degradation of new ALK inhibitor: Entrectinib by LC-MS/MS. Clin. Chim. Acta 2018, 485, 298–304. [Google Scholar] [CrossRef]
- Amer, S.M.; Kadi, A.A.; Darwish, H.W.; Attwa, M.W. LC-MS/MS method for the quantification of masitinib in RLMs matrix and rat urine: Application to metabolic stability and excretion rate. Chem. Cent. J. 2017, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Attwa, M.W.; Kadi, A.A. Sapitinib: Reactive intermediates and bioactivation pathways characterized by LC-MS/MS. RSC Adv. 2019, 9, 32995–33006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, P.; Swaisland, H.C.; Malone, M.D.; Sarda, S.; Ghiorghiu, S.; Wilbraham, D. Metabolic disposition of AZD8931, an oral equipotent inhibitor of EGFR, HER2 and HER3 signalling, in rat, dog and man. Xenobiotica 2014, 44, 1083–1098. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.V.; Padmalatha, K.; Madhavi, G. In vitro Metabolic Stability of Drugs and Applications of LC-MS in Metabolite Profiling. Drug Metab. 2021, 10, 77. [Google Scholar] [CrossRef]
- Wadhwa, G.; Krishna, K.V.; Taliyan, R.; Tandon, N.; Yadav, S.S.; Katiyar, C.; Dubey, S.K. Pre-clinical pharmacokinetic and pharmacodynamic modelling study of 4-hydroxyisoleucine using validated ultra-performance liquid chromatography-tandem mass spectrometry. RSC Adv. 2020, 10, 5525–5532. [Google Scholar] [CrossRef]
- Baranczewski, P.; Stańczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharm. Rep. 2006, 58, 453–472. [Google Scholar]
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef]
- Tan, L.; Kirchmair, J. Software for metabolism prediction. Drug Metab. Predict. 2014, 27–52. [Google Scholar] [CrossRef]
- Hunt, P.A.; Segall, M.D.; Tyzack, J.D. WhichP450: A multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J. Comput. Mol. Des. 2018, 32, 537–546. [Google Scholar] [CrossRef]
- Shin, Y.G.; Le, H.; Khojasteh, C.; ECA Hop, C. Comparison of metabolic soft spot predictions of CYP3A4, CYP2C9 and CYP2D6 substrates using MetaSite and StarDrop. Comb. Chem. High Throughput Screen. 2011, 14, 811–823. [Google Scholar] [CrossRef]
- McNaney, C.A.; Drexler, D.M.; Hnatyshyn, S.Y.; Zvyaga, T.A.; Knipe, J.O.; Belcastro, J.V.; Sanders, M. An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. ASSAY Drug Dev. Technol. 2008, 6, 121–129. [Google Scholar] [CrossRef]
- Leahy, D.E. Integrating invitro ADMET data through generic physiologically based pharmacokinetic models. Expert Opin. Drug Metab. Toxicol. 2006, 2, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation EGFR-TKI naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019, 9, 4862–4869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadi, A.A.; Darwish, H.W.; Abuelizz, H.A.; Alsubi, T.A.; Attwa, M.W. Identification of reactive intermediate formation and bioactivation pathways in Abemaciclib metabolism by LC-MS/MS: In vitro metabolic investigation. R. Soc. Open Sci. 2019, 6, 181714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S.; Alhazmi, H.A. Metabolic stability assessment of new parp inhibitor talazoparib using validated lc–ms/ms methodology: In silico metabolic vulnerability and toxicity studies. Drug Des. Dev. Ther. 2020, 14, 783–793. [Google Scholar] [CrossRef]
- Busby, W.F., Jr.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999, 27, 246–249. [Google Scholar]
- Störmer, E.; Roots, I.; Brockmöller, J. Benzydamine N-oxidation as an index reaction reflecting FMO activity in human liver microsomes and impact of FMO3 polymorphisms on enzyme activity. Br. J. Clin. Pharm. 2000, 50, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Fouin-Fortunet, H.; Tinel, M.; Descatoire, V.; Letteron, P.; Larrey, D.; Geneve, J.; Pessayre, D. Inactivation of cytochrome P-450 by the drug methoxsalen. J. Pharm. Exp. 1986, 236, 237–247. [Google Scholar]
- Meesters, R.; Voswinkel, S. Bioanalytical method development and validation: From the USFDA 2001 to the USFDA 2018 guidance for industry. J. Appl. Bioanal. 2018, 4, 67–73. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules 2020, 25, 5004. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef] [Green Version]
- Attwa, M.W.; Al-Shakliah, N.S.; AlRabiah, H.; Kadi, A.A.; Abdelhameed, A.S. Estimation of zorifertinib metabolic stability in human liver microsomes using LC-MS/MS. J. Pharm. Biomed. Anal. 2022, 211, 114626. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Abdelhameed, A.S.; Alsaif, N.A.; Kadi, A.A.; AlRabiah, H. A validated LC-MS/MS analytical method for the quantification of pemigatinib: Metabolic stability evaluation in human liver microsomes. RSC Adv. 2022, 12, 20387–20394. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, G.A.E.; Kadi, A.A.; AlMasoud, N.; Attwa, M.W.; Al-Shakliah, N.S.; AlRabiah, H. LC-MS/MS method for the quantification of the anti-cancer agent infigratinib: Application for estimation of metabolic stability in human liver microsomes. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1179, 122806. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.W.; Abdelhameed, A.S.; Al-Shakliah, N.S.; Kadi, A.A. Lc-ms/ms estimation of the anti-cancer agent tandutinib levels in human liver microsomes: Metabolic stability evaluation assay. Drug Des. Dev. Ther. 2020, 14, 4439–4449. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | ACN | Methanol | Protein Precipitation | Solid Phase Extraction | PFP Column | C18 or C8 Column |
|---|---|---|---|---|---|---|
| SPT | 2.17 min | 2.81 min | High recovery | Low recovery | 2.17 min | 3.75 min |
| Good | Tailed | Reproducible | Unreproducible | Perfect | Tailed | |
| FGT | 1.24 min | 2.64 min | High recovery | Low recovery | 1.24 min | 2.14 min |
| Good | Overlapped | Reproducible | Unreproducible | Perfect | Perfect |
| SPT Nominal Concentrations (ng/mL) | Mean | SD | Precision (RSD %) | Accuracy (RE %) | Recovery |
|---|---|---|---|---|---|
| 1(LLQC) | 1.06 | 0.02 | 1.92 | 5.96 | 105.96 |
| 3(LQC) | 3.11 | 0.12 | 3.72 | 3.59 | 103.59 |
| 15 | 15.37 | 0.18 | 1.19 | 2.45 | 102.45 |
| 50 | 50.40 | 1.41 | 2.80 | 0.81 | 100.81 |
| 100 | 152.07 | 1.56 | 1.02 | 1.38 | 101.38 |
| 300 | 295.03 | 2.48 | 0.84 | −1.66 | 98.34 |
| 500 | 502.85 | 7.92 | 1.57 | 0.57 | 100.57 |
| 900 (MQC) | 894.72 | 2.63 | 0.29 | −0.59 | 99.41 |
| 1500 | 1520.98 | 19.26 | 1.27 | 1.40 | 101.40 |
| 2400 (HQC) | 2394.09 | 17.45 | 0.73 | −0.25 | 99.75 |
| 3000 | 3027.67 | 42.18 | 1.39 | 0.92 | 100.92 |
| % Recovery | 101.33 ± 2.11 |
| STP in HLM Matrix (ng/mL) | Intraday Assay (Twelve Replicates in the Same Day) | Interday Assay (Six Replicates Over Three Consecutive Days) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 (LLQC) | 3 (LQC) | 900 (MQC) | 2400 (HQC) | 1 (LLQC) | 3 (LQC) | 900 (MQC) | 2400 (HQC) | |
| Mean | 1.06 | 3.11 | 894.72 | 2394.09 | 1.03 | 3.22 | 886.99 | 2365.33 |
| SD | 0.02 | 0.12 | 2.63 | 17.45 | 0.06 | 0.20 | 6.17 | 17.61 |
| Precision (RSD %) | 1.92 | 3.72 | 0.29 | 0.73 | 6.31 | 6.29 | 0.70 | 0.74 |
| Accuracy (RE %) | 5.96 | 3.59 | −0.59 | −0.25 | 2.57 | 7.28 | −1.45 | −1.44 |
| Recovery (%) | 105.96 | 103.59 | 99.41 | 99.75 | 102.57 | 107.28 | 98.55 | 98.56 |
| Analyte | Concentration (ng/mL) | Freeze–Thaw Stability (3 Cycles, −80 °C) | Short-Term Stability (4 h at Room T) | Long-Term Stability (−80 °C for 28 d) | Autosampler Stability (24 h at 15 °C) |
|---|---|---|---|---|---|
| SPT | LQC (3) | 98.8 ± 2.4 | 99.4 ± 2.5 | 98.6 ± 2.3 | 99.7 ± 2.7 |
| HQC (2400) | 101.3 ± 2.2 | 99.2 ± 2.7 | 100.5 ± 2.8 | 99.8 ± 2.5 |
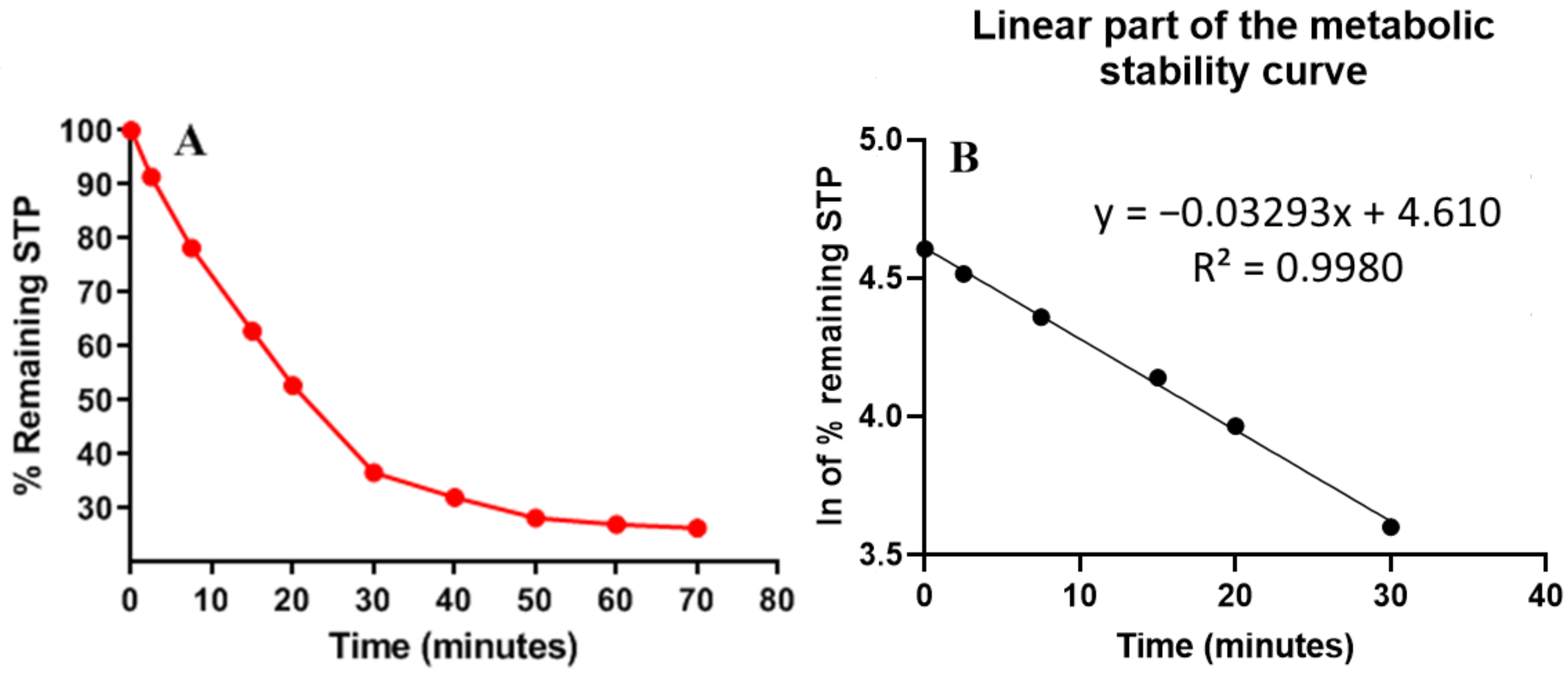
| Time (min) | Mean a (ng/mL) | X b | ln X | Linearity Parameters |
|---|---|---|---|---|
| 0 | 584 | 100.00 | 4.61 | Regression equation: y = −0.0329x + 4.6097 |
| 2.5 | 534 | 91.44 | 4.52 | |
| 7.5 | 457 | 78.25 | 4.36 | R² = 0.998 |
| 15 | 367 | 62.84 | 4.14 | |
| 20 | 308 | 52.74 | 3.97 | Slope: −0.0329 |
| 30 | 214 | 36.64 | 3.60 | |
| 40 | 187 | 32.02 | 3.47 | t1/2: 21.07 min |
| 50 | 165 | 28.25 | 3.34 | Clint: 38.48 mL/min/kg |
| 60 | 158 | 27.05 | 3.30 | |
| 70 | 154 | 26.37 | 3.27 |
| Analyte | Rt | Ion Mode | Precursor (m/z) | Quantification Traces (m/z) | Qualification Traces (m/z) | Cone Voltage (V) | Collision Energy (CE, eV) |
|---|---|---|---|---|---|---|---|
| SPT | 2.17 | +ve | 474.0 | 320.0 | 149.9 | 38 | 24/28 |
| FGT (IS) | 1.24 | +ve | 426.0 | 291.0 | 323.0 | 38 | 24/38 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attwa, M.W.; AlRabiah, H.; Mostafa, G.A.E.; Kadi, A.A. Development of an LC-MS/MS Method for Quantification of Sapitinib in Human Liver Microsomes: In Silico and In Vitro Metabolic Stability Evaluation. Molecules 2023, 28, 2322. https://doi.org/10.3390/molecules28052322
Attwa MW, AlRabiah H, Mostafa GAE, Kadi AA. Development of an LC-MS/MS Method for Quantification of Sapitinib in Human Liver Microsomes: In Silico and In Vitro Metabolic Stability Evaluation. Molecules. 2023; 28(5):2322. https://doi.org/10.3390/molecules28052322
Chicago/Turabian StyleAttwa, Mohamed W., Haitham AlRabiah, Gamal A. E. Mostafa, and Adnan A. Kadi. 2023. "Development of an LC-MS/MS Method for Quantification of Sapitinib in Human Liver Microsomes: In Silico and In Vitro Metabolic Stability Evaluation" Molecules 28, no. 5: 2322. https://doi.org/10.3390/molecules28052322
APA StyleAttwa, M. W., AlRabiah, H., Mostafa, G. A. E., & Kadi, A. A. (2023). Development of an LC-MS/MS Method for Quantification of Sapitinib in Human Liver Microsomes: In Silico and In Vitro Metabolic Stability Evaluation. Molecules, 28(5), 2322. https://doi.org/10.3390/molecules28052322