
Substituent Effects in Tetrel Bonds Involving Aromatic Silane Derivatives: An ab initio Study




Abstract
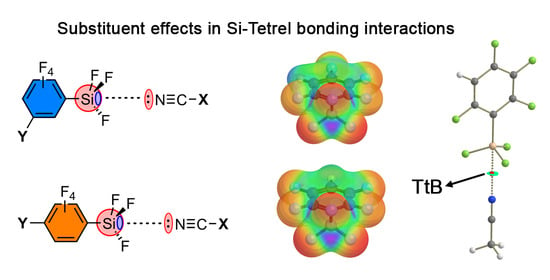
:
1. Introduction
2. Results and Discussion
2.1. ESP Analysis
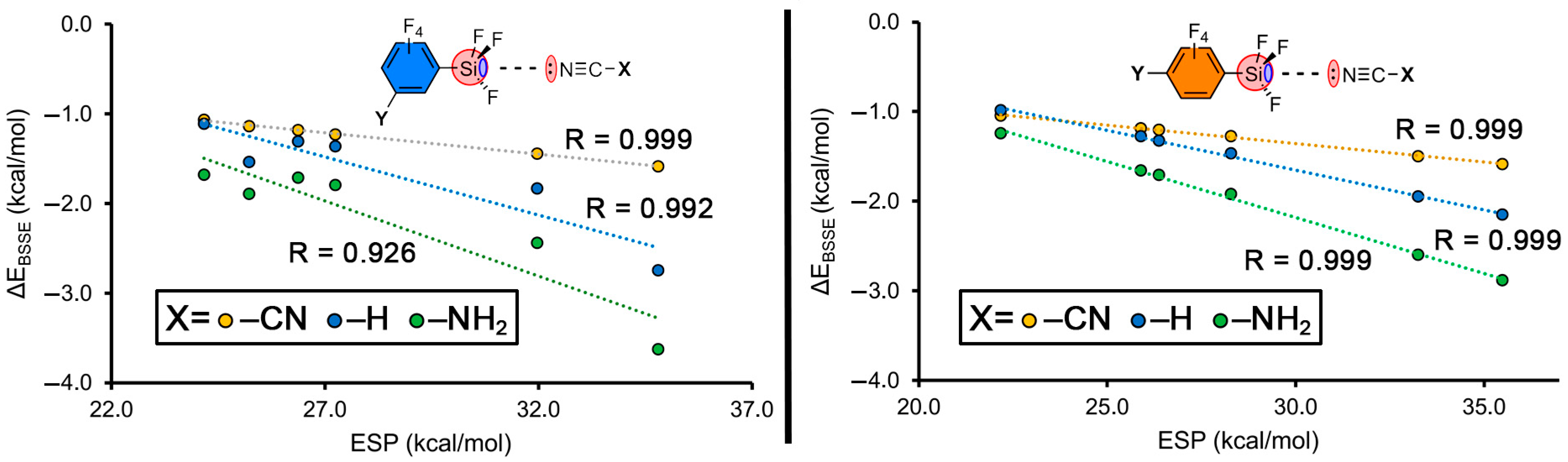
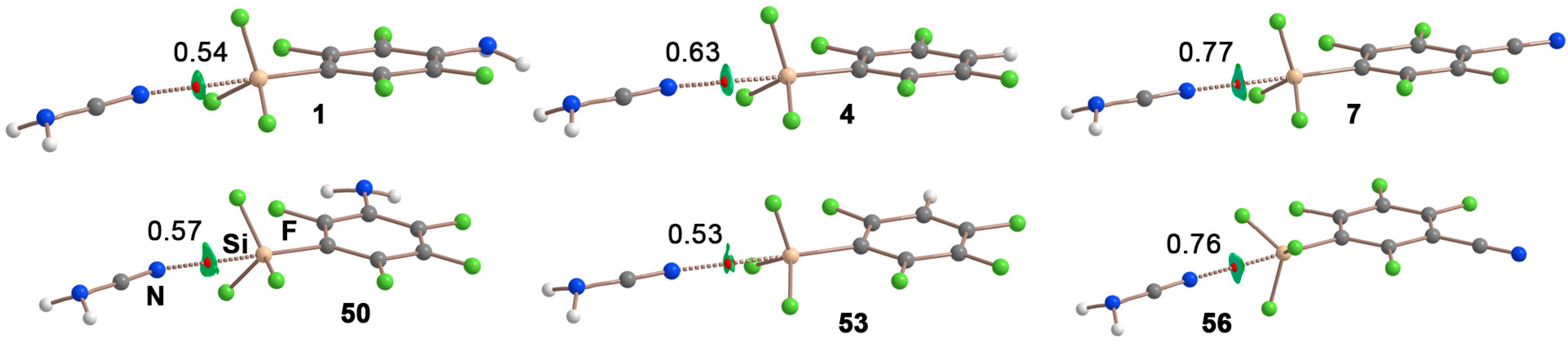
2.2. Energetic Study
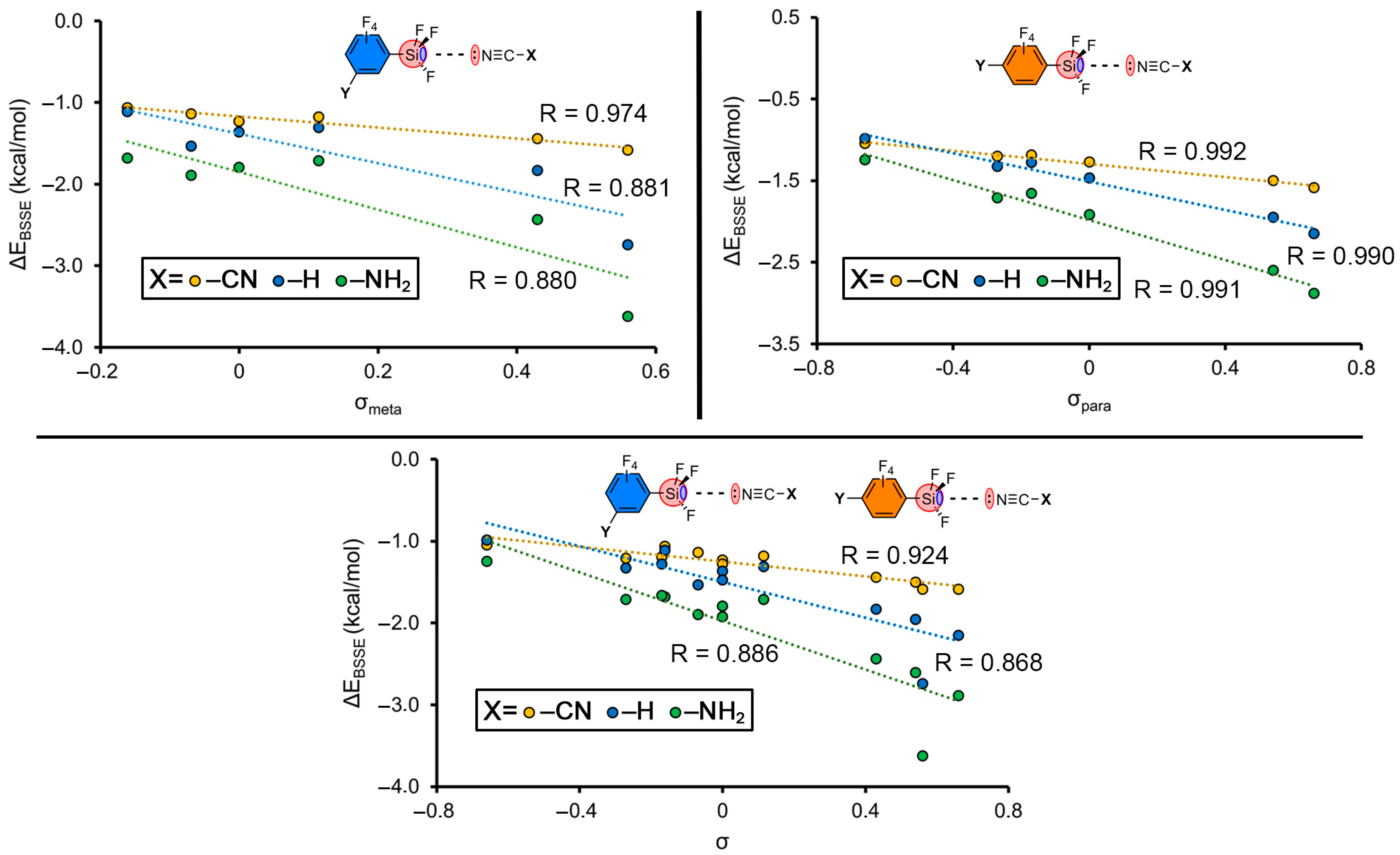
2.3. Hammett’s Representations
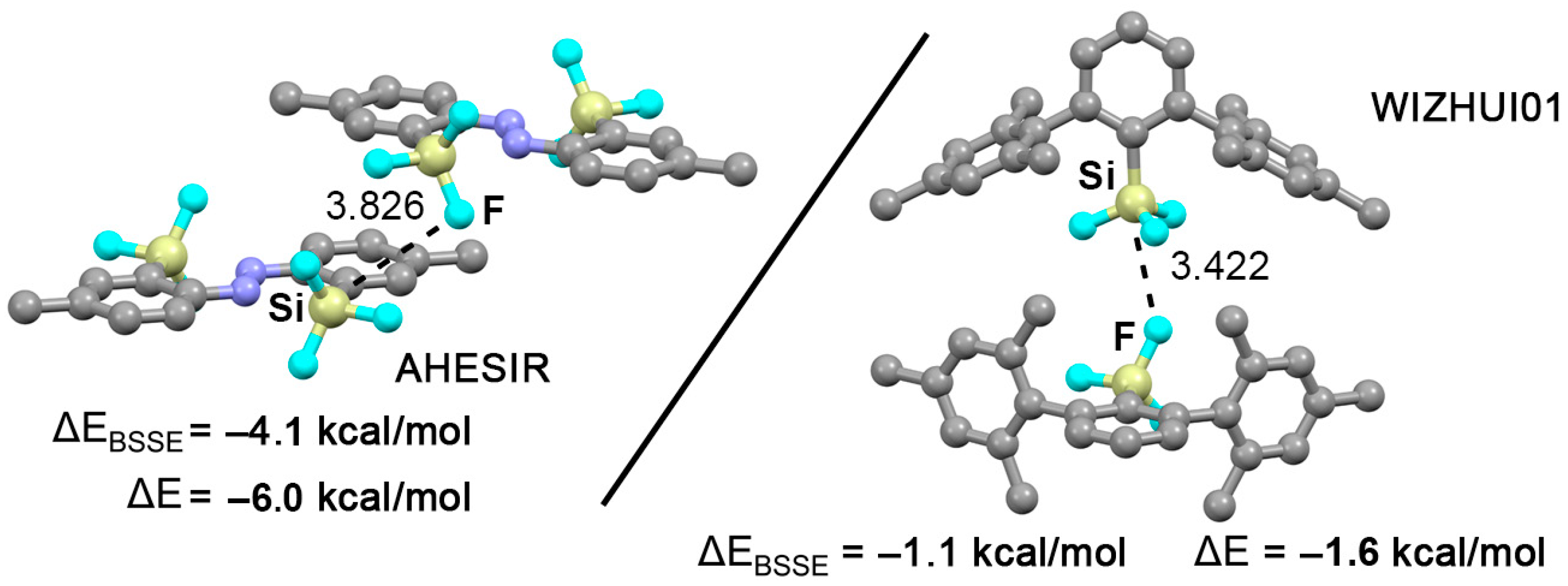
2.4. CSD Search
3. Conclusions
4. Materials and Methods
4.1. General Considerations
4.2. CSD Survey
- dSi···A ≤ sum of vdW radii + 0.5 Å.
- C/X–Si···A between 160 and 180 degrees (X = C, F, Cl, Br and I and A = any atom).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehn, J.M. Supramolecular Chemistry: Concepts and Perspectives, 1st ed.; Wiley VCH: Weinheim, Germany, 1995. [Google Scholar]
- Schneider, H.J. Supramolecular Systems in Biomedical Fields, 1st ed.; RSC Publishing: Cambridge, UK, 2013. [Google Scholar]
- Steed, A.W.; Atwood, J.L. Supramolecular Chemistry, 1st ed.; John Wily & Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Cragg, P.J. Supramolecular Chemistry: From Biological Inspiration to Biomedical Applications, 1st ed.; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Politzer, P.; Murray, J. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen bonds: A new molecular linker? Chem. Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. NO3− anions can act as Lewis acid in the solid state. Nat. Commun. 2017, 8, 14522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 28, 5371–5376. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Sharko, A.V.; Senchyk, G.A.; Rusanov, E.B.; Frontera, A.; Domasevitch, K.V. π–hole interactions at work: Crystal engineering with nitro-derivatives. Cryst. Eng. Comm. 2017, 19, 1933–1937. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. π-hole interactions involving nitro compounds: Directionality of nitrate esters. Cryst. Growth Des. 2016, 16, 5520–5524. [Google Scholar] [CrossRef]
- Hazari, A.; Das, L.K.; Bauzá, A.; Frontera, A.; Ghosh, A. The influence of H-bonding on the ‘ambidentate’coordination behaviour of the thiocyanate ion to Cd (II): A combined experimental and theoretical study. Dalton Trans. 2014, 43, 8007–8015. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. Chem. Phys. Chem. 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Riwar, L.J.; Trapp, N.; Root, K.; Zenobi, R.; Diederich, F. Supramolecular Capsules: Strong versus Weak Chalcogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 17259–17264. [Google Scholar] [CrossRef]
- Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S.M. Chalcogen Bonding Catalysis of a Nitro-Michael Reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borissov, A.; Marques, I.; Lim, J.Y.C.; Félix, V.; Smith, M.D.; Beer, P.D. Anion Recognition in Water by Charge-Neutral Halogen and Chalcogen Bonding Foldamer Receptors. J. Am. Chem. Soc. 2019, 141, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Mallada, B.; Gallardo, A.; Lamanec, M.; de la Torre, B.; Špirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, J.; Ling, K.B.; Cockroft, S.L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [Green Version]
- Macchione, M.; Goujon, A.; Strakova, K.; Humeniuk, H.V.; Licari, G.; Tajkhorshid, E.; Sakai, N.; Matile, S. A Chalcogen-Bonding Cascade Switch for Planarizable Push-Pull Probes. Angew. Chem. Int. Ed. 2019, 58, 15752–15756. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An overview of strengths and directionalities of noncovalent interactions: σ-holes and π-holes. Crystals 2019, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Naming interactions from the electrophilic site. Cryst. Growth Des. 2014, 14, 2697–2702. [Google Scholar] [CrossRef]
- Terraneo, G.; Resnati, G. Bonding Matters. Cryst. Growth Des. 2017, 17, 1439–1440. [Google Scholar] [CrossRef] [Green Version]
- Scheiner, S. Steric crowding in tetrel bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef] [Green Version]
- Heywood, V.L.; Alford, T.P.J.; Roeleveld, J.J.; Deprez, S.J.L.; Verhoofstad, A.; van der Vlugt, J.I.; Domingos, S.R.; Schnell, M.; Davis, A.P.; Mooibroek, T.J. Observations of tetrel bonding between sp3-carbon and THF. Chem. Sci. 2020, 11, 5289–5293. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W.; Wang, Y.-B. Tetrel bonding on Graphene. Comp. Theor. Chem. 2019, 1147, 8–12. [Google Scholar] [CrossRef]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213720. [Google Scholar] [CrossRef]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent Carbon-Bonding Interactions in Proteins. Angew. Chem. Int. Ed. 2018, 57, 16496. [Google Scholar] [CrossRef]
- Dutta, J.; Sahoo, D.K.; Jena, S.; Tulsiyan, K.D.; Biswal, H.S. Non-covalent interactions with inverted carbon: A carbo-hydrogen bond or a new type of hydrogen bond? Phys. Chem. Chem. Phys. 2020, 22, 8988–8997. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Mod. 2012, 18, 541–548. [Google Scholar] [CrossRef]
- Burgi, H.B.; Dunitz, J.D.; Shefter, E. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar]
- Jönsson, B.; Karlström, G.; Wennerström, H. Ab initio molecular orbital calculations on the water-carbon dioxide system: Molecular complexes. Chem. Phys. Lett. 1975, 30, 58–59. [Google Scholar] [CrossRef]
- Peterson, K.I.; Klemperer, W. Structure and internal rotation of H2O−CO2, HDO−CO2 and D2O−CO2 van der Waals complexes. J. Chem. Phys. 1984, 80, 2439–2445. [Google Scholar] [CrossRef]
- Peng, Y.P.; Sharpe, S.W.; Shin, S.K.; Wittig, C.; Beaudet, R.A. Infrared spectroscopy of CO2–D(H)Br: Molecular structure and its reliability. J. Chem. Phys. 1992, 97, 5392. [Google Scholar]
- Leopold, K.R.; Fraser, G.T.; Klemperer, W. Rotational spectrum and structure of the complex HCNCO2. J. Chem. Phys. 1984, 80, 1039. [Google Scholar] [CrossRef]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular Complexes between Silicon Derivatives and Electron-Rich Groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Fruchier, A.; Macquarrie, D.J.; Virgili, A. Aminopropylsilanes versus silatranes: An experimental and theoretical study. J. Organomet. Chem. 2001, 625, 148–153. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furanTF3 (T= C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Concha, M.C.; Politzer, P. Molecular surface electrostatic potentials as guides to Si-O-N angle contraction: Tunable σ-holes. J. Mol. Model. 2011, 7, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Patia, S.; Rappoport, Z. (Eds.) The Chemistry of Functional Groups. In The Chemistry of Organic Germanium, Tin and Lead Compounds; Wiley: Hoboken, NJ, USA, 1995; Volume 19. [Google Scholar]
- Parr, J. Comprehensive Coordination Chem II; McCleverty, J.A., Meye, T., Jr., Eds.; Elsevier Pergamon: Oxford, UK, 2004; Volume 3, p. 545. [Google Scholar]
- Sato, T. Comprehensive Organometallic Chem II—8—Tin; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Volume 11, p. 355. [Google Scholar]
- Pinhey, J.T. 11—Lead. In Comprehensive Organometallic Chem II; Abel, E.W., Stone, F.G.A., Wilkinson, G., Eds.; Pergamon Press: Oxford, UK, 1995; Volume 11, p. 461. [Google Scholar]
- Greenberg, A.; Wu, G. Structural relationships in silatrane molecules. Struct. Chem. 1990, 1, 79–85. [Google Scholar] [CrossRef]
- Hencsei, P. Evaluation of silatrane structures by correlation relationships. Struct. Chem. 1991, 2, 21–26. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Barishok, V.P.; Petukhov, L.P.; Rahklin, R.G.; Pestunovich, V.A. 1-Halosilatranes. J. Organomet. Chem. 1988, 358, 39–55. [Google Scholar] [CrossRef]
- Lukevics, E.; Dimens, V.; Pokrovska, N.; Zicmane, I.; Popelis, J.; Kemme, A. Addition of nitrile oxides to 2,3-dihydrofurylsilanes. Crystal and molecular structure of tetrahydrofuro-[2,3-d]-isoxazolylsilanes. J. Organomet. Chem. 1999, 586, 200–207. [Google Scholar] [CrossRef]
- Franconetti, A.; Frontera, A. “Like–like” tetrel bonding interactions between Sn centres: A combined ab initio and CSD study. Dalton Trans. 2019, 48, 11208–11216. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Yang, X.; Li, Q. Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition. Molecules 2021, 26, 7231. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Frontera, A.; Deyà, P.M. Substituent effects in halogen bonding complexes between aromatic donors and acceptors: A comprehensive ab initio study. Phys. Chem. Chem. Phys. 2011, 13, 20371–20379. [Google Scholar] [CrossRef]
- Kano, N.; Yamamura, M.; Kawashima, T. 2,2′-Disilylazobenzenes featuring double intramolecular nitrogen⋯silicon coordination: A photoisomerizable fluorophore. Dalton Trans. 2015, 44, 16256–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, A.; Lork, E.; Beckmann, J. A monoclinic polymorph of 2,6-Mes2 C6 H3 SiF3. Main Group Met. Chem. 2014, 37, 153. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Bauzá, A.; Alkorta, I.; Frontera, A.; Elguero, J. On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors. J. Chem. Theory Comput. 2013, 9, 5201–5210. [Google Scholar] [CrossRef] [Green Version]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic Structure Calculations on Workstation Computers—The Program System turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Boys, S.B.; Bernardy, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Todd, A.; Keith, T.K. AIMAll, version 13.05.06; Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Cryst. 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]








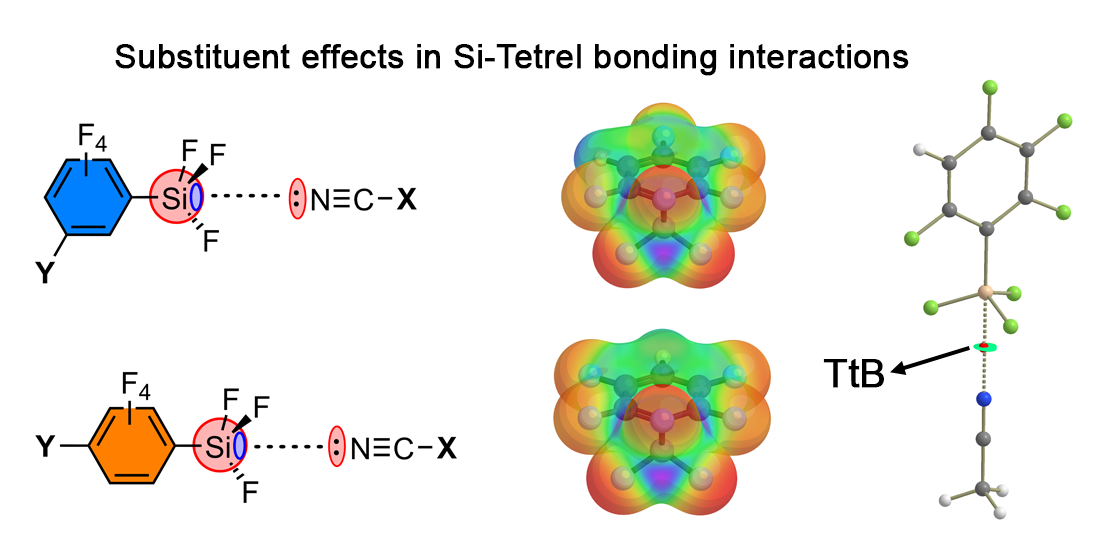{kind=link}
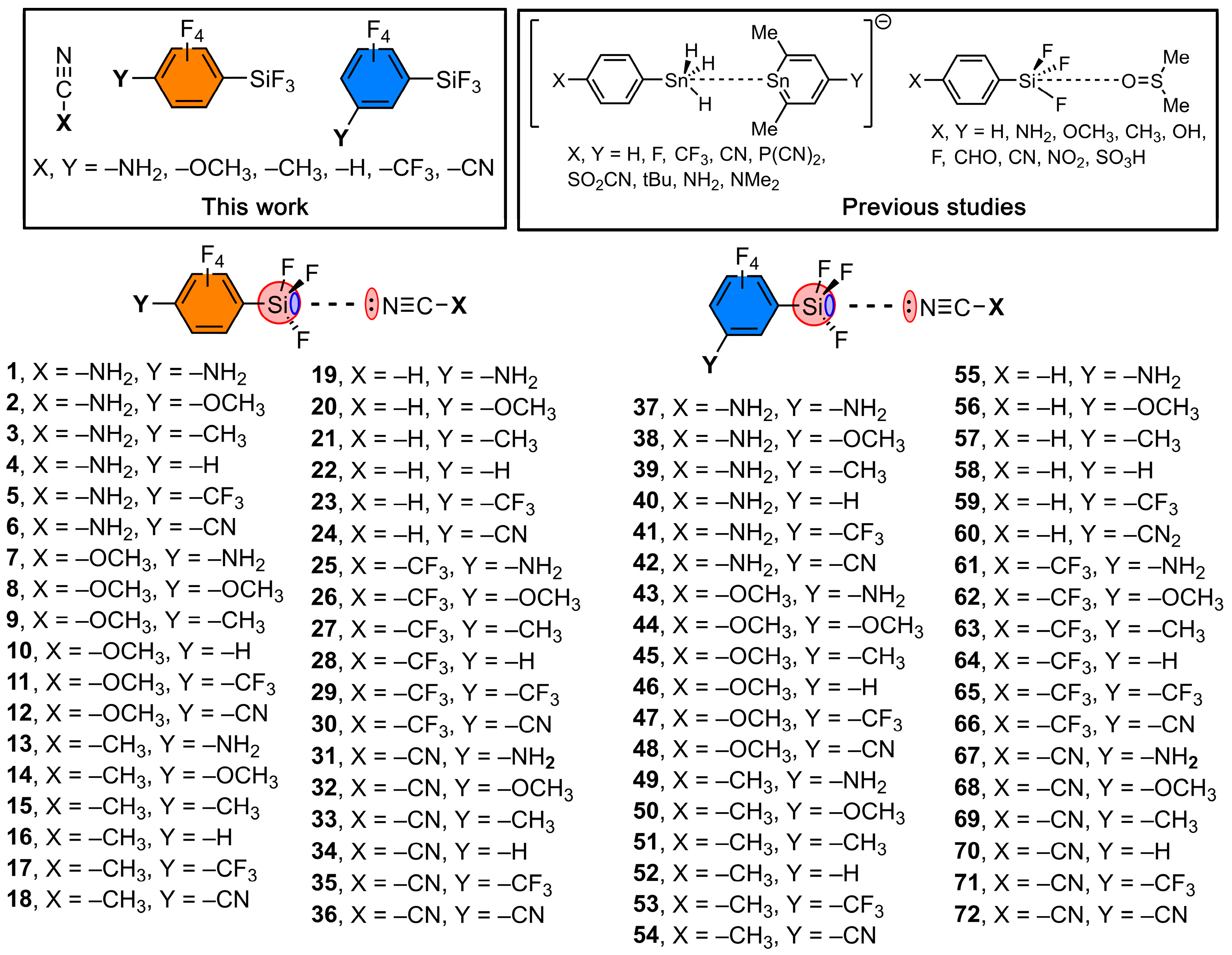{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
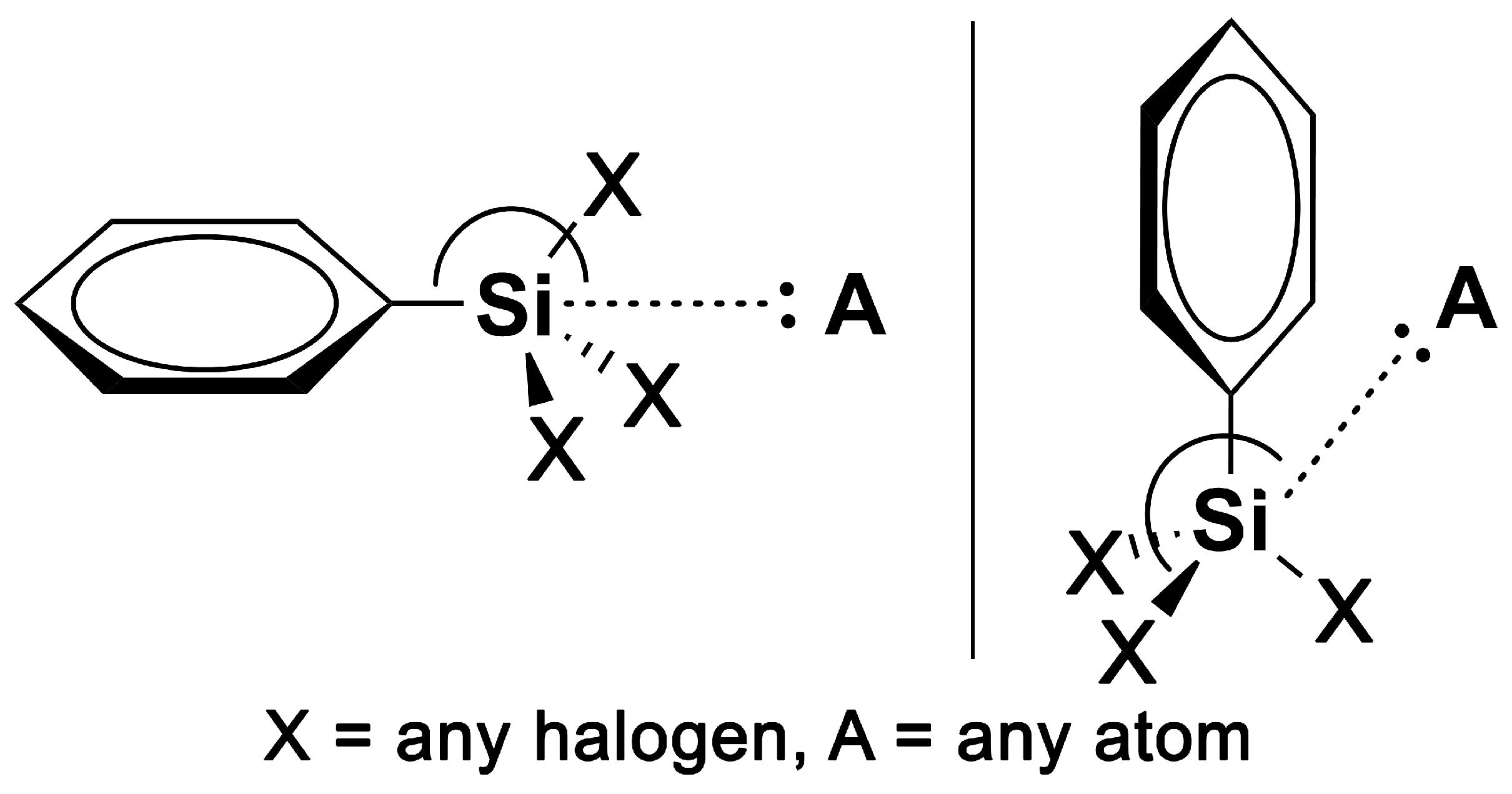{kind=link}
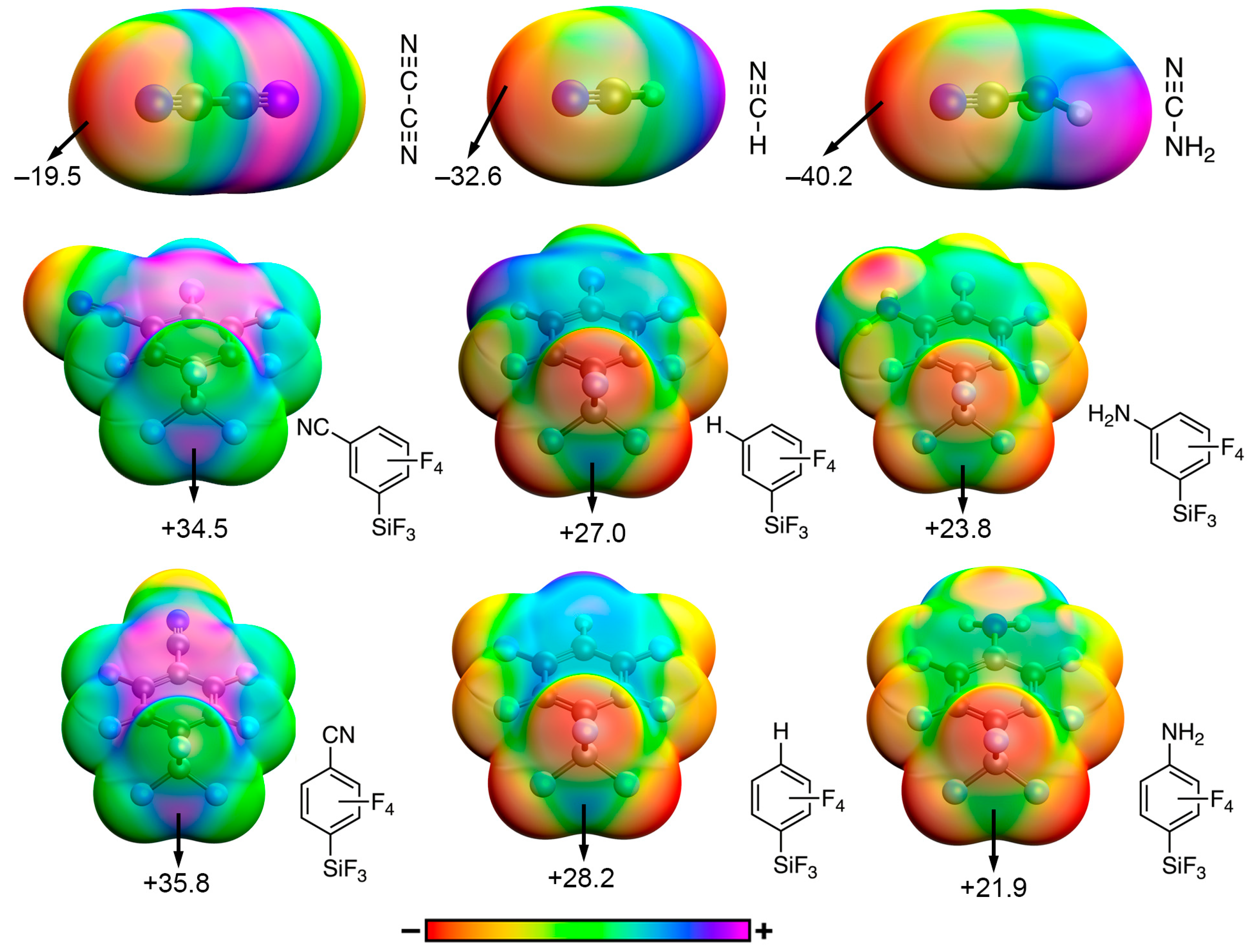
| TtB Donor (para substitution) | ESP (kcal/mol) |
|---|---|
| –CF3 | +33.3 |
| –CN | +35.8 |
| –H | +28.2 |
| –CH3 | +25.9 |
| –NH2 | +21.9 |
| –OCH3 | +26.4 |
| TtB donor (meta substitution) | ESP (kcal/mol) |
| –CF3 | +32.0 |
| –CN | +34.5 |
| –H | +27.0 |
| –CH3 | +25.2 |
| –NH2 | +23.8 |
| –OCH3 | +26.4 |
| TtB acceptor | ESP (kcal/mol) |
| –CF3 | −23.3 |
| –CN | −19.5 |
| –H | −32.6 |
| –CH3 | −38.3 |
| –NH2 | −40.2 |
| –OCH3 | −39.2 |
| Complex | ΔE | ΔEBSSE | d | ρ × 100 |
|---|---|---|---|---|
| 1 (X = –NH2, Y = –NH2) | −1.9 | −1.2 | 3.159 | 0.72 |
| 2 (X = –NH2, Y = –OCH3) | −2.4 | −1.7 | 3.114 | 0.77 |
| 3 (X = –NH2, Y = –CH3) | −2.4 | −1.7 | 3.119 | 0.77 |
| 4 (X =–NH2, Y = –H) | −2.7 | −1.9 | 3.095 | 0.79 |
| 5 (X = –NH2, Y = –CF3) | −3.4 | −2.6 | 3.030 | 0.89 |
| 6 (X = –NH2, Y = –CN) | −3.7 | −2.9 | 3.024 | 0.93 |
| 7 (X = –OCH3, Y = –NH2) | −1.9 | −1.2 | 3.173 | 0.69 |
| 8 (X = –OCH3, Y = –OCH3) | −2.4 | −1.7 | 3.142 | 0.72 |
| 9 (X = –OCH3, Y = –CH3) | −2.3 | −1.6 | 3.132 | 0.74 |
| 10 (X = –OCH3, Y = –H) | −2.6 | −1.9 | 3.108 | 0.77 |
| 11 (X = –OCH3, Y = –CF3) | −3.3 | −2.5 | 3.046 | 0.86 |
| 12 (X = –OCH3, Y = –CN) | −3.6 | −2.8 | 3.024 | 0.89 |
| 13 (X = –CH3, Y = –NH2) | −1.9 | −1.2 | 3.182 | 0.69 |
| 14 (X = –CH3, Y = –OCH3) | −2.3 | −1.6 | 3.139 | 0.74 |
| 15 (X = –CH3, Y = –CH3) | −2.3 | −1.6 | 3.143 | 0.74 |
| 16 (X = –CH3, Y = –H) | −2.5 | −1.8 | 3.121 | 0.77 |
| 17 (X = –CH3, Y = –CF3) | −3.2 | −2.4 | 3.060 | 0.85 |
| 18 (X = –CH3, Y = –CN) | −3.5 | −2.7 | 3.039 | 0.88 |
| 19 (X = –H, Y = –NH2) | −1.5 | −1.0 | 3.259 | 0.42 |
| 20 (X = –H, Y = –OCH3) | −1.9 | −1.3 | 3.221 | 0.64 |
| 21 (X = –H, Y = –CH3) | −1.9 | −1.3 | 3.218 | 0.64 |
| 22 (X = –H, Y = –H) | −2.1 | −1.5 | 3.206 | 0.66 |
| 23 (X = –H, Y = –CF3) | −2.6 | −2.0 | 3.155 | 0.71 |
| 24 (X = –H, Y = –CN) | −2.8 | −2.1 | 3.138 | 0.74 |
| 25 (X = –CF3, Y = –NH2) | −1.5 | −1.0 | 3.310 | 0.37 |
| 26 (X = –CF3, Y = –OCH3) | −1.8 | −1.2 | 3.268 | 0.58 |
| 27 (X = –CF3, Y = –CH3) | −1.7 | −1.1 | 3.283 | 0.57 |
| 28 (X = –CF3, Y = –H) | −1.8 | −1.3 | 3.267 | 0.58 |
| 29 (X = –CF3, Y = –CF3) | −2.2 | −1.6 | 3.228 | 0.62 |
| 30 (X = –CF3, Y = –CN) | −2.3 | −1.7 | 3.214 | 0.47 |
| 31 (X = –CN, Y = –NH2) | −1.6 | −1.0 | 3.324 | 0.69 |
| 32 (X = –CN, Y = –OCH3) | −1.8 | −1.2 | 3.297 | 0.74 |
| 33 (X = –CN, Y = –CH3) | −1.8 | −1.2 | 3.299 | 0.74 |
| 34 (X = –CN, Y = –H) | −1.8 | −1.3 | 3.286 | 0.77 |
| 35 (X = –CN, Y = –CF3) | −2.1 | −1.5 | 3.250 | 0.85 |
| 36 (X = –CN, Y = –CN) | −2.2 | −1.6 | 3.238 | 0.89 |
| Complex | ΔE | ΔEBSSE | d | ρ × 100 |
|---|---|---|---|---|
| 37 (X = –NH2, Y = –NH2) | −2.4 | −1.7 | 3.135 | 0.74 |
| 38 (X = –NH2, Y = –OCH3) | −2.4 | −1.7 | 3.112 | 0.77 |
| 39 (X = –NH2, Y = –CH3) | −2.6 | −1.9 | 3.131 | 0.75 |
| 40 (X = –NH2, Y = –H) | −2.5 | −1.8 | 3.162 | 0.71 |
| 41 (X = –NH2, Y = –CF3) | −3.2 | −2.4 | 3.048 | 0.87 |
| 42 (X = –NH2, Y = –CN) | −3.6 | −3.6 | 3.014 | 0.92 |
| 43 (X = –OCH3, Y = –NH2) | −2.3 | −1.6 | 3.160 | 0.71 |
| 44 (X = –OCH3, Y = –OCH3) | −2.7 | −2.0 | 3.128 | 0.75 |
| 45 (X = –OCH3, Y = –CH3) | −2.6 | −1.8 | 3.141 | 0.73 |
| 46 (X = –OCH3, Y = –H) | −2.4 | −1.7 | 3.142 | 0.73 |
| 47 (X = –OCH3, Y = –CF3) | −3.1 | −2.4 | 3.073 | 0.82 |
| 48 (X = –OCH3, Y = –CN) | −3.5 | −2.7 | 3.056 | 0.84 |
| 49 (X = –CH3, Y = –NH2) | −2.0 | −1.3 | 3.176 | 0.70 |
| 50 (X = –CH3, Y = –OCH3) | −2.3 | −1.6 | 3.164 | 0.71 |
| 51 (X = –CH3, Y = –CH3) | −2.1 | −1.5 | 3.157 | 0.72 |
| 52 (X = –CH3, Y = –H) | −2.4 | −1.7 | 3.121 | 0.77 |
| 53 (X = –CH3, Y = –CF3) | −3.0 | −2.3 | 3.070 | 0.84 |
| 54 (X = –CH3, Y = –CN) | −3.4 | −2.6 | 3.045 | 0.88 |
| 55 (X = –H, Y = –NH2) | −1.7 | −1.1 | 3.258 | 0.60 |
| 56 (X = –H, Y = –OCH3) | −1.9 | −1.3 | 3.232 | 0.63 |
| 57 (X = –H, Y = –CH3) | −2.1 | −1.5 | 3.238 | 0.62 |
| 58 (X = –H, Y = –H) | −1.9 | −1.4 | 3.223 | 0.64 |
| 59 (X = –H, Y = –CF3) | −2.4 | −1.8 | 3.168 | 0.70 |
| 60 (X = –H, Y = –CN) | −2.7 | −2.7 | 3.138 | 0.74 |
| 61 (X = –CF3, Y = –NH2) | −1.9 | −1.3 | 3.307 | 0.54 |
| 62 (X = –CF3, Y = –OCH3) | −1.7 | −1.2 | 3.287 | 0.56 |
| 63 (X = –CF3, Y = –CH3) | −2.0 | −1.4 | 3.290 | 0.56 |
| 64 (X = –CF3, Y = –H) | −1.8 | −1.2 | 3.280 | 0.57 |
| 65 (X = –CF3, Y = –CF3) | −2.1 | −1.5 | 3.229 | 0.62 |
| 66 (X = –CF3, Y = –CN) | −2.3 | −1.7 | 3.214 | 0.64 |
| 67 (X = –CN, Y = –NH2) | −1.6 | −1.1 | 3.341 | 0.51 |
| 68 (X = –CN, Y = –OCH3) | −1.7 | −1.2 | 3.302 | 0.55 |
| 69 (X = –CN, Y = –CH3) | −1.7 | −1.1 | 3.318 | 0.53 |
| 70 (X = –CN, Y = –H) | −1.8 | −1.2 | 3.308 | 0.54 |
| 71 (X = –CN, Y = –CF3) | −2.0 | −1.4 | 3.277 | 0.57 |
| 72 (X = –CN, Y = –CN) | −2.2 | −1.6 | 3.245 | 0.60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burguera, S.; Frontera, A.; Bauzá, A. Substituent Effects in Tetrel Bonds Involving Aromatic Silane Derivatives: An ab initio Study. Molecules 2023, 28, 2385. https://doi.org/10.3390/molecules28052385
Burguera S, Frontera A, Bauzá A. Substituent Effects in Tetrel Bonds Involving Aromatic Silane Derivatives: An ab initio Study. Molecules. 2023; 28(5):2385. https://doi.org/10.3390/molecules28052385
Chicago/Turabian StyleBurguera, Sergi, Antonio Frontera, and Antonio Bauzá. 2023. "Substituent Effects in Tetrel Bonds Involving Aromatic Silane Derivatives: An ab initio Study" Molecules 28, no. 5: 2385. https://doi.org/10.3390/molecules28052385
APA StyleBurguera, S., Frontera, A., & Bauzá, A. (2023). Substituent Effects in Tetrel Bonds Involving Aromatic Silane Derivatives: An ab initio Study. Molecules, 28(5), 2385. https://doi.org/10.3390/molecules28052385