
On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms


Abstract
:1. Introduction
2. Computational Tools to Analyze the Bonding
3. The General Bonding Picture
4. Types of Noble Gas Compounds
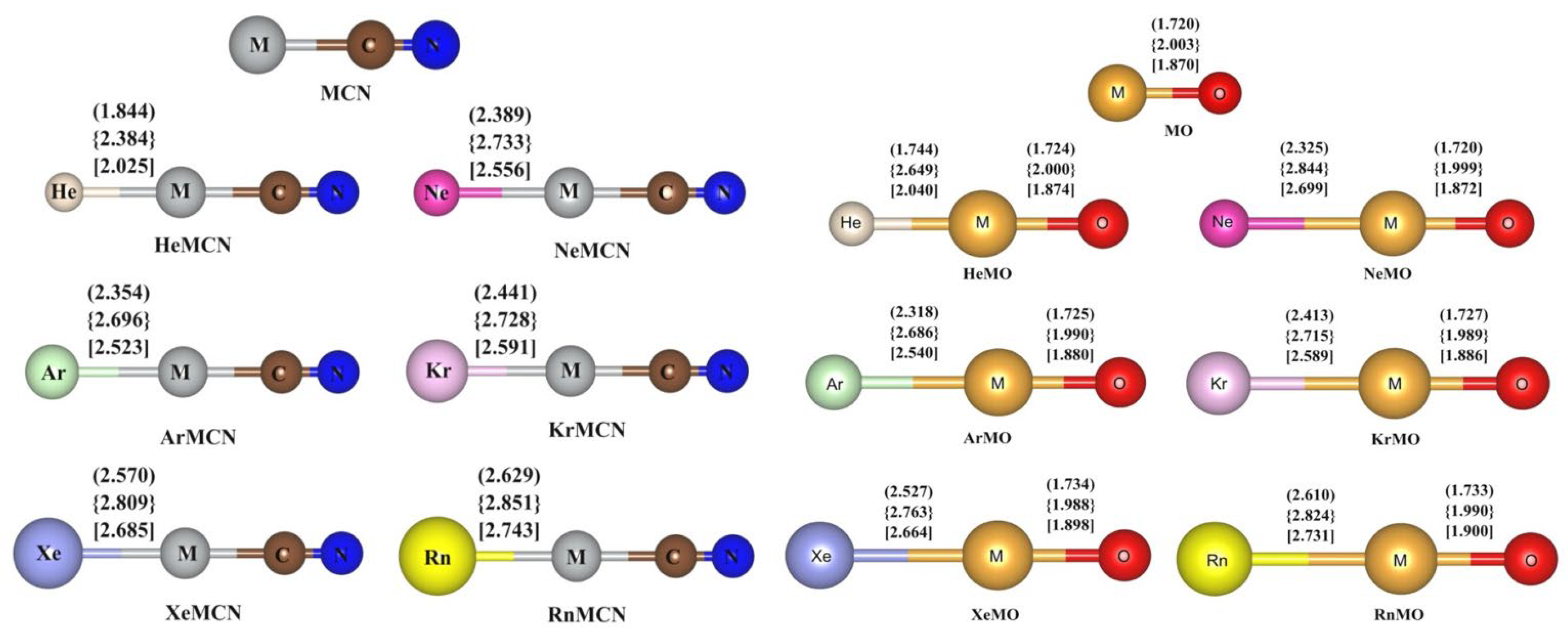
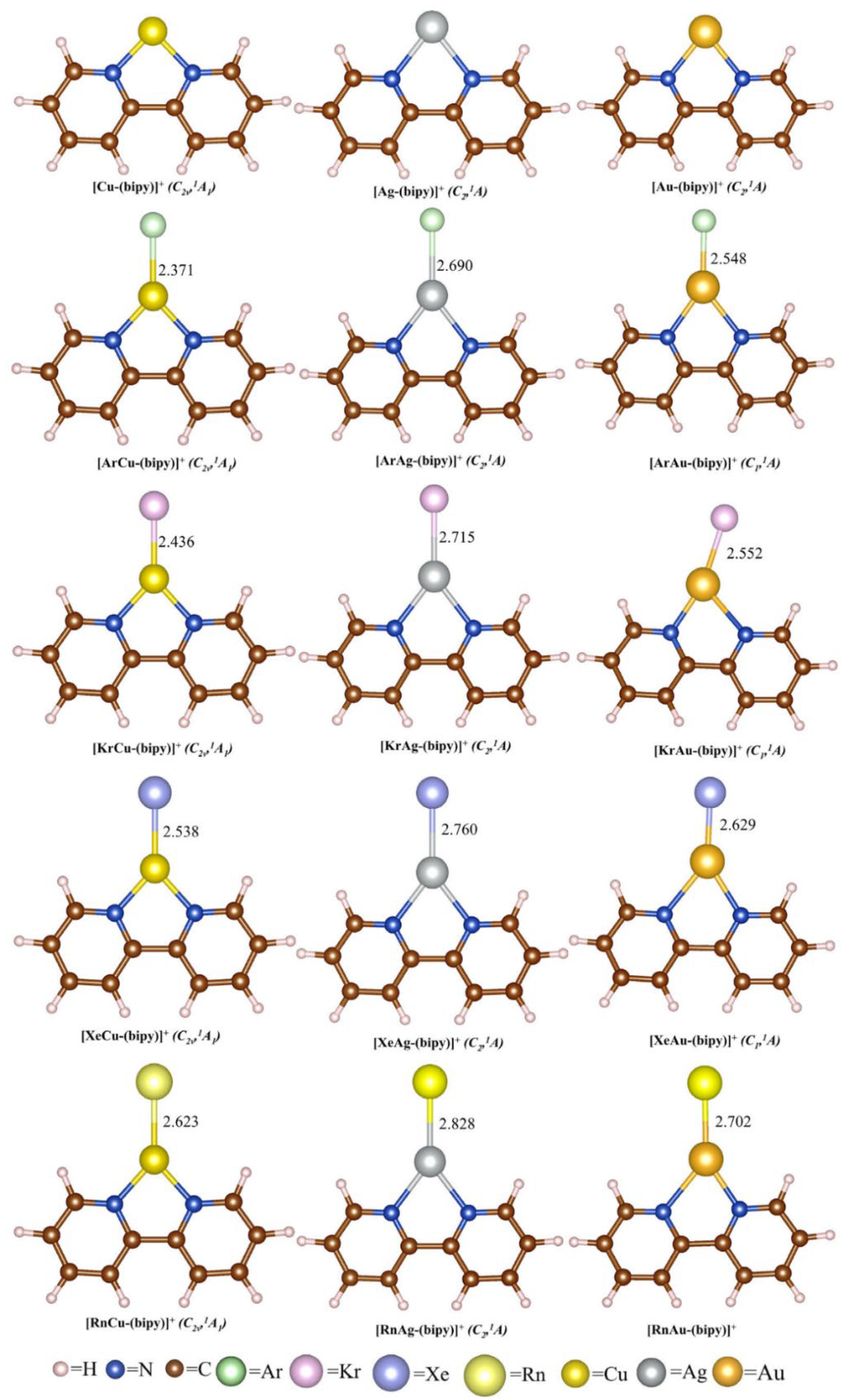
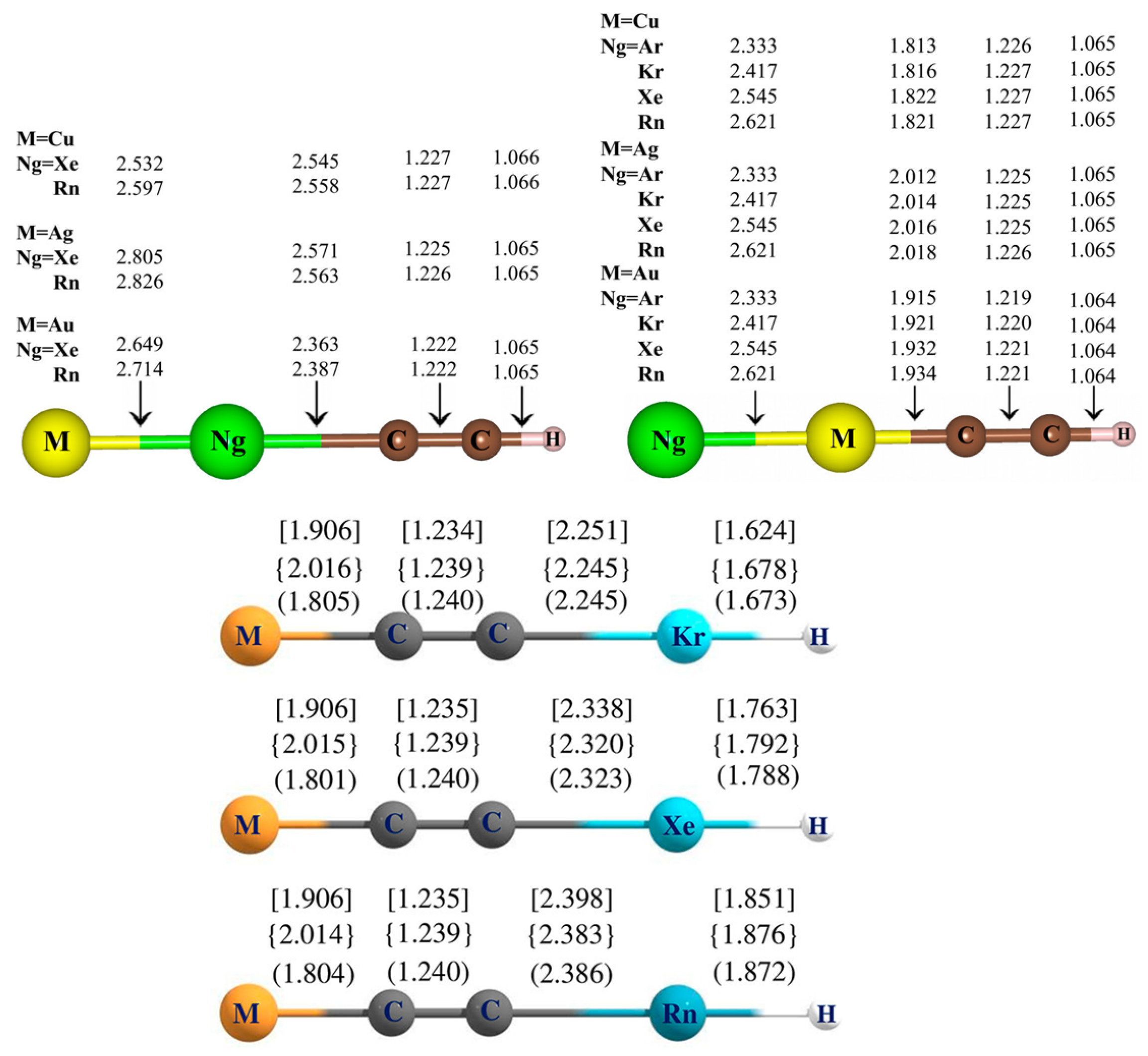
5. Case Studies of Noble Gas−Noble Metal Binding

6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kossel, W. Über Molekülbildung als Frage des Atombaus. Ann. Der Phys. 1916, 354, 229–362. [Google Scholar] [CrossRef] [Green Version]
- Von Antropoff, A. Die Wertigkeit der Edelgase und ihre Stellung im periodischen System. II. Angew. Chem. Int. Ed. 1924, 37, 695–696. [Google Scholar] [CrossRef]
- Pauling, L. The formulas of antimonic acid and the antimonates. J. Am. Chem. Soc. 1933, 55, 1895–1900. [Google Scholar] [CrossRef]
- Bartlett, N.; Lohmann, D. 1005. Fluorides of the noble metals. Part II. Dioxygenyl hexafluoroplatinate (V), O2+[PtF6]-. J. Am. Chem. Soc. 1962, 5253–5261. [Google Scholar] [CrossRef]
- Bartlett, N. Xenon hexafluoroplatinate (V) Xe+[PtF6]-. Proc. Chem. Soc. Lond. 1962, 218, 2933. [Google Scholar]
- Hargittai, I. Neil Bartlett and the first noble-gas compound. Struct. Chem. 2009, 20, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Grandinetti, F. 60 years of chemistry of the noble gases. Nature 2022, 606, 659–661. [Google Scholar] [CrossRef]
- Graham, L.; Graudejus, O.; Jha, N.K.; Bartlett, N. Concerning the nature of XePtF6. Coord. Chem. Rev. 2000, 197, 321–334. [Google Scholar] [CrossRef]
- Streng, A.; Kirshenbaum, A.; Streng, L.; Grosse, A. Preparation of Rare-Gas Fluorides and Oxyfluorides by the Electric Discharge Method and their Properties. In Noble Gas Compounds; Hyman, H.H., Ed.; The University of Chicago Press: Chicago, IL, USA, 1963; pp. 73–80. [Google Scholar]
- Lehmann, J.F.; Mercier, H.P.; Schrobilgen, G.J. The chemistry of krypton. Coord. Chem. Rev. 2002, 233, 1–39. [Google Scholar] [CrossRef]
- Claassen, H.H.; Selig, H.; Malm, J.G. Xenon tetrafluoride. J. Am. Chem. Soc. 1962, 84, 3593. [Google Scholar] [CrossRef]
- Slivnik, J.; Brcic, B.; Volavsek, B.; Marsel, J.; Vrscaj, V.; Smalc, A.; Frlec, B.; Zemljic, Z. Über die Synthese von XeF6. Croat. Chem. Acta 1962, 34, 253. [Google Scholar]
- Turner, J.; Pimentel, G.C. Krypton fluoride: Preparation by the matrix isolation technique. Science 1963, 140, 974–975. [Google Scholar] [CrossRef]
- Nelson, L.Y.; Pimentel, G.C. Infrared detection of xenon dichloride. Inorg. Chem. 1967, 6, 1758–1759. [Google Scholar] [CrossRef]
- Bartlett, N.; Wechsberg, M. The Xenon Difluoride Complexes XeF2 · XeOF4; XeF2 · XeF6 · AsF5 and XeF2 · 2XeF6 · 2AsF5 and Their Relevance to Bond Polarity and Fluoride Ion Donor Ability of XeF2 and XeF6. Z. Anorg. Allg. Chem. 1951, 455, 5–17. [Google Scholar]
- Holloway, J.H.; Hope, E.G. Recent advances in noble-gas chemistry. Adv. Inorg. Chem. 1998, 46, 51–100. [Google Scholar]
- Stein, L. Ionic radon solutions. Science 1970, 168, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Khriachtchev, L.; Pettersson, M.; Runeberg, N.; Lundell, J.; Räsänen, M. A stable argon compound. Nature 2000, 406, 874. [Google Scholar] [CrossRef]
- Frenking, G. Another noble gas conquered. Nature 2000, 406, 836. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, X. Infrared Spectra of NgBeS (Ng = Ne, Ar, Kr, Xe) and BeS2 in Noble-Gas Matrices. J. Phys. Chem. A 2013, 117, 1508–1513. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, M.; Zhou, M.; Andrada, D.M.; Frenking, G. Experimental and Theoretical Studies of the Infrared Spectra and Bonding Properties of NgBeCO3 and a Comparison with NgBeO (Ng = He, Ne, Ar, Kr, Xe). J. Phys. Chem. A 2014, 119, 2543–2552. [Google Scholar] [CrossRef]
- Yu, W.; Liu, X.; Xu, B.; Xing, X.; Wang, X. Infrared Spectra of Novel NgBeSO2 Complexes (Ng = Ne, Ar, Kr, Xe) in Low Temperature Matrixes. J. Phys. Chem. A 2016, 120, 8590–8598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, W.L.; Zhao, L.; Chen, M.; Zhou, M.; Li, J.; Frenking, G. A Very Short Be-Be Distance but No Bond: Synthesis and Bonding Analysis of Ng-Be2O2-Ng0 (Ng, Ng = Ne, Ar, Kr, Xe). Chem. Eur. J. 2017, 23, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Oganov, A.R.; Goncharov, A.F.; Stavrou, E.; Lobanov, S.; Saleh, G.; Qian, G.-R.; Zhu, Q.; Gatti, C.; Deringer, V.L.; et al. A stable compound of helium and sodium at high pressure. Nat. Chem. 2017, 9, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Pyykko, P. Predicted chemical bonds between rare gases and Au. J. Am. Chem. Soc. 1995, 117, 2067–2070. [Google Scholar] [CrossRef]
- Schr€oder, D.; Schwarz, H.; Hrusak, J.; Pyykko, P. Cationic gold (I) complexes of xenon and of ligands containing the donor atoms oxygen, nitrogen, phosphorus, and sulfur. Inorg. Chem. 1998, 37, 624–632. [Google Scholar] [CrossRef]
- Read, J.P.; Buckingham, A.D. Covalency in ArAu+ and Related Species? J. Am. Chem. Soc. 1997, 119, 9010–9013. [Google Scholar] [CrossRef]
- Evans, C.J.; Lesarri, A.; Gerry, M.C.L. Noble Gas—Metal Chemical Bonds. Microwave Spectra, Geometries, and Nuclear Quadrupole Coupling Constants of Ar-AuCl and Kr-AuCl. J. Am. Chem. Soc. 2000, 122, 6100–6105. [Google Scholar] [CrossRef]
- Seidel, S.; Seppelt, K. Xenon as a complex ligand: The tetra Xenono Gold (II) cation in AuXe42+(Sb2F11−)2. Science 2000, 290, 117–118. [Google Scholar] [CrossRef]
- Cooke, S.A.; Gerry, M.C.L. Insights into the Xenon–Silver Halide Interaction from a Rotational Spectroscopic Study of XeAgF and XeAgCl. Phys. Chem. Chem. Phys. 2004, 6, 3248–3256. [Google Scholar] [CrossRef]
- Cooke, S.A.; Gerry, M.C.L. XeAuF. J. Am. Chem. Soc. 2004, 126, 17000–17008. [Google Scholar] [CrossRef]
- Thomas, J.M.; Walker, N.R.; Cooke, S.A.; Gerry, M.C.L. Microwave Spectra and Structures of KrAuF, KrAgF, and KrAgBr; 83Kr Nuclear Quadrupole Coupling and the Nature of Noble Gas−Noble Metal Halide Bonding. J. Am. Chem. Soc. 2004, 126, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.M.; Cooke, S.A.; Gerry, M.C.L. Rotational Spectra, Structures, Hyperfine Constants, and the Nature of the Bonding of KrCuF and KrCuCl. Inorg. Chem. 2004, 43, 3871–3881. [Google Scholar] [CrossRef]
- Michaud, J.M.; Gerry, M.C.L. XeCu Covalent Bonding in XeCuF and XeCuCl, Characterized by Fourier Transform Microwave Spectroscopy Supported by Quantum Chemical Calculations. J. Am. Chem. Soc. 2006, 128, 7613–7621. [Google Scholar] [CrossRef] [PubMed]
- Lantto, P.; Vaara, J. Calculations of nuclear quadrupole coupling in noble gas–noble metal fluorides: Interplay of relativistic and electron correlation effects. J. Chem. Phys. 2006, 125, 174315. [Google Scholar] [CrossRef]
- Mou, C.H.; Witek, H.A. Theoretical study of noble-gas containing metal halides. J. Chem. Phys. 2008, 129, 244310. [Google Scholar] [CrossRef]
- Zou, W.; Liu, Y.; Boggs, J.E. Theoretical study of RgMF (Rg= He, Ne; M= Cu, Ag, Au): Bonded structures of helium. Chem. Phys. Lett. 2009, 482, 207–210. [Google Scholar] [CrossRef]
- Chen, R.; Zhu, H.; Xie, D.Q.; GuoSen, G.S. Theoretical prediction of the noble gas complexes HeAuF and NeAuF. Sci. Chin. Ser. B Chem. 2009, 52, 1987–1990. [Google Scholar] [CrossRef]
- Evans, C.J.; Wright, T.G.; Gardner, A.M. Geometries and Bond Energies of the He− MX, Ne− MX, and Ar− MX (M= Cu, Ag, Au; X= F, Cl) Complexes. J. Phys. Chem. A 2010, 114, 4446–4454. [Google Scholar] [CrossRef]
- Beyhan, S.M.; Gotz, A.W.; Jacob, C.R.; Visscher, L. The weak covalent bond in NgAuF (Ng = Ar, Kr, Xe): A challenge for subsystem density functional theory. J. Chem. Phys. 2010, 132, 044114. [Google Scholar] [CrossRef]
- Wang, X.; Andrews, L.; Brosi, F.; Riedel, S. Matrix Infrared Spectroscopy and Quantum-Chemical Calculations for the Coinage-Metal Fluorides: Comparisons of Ar-AuF, Ne-AuF, and Molecules MF2 and MF3. Chem. Eur. J. 2013, 19, 1397–1409. [Google Scholar] [CrossRef]
- Zhang, P.X.; Zhao, Y.F.; Hao, F.Y.; Zhang, G.H.; Song, X.D.; Li, X.Y. Bonding analysis for NgMOH (Ng= Ar, Kr and Xe; M= Cu and Ag). Mol. Phys. 2008, 106, 1007–1014. [Google Scholar] [CrossRef]
- Zhang, P.X.; Zhao, Y.F.; Hao, F.Y.; Li, X.Y. Bonding analysis for NgAuOH (Ng= Kr, Xe). Int. J. Quantum Chem. 2008, 108, 937–944. [Google Scholar] [CrossRef]
- Ghanty, T.K. Insertion of noble-gas atom (Kr and Xe) into noble-metal molecules (AuF and AuOH): Are they stable? J. Chem. Phys. 2005, 123, 074323. [Google Scholar] [CrossRef]
- Ghanty, T.K. How strong is the interaction between a noble gas atom and a noble metal atom in the insertion compounds MNgF (M=Cu and Ag, and Ng=Ar, Kr, and Xe)? J. Chem. Phys. 2006, 124, 124304. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. Molecular fragments or chemical bonds. Acc. Chem. Res. 1975, 8, 34–40. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Fonseca, G.C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Haaland, A.; Helgaker, T.U.; Ruud, K.; Shorokhov, D.J. Should Gaseous BF3 and SiF4Be Described as Ionic Compounds? J. Chem. Educ. 2000, 77, 1076. [Google Scholar] [CrossRef]
- De Proft, F.; Van Alsenoy, C.; Peeters, A.; Langenaeker, W.; Geerlings, P. Atomic charges, dipole moments, and Fukui functions using the Hirshfeld partitioning of the electron density. J. Comput. Chem. 2002, 23, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Introduction To Computational Chemistry; John Wiley and Sons: New York, NY, USA, 1999. [Google Scholar]
- Brom, J.M.; Schmitz, B.J.; Thompson, J.D.; Cramer, C.J.; Truhlar, D.G. A Class IV Charge Model for Boron Based on Hybrid Density Functional Theory. J. Phys. Chem. A 2003, 107, 6483–6488. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Matta, C.F. Atoms in molecules as non-overlapping, bounded, space-filling open quantum systems. Found. Chem. 2013, 15, 253–276. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Zou, P.F. An atomic population as the expectation value of a quantum observable. Chem. Phys. Lett. 1992, 191, 54–58. [Google Scholar]
- Borocci, S.; Grandinetti, F.; Sanna, N. Noble-gas compounds: A general procedure of bonding analysis. J. Chem. Phys. 2022, 156, 014104. [Google Scholar] [CrossRef]
- Makarewicz, E.; Gordon, A.J.; Berski, S. Nature of the Bonding in the AuNgX (Ng = Ar, Kr, Xe; X = F, Cl, Br, I) Molecules. Topological Study on Electron Density and the Electron Localization Function (ELF). J. Phys. Chem. A 2015, 119, 2401–2412. [Google Scholar] [CrossRef]
- Borocci, S.; Giordani, M.; Grandinetti, F. Bonding Motifs of Noble-Gas Compounds as Described by the Local Electron Energy Density. J. Phys. Chem. A 2015, 119, 6528–6541. [Google Scholar] [CrossRef]
- Borocci, S.; Grandinetti, F.; Nunzi, F.; Sanna, N. Classifying the chemical bonds involving the noble-gas atoms. New J. Chem. 2020, 44, 14536–14550. [Google Scholar] [CrossRef]
- Michalak, A.; Mitoraj, M.; Ziegler, T. Bond orbitals from chemical valence theory. J. Phys. Chem. A 2008, 112, 1933–1939. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar]
- Pan, S.; Saha, R.; Chattaraj, P.K. Exploring the nature of silicon-noble gas bonds in H3SiNgNSi and HSiNgNSi compounds (Ng = Xe, Rn). Int. J. Mol. Sci. 2015, 16, 6402–6418. [Google Scholar] [CrossRef] [Green Version]
- Tonner, R.; Frenking, G. Divalent carbon (0) chemistry, part 1: Parent compounds. Chem. Eur. J. 2008, 14, 3260–3272. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Autschbach, J.; Bashford, D.; B_erces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; et al. ADF2013.01; SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Pal, R.; Patra, S.G.; Chattaraj, P.K. Can a chemical bond be exclusively covalent or ionic? J. Chem. Sci. 2022, 134, 108. [Google Scholar] [CrossRef]
- Li, T.H.; Liu, Y.L.; Lin, R.J.; Yeh, T.Y.; Hu, W.P. On the stability of noble gas molecules. Chem. Phys. Lett. 2007, 434, 38–41. [Google Scholar] [CrossRef]
- Krapp, A.; Frenking, G. Is this a chemical bond? a theoretical study of Ng2@C60 (Ng= He, Ne, Ar, Kr, Xe). Chem. Eur. J. 2007, 13, 8256–8270. [Google Scholar] [CrossRef]
- Khatua, M.; Pan, S.; Chattaraj, P.K. Movement of Ng2 molecules confined in a C60 cage: An ab initio molecular dynamics study. Chem. Phys. Lett. 2014, 610, 351–356. [Google Scholar] [CrossRef]
- Pan, S.; Gupta, A.; Saha, R.; Merino, G.; Chattaraj, P.K. A coupled-cluster study on the noble gas binding ability of metal cyanides versus metal halides (metal = Cu, Ag, Au). J. Comput. Chem. 2015, 36, 2168–2176. [Google Scholar] [CrossRef]
- Pan, S.; Saha, R.; Kumar, A.; Gupta, A.; Merino, G.; Chattaraj, P.K. A noble interaction: An assessment of noble gas binding ability of metal oxides (metal = Cu, Ag, Au). Int. J. Quantum Chem. 2016, 116, 1016–1024. [Google Scholar] [CrossRef]
- Ghara, M.; Pan, S.; Deb, J.; Kumar, A.; Sarkar, U.; Chattaraj, P.K. A computational study on structure, stability and bonding in Noble Gas bound metal Nitrates, Sulfates and Carbonates (metal = Cu, Ag, Au). J. Chem. Sci. 2016, 128, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Pople, J.A.; Head-Gordon, M.; Raghavachari, K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J. Chem. Phys. 1987, 87, 5968–5975. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Dunning Jr, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H., Jr. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+ H2→ H2+ H reaction. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar]
- Jana, G.; Saha, R.; Pan, S.; Kumar, A.; Merino, G.; Chattaraj, P.K. Noble gas binding ability of metal-bipyridine monocationic complexes (metal = Cu, Ag, Au): A computational study. ChemistrySelect 2016, 1, 5842–5849. [Google Scholar] [CrossRef]
- Jana, G.; Pan, S.; Merino, G.; Chattaraj, P.K. MNgCCH (M = Cu, Ag, Au; Ng = Xe, Rn): The First Set of Compounds with M–Ng–C Bonding Motif. J. Phys. Chem. A 2017, 121, 6491–6499. [Google Scholar] [CrossRef]
- Jana, G.; Pan, S.; Merino, G.; Chattaraj, P.K. Noble Gas Inserted Metal Acetylides (Metal = Cu, Ag, Au). J. Phys. Chem. A 2018, 122, 7391–7401. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Hybrid Meta Density Functional Theory Methods for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions: The MPW1B95 and MPWB1K Models and Comparative Assessments for Hydrogen Bonding and van der Waals Interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Moller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning Jr, T.H. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.G. The principle of maximum hardness. Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar] [CrossRef]
- Pearson, R.G. Maximum Chemical and Physical Hardness. J. Chem. Educ. 1999, 76, 267. [Google Scholar] [CrossRef]
- Pan, S.; Solà, M.; Chattaraj, P.K. On the validity of the maximum hardness principle and the minimum electrophilicity principle during chemical reactions. J. Phys. Chem. A 2013, 117, 1843–1852. [Google Scholar] [CrossRef]
- Chamorro, E.; Chattaraj, P.K.; Fuentealba, P. Variation of the electrophilicity index along the reaction path. J. Phys. Chem. A 2003, 107, 7068–7072. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Elango, M.; Subramanian, V.; Chattaraj, P.K. Variation of electrophilicity during molecular vibrations and internal rotations. Theor. Chem. Acc. 2005, 113, 257–266. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. A minimum electrophilicity perspective of the HSAB principle. Indian J. Phys. 2007, 81, 871–879. [Google Scholar]
- Miranda-Quintana, R.A.; Chattaraj, P.K.; Ayers, P.W. Finite temperature grand canonical ensemble study of the minimum electrophilicity principle. J. Chem. Phys. 2017, 147, 124103. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| System | BCP | ρ(rc) | ∇2ρ(rc) | G(rc) | V(rc) | H(rc) | G(rc)/ρ(rc) | ELF |
|---|---|---|---|---|---|---|---|---|
| HF | H-F | 0.412 | −0.339 | 0.999 | −0.105 | −0.947 | 2.425 | 0.977 |
| HCl | H-Cl | 0.310 | −0.157 | 0.143 | −0.394 | −0.394 | 0.461 | 0.999 |
| HBr | H-Br | 0.193 | −0.420 | 0.530 | −0.211 | −0.158 | 2.747 | 0.924 |
| LiF | Li-F | 0.079 | 0.705 | 0.163 | −0.149 | 0.014 | 2.056 | 0.062 |
| LiCl | Li-Cl | 0.047 | 0.274 | 0.065 | −0.062 | 0.003 | 1.391 | 0.067 |
| LiBr | Li-Br | 0.040 | 0.198 | 0.048 | −0.046 | 0.002 | 1.210 | 0.070 |
| NaF | Na-F | 0.054 | 0.440 | 0.101 | −0.091 | 0.009 | 1.865 | 0.046 |
| NaCl | Na-Cl | 0.035 | 0.200 | 0.045 | −0.040 | 0.005 | 1.279 | 0.054 |
| NaBr | Na-Br | 0.030 | 0.150 | 0.034 | −0.030 | 0.004 | 1.109 | 0.059 |
| KF | K-F | 0.052 | 0.291 | 0.071 | −0.068 | 0.002 | 1.353 | 0.081 |
| KCl | K-Cl | 0.031 | 0.131 | 0.031 | −0.285 | 0.002 | 0.984 | 0.077 |
| KBr | K-Br | 0.027 | 0.099 | 0.023 | −0.022 | 0.002 | 0.868 | 0.080 |
| MgF2 | Mg-F | 0.082 | 0.759 | 0.179 | −0.168 | 0.011 | 2.177 | 0.058 |
| MgCl2 | Mg-Cl | 0.055 | 0.311 | 0.077 | −0.076 | 0.001 | 1.392 | 0.082 |
| MgBr2 | Mg-Br | 0.048 | 0.222 | 0.056 | −0.057 | −0.001 | 1.171 | 0.095 |
| CaF2 | Ca-F | 0.045 | 0.150 | 0.041 | −0.044 | −0.003 | 0.909 | 0.109 |
| CaCl2 | Ca-Cl | 0.052 | 0.203 | 0.054 | −0.058 | −0.004 | 1.042 | 0.129 |
| CaBr2 | Ca-Br | 0.045 | 0.150 | 0.041 | −0.044 | −0.003 | 0.909 | 0.138 |
| H2 | H-H | 0.250 | −0.982 | 0.548 | −0.246 | −0.246 | 2.195 | 0.999 |
| N2 | N-N | 0.720 | −0.287 | 0.629 | −0.198 | −0.135 | 0.875 | 0.874 |
| O2 | O-O | 0.546 | −0.960 | 0.492 | −0.122 | −0.732 | 0.900 | 0.820 |
| F2 | F-F | 0.294 | 0.564 | 0.287 | −0.433 | −0.146 | 0.977 | 0.628 |
| Cl2 | Cl-Cl | 0.154 | −0.205 | 0.711 | −0.147 | −0.763 | 4.634 | 0.759 |
| CO | C-O | 0.508 | 0.724 | 0.114 | −0.209 | −0.955 | 0.223 | 0.401 |
| H2O | O-H | 0.362 | −0.241 | 0.729 | −0.749 | −0.676 | 2.015 | 0.981 |
| NH3 | N-H | 0.337 | −0.157 | 0.603 | −0.514 | −0.454 | 1.790 | 0.984 |
| CH4 | C-H | 0.278 | −0.947 | 0.428 | −0.322 | −0.279 | 1.542 | 0.984 |
| C2H4 | C-C | 0.359 | −0.112 | 0.147 | −0.574 | −0.428 | 0.409 | 0.926 |
| C2H2 | C-C | 0.426 | −0.125 | 0.314 | −0.940 | −0.626 | 0.737 | 0.830 |
| ArCuNO3 | Ar-Cu | 0.042 | 0.209 | 0.056 | −0.059 | −0.003 | 1.312 | 0.066 |
| Cu-N | 0.069 | 0.322 | 0.096 | −0.111 | −0.015 | 1.397 | 0.106 | |
| KrCuNO3 | Kr-Cu | 0.053 | 0.188 | 0.053 | −0.058 | −0.006 | 1.000 | 0.096 |
| Cu-N | 0.068 | 0.320 | 0.095 | −0.110 | −0.015 | 1.394 | 0.106 | |
| XeCuNO3 | Xe-Cu | 0.049 | 0.157 | 0.047 | −0.055 | −0.008 | 0.957 | 0.139 |
| Cu-N | 0.068 | 0.317 | 0.094 | −0.109 | −0.148 | 1.391 | 0.105 | |
| RnCuNO3 | Rn-Cu | 0.047 | 0.136 | 0.042 | −0.049 | −0.007 | 0.889 | 0.149 |
| Cu-O | 0.067 | 0.315 | 0.093 | −0.108 | −0.015 | 1.389 | 0.104 | |
| ArAgNO3 | Ar-Ag | 0.026 | 0.110 | 0.028 | −0.028 | 0.000 | 1.053 | 0.055 |
| Ag-N | 0.057 | 0.247 | 0.068 | −0.075 | −0.007 | 1.202 | 0.111 | |
| KrAgNO3 | Kr-Ag | 0.033 | 0.116 | 0.031 | −0.339 | −0.002 | 0.943 | 0.090 |
| Ag-N | 0.057 | 0.245 | 0.068 | −0.074 | −0.007 | 1.202 | 0.110 | |
| XeAgNO3 | Xe-Ag | 0.039 | 0.111 | 0.033 | −0.037 | −0.005 | 0.833 | 0.137 |
| Ag-N | 0.056 | 0.244 | 0.068 | −0.074 | −0.007 | 1.201 | 0.110 | |
| RnAgNO3 | Rn-Ag | 0.039 | 0.100 | 0.030 | −0.036 | −0.005 | 0.776 | 0.153 |
| Ag-N | 0.056 | 0.243 | 0.067 | −0.074 | −0.006 | 1.200 | 0.109 | |
| Ar2Ag2SO4 | Ar-Ag | 0.024 | 0.102 | 0.025 | −0.025 | 0.002 | 1.037 | 0.051 |
| Ag-S | 0.055 | 0.245 | 0.067 | −0.073 | −0.006 | 1.221 | 0.104 | |
| Kr2Ag2SO4 | Kr-Ag | 0.032 | 0.110 | 0.030 | −0.318 | −0.214 | 0.937 | 0.085 |
| Ag-S | 0.055 | 0.245 | 0.067 | −0.073 | −0.006 | 1.220 | 0.104 | |
| Xe2Ag2SO4 | Xe-Ag | 0.038 | 0.109 | 0.032 | −0.036 | −0.004 | 0.834 | 0.132 |
| Ag-S | 0.055 | 0.243 | 0.067 | −0.073 | −0.006 | 1.218 | 0.104 | |
| Rn2Ag2SO4 | Rn-Ag | 0.038 | 0.098 | 0.030 | −0.035 | −0.005 | 0.778 | 0.148 |
| Ag-S | 0.055 | 0.242 | 0.066 | −0.072 | −0.006 | 1.217 | 0.103 |
| System | Bonds | ΔEpauli | ΔEelstat | ΔEorb | ΔEdisp | ΔEint | %Covalent | %Ionic |
|---|---|---|---|---|---|---|---|---|
| HF | H-F | 0.0 | −232.76 | −145.92 | −0.11 | −378.79 | 38.53 | 61.47 |
| HCl | H-Cl | 0.0 | −159.91 | −178.63 | −0.35 | −338.89 | 52.76 | 47.24 |
| HBr | H-Br | −0.01 | −141.69 | −184.5 | −0.43 | −326.61 | 56.56 | 43.44 |
| LiF | Li-F | 41.38 | −210.53 | −24.01 | −0.28 | −193.44 | 10.24 | 89.76 |
| LiCl | Li-Cl | 29.16 | −160.53 | −26.33 | −0.91 | −158.62 | 14.09 | 85.91 |
| LiBr | Li-Br | 27.72 | −147.42 | −26.89 | −1.07 | −147.66 | 15.43 | 84.57 |
| NaF | Na-F | 31.09 | −179.91 | −12.91 | −0.34 | −162.06 | 6.70 | 93.30 |
| NaCl | Na-Cl | 25.45 | −146.21 | −14.95 | −1.03 | −136.74 | 9.28 | 90.72 |
| NaBr | Na-Br | 24.6 | −137.15 | −16.36 | −1.19 | −130.09 | 10.66 | 89.34 |
| KF | K-F | 39.61 | −163.92 | −21.37 | −0.43 | −146.11 | 11.53 | 88.47 |
| KCl | K-Cl | 29.18 | −132.33 | −15.93 | −1.21 | −120.29 | 10.74 | 89.26 |
| KBr | K-Br | 28.28 | −125.09 | −15.59 | −1.36 | −113.75 | 11.08 | 88.92 |
| MgF2 | Mg-F | 95.84 | −734.88 | −82.7 | −0.81 | −722.56 | 10.12 | 89.88 |
| MgCl2 | Mg-Cl | 77.01 | −582.58 | −120.12 | −2.52 | −628.21 | 17.09 | 82.91 |
| MgBr2 | Mg-Br | 69.11 | −528.17 | −141.21 | −2.93 | −603.2 | 21.10 | 78.90 |
| CaF2 | Ca-F | 121.9 | −666.31 | −79.32 | −0.98 | −624.71 | 10.64 | 89.36 |
| CaCl2 | Ca-Cl | 101.73 | −542.27 | −92.56 | −2.82 | −535.92 | 14.58 | 85.42 |
| CaBr2 | Ca-Br | 95.85 | −501.53 | −103.75 | −0.08 | −509.51 | 17.14 | 82.86 |
| H2 | H-H | 226.24 | 6.19 | −410.2 | −0.09 | −177.86 | 101.53 | −1.53 |
| N2 | N-N | 1683.65 | −320.32 | −1830.98 | −0.47 | −468.11 | 85.11 | 14.89 |
| O2 | O-O | 956.56 | −239.07 | −849.78 | −0.29 | −132.58 | 78.04 | 21.96 |
| F2 | F-F | 335.12 | −98.82 | −381.46 | −0.17 | −145.34 | 79.42 | 20.58 |
| Cl2 | Cl-Cl | 298.94 | −109.56 | −303.5 | −1.29 | −115.41 | 73.48 | 26.52 |
| CO | C-O | 1362.11 | −277.75 | −1502.04 | −0.46 | −418.14 | 84.39 | 15.61 |
| H2O | O-H | 409.15 | −69.44 | −534.56 | −0.21 | −195.04 | 88.50 | 11.50 |
| NH3 | N-H | 418.92 | −84.04 | −514.21 | −0.3 | −179.63 | 85.95 | 14.05 |
| CH4 | C-H | 387.57 | −55.77 | −511.01 | −0.33 | −179.53 | 90.16 | 9.84 |
| C2H4 | C-C | 1154.17 | −453.35 | −908.61 | −1.42 | −209.21 | 66.71 | 33.29 |
| C2H2 | C-C | 1373.8 | −463.1 | −1210.62 | −0.89 | −300.81 | 72.33 | 27.67 |
| ArCuNO3 | Ar-Cu | 18 | −13.27 | −11.36 | −1.38 | −8.01 | 46.12 | 53.88 |
| Cu-N | 67.77 | −186.74 | −47.74 | −2.15 | −168.87 | 20.36 | 79.64 | |
| KrCuNO3 | Kr-Cu | 22.86 | −17.78 | −15.01 | −1.7 | −11.63 | 45.78 | 54.22 |
| Cu-N | 67.03 | −182.95 | −47.83 | −2.24 | −166 | 20.73 | 79.27 | |
| XeCuNO3 | Xe-Cu | 28.91 | −23.59 | −18.59 | −2.19 | −15.45 | 44.07 | 55.93 |
| Cu-N | 66.82 | −178.15 | −48.15 | −2.36 | −161.84 | 21.28 | 78.72 | |
| RnCuNO3 | Rn-Cu | 29.16 | −24.27 | −18.69 | −2.31 | −16.12 | 43.51 | 56.49 |
| Cu-O | 66.98 | −175.24 | −48.22 | −2.41 | −158.89 | 21.58 | 78.42 | |
| ArAgNO3 | Ar-Ag | 81.38 | −6.34 | −5.43 | −1.46 | 68.15 | 46.13 | 53.87 |
| Ag-N | 60.16 | −167.49 | −38.87 | −2.38 | −148.59 | 18.84 | 81.16 | |
| KrAgNO3 | Kr-Ag | 14.98 | −11.05 | −9.14 | −1.8 | −7.01 | 45.27 | 54.73 |
| Ag-N | 59.13 | −164.45 | −38.93 | −2.5 | −146.74 | 19.14 | 80.86 | |
| XeAgNO3 | Xe-Ag | 23.11 | −17.96 | −13.1 | −2.32 | −10.26 | 42.18 | 57.82 |
| Ag-N | 58.81 | −160.71 | −39.21 | −2.57 | −143.68 | 19.61 | 80.39 | |
| RnAgNO3 | Rn-Ag | 25.04 | −19.98 | −13.96 | −2.43 | −11.33 | 41.13 | 58.87 |
| Ag-N | 58.55 | −158.24 | −39.25 | −2.61 | −141.55 | 19.87 | 80.13 | |
| Ar2Ag2SO4 | Ar-Ag | 8.27 | −5.82 | −4.55 | −1.5 | −3.6 | 43.88 | 56.12 |
| Ag-S | 135.09 | −522.47 | −73.49 | −6.9 | −467.78 | 12.33 | 87.67 | |
| Kr2Ag2SO4 | Kr-Ag | 14.25 | −10.55 | −8.04 | −1.88 | −6.22 | 43.25 | 56.75 |
| Ag-S | 134.31 | −511.84 | −76.42 | −7.27 | −461.22 | 12.99 | 87.01 | |
| Xe2Ag2SO4 | Xe-Ag | 22.95 | −17.88 | −11.83 | −2.45 | −9.2 | 39.82 | 60.18 |
| Ag-S | 133.17 | −496.82 | −80.74 | −7.52 | −451.9 | 13.98 | 86.02 | |
| Rn2Ag2SO4 | Rn-Ag | 24.84 | −19.85 | −12.44 | −2.59 | −10.05 | 38.53 | 61.47 |
| Ag-S | 132.96 | −488.06 | −82.97 | −7.65 | −445.72 | 14.53 | 85.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, R.; Chattaraj, P.K. On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms. Molecules 2023, 28, 3253. https://doi.org/10.3390/molecules28073253
Pal R, Chattaraj PK. On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms. Molecules. 2023; 28(7):3253. https://doi.org/10.3390/molecules28073253
Chicago/Turabian StylePal, Ranita, and Pratim Kumar Chattaraj. 2023. "On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms" Molecules 28, no. 7: 3253. https://doi.org/10.3390/molecules28073253
APA StylePal, R., & Chattaraj, P. K. (2023). On the Nature of the Partial Covalent Bond between Noble Gas Elements and Noble Metal Atoms. Molecules, 28(7), 3253. https://doi.org/10.3390/molecules28073253