
Glycoprotein In Vitro N-Glycan Processing Using Enzymes Expressed in E. coli


Abstract
:1. Introduction
2. Results and Discussion
2.1. Glycoprotein In Vitro N-Glycan Processing Route Design and Enzyme Selection
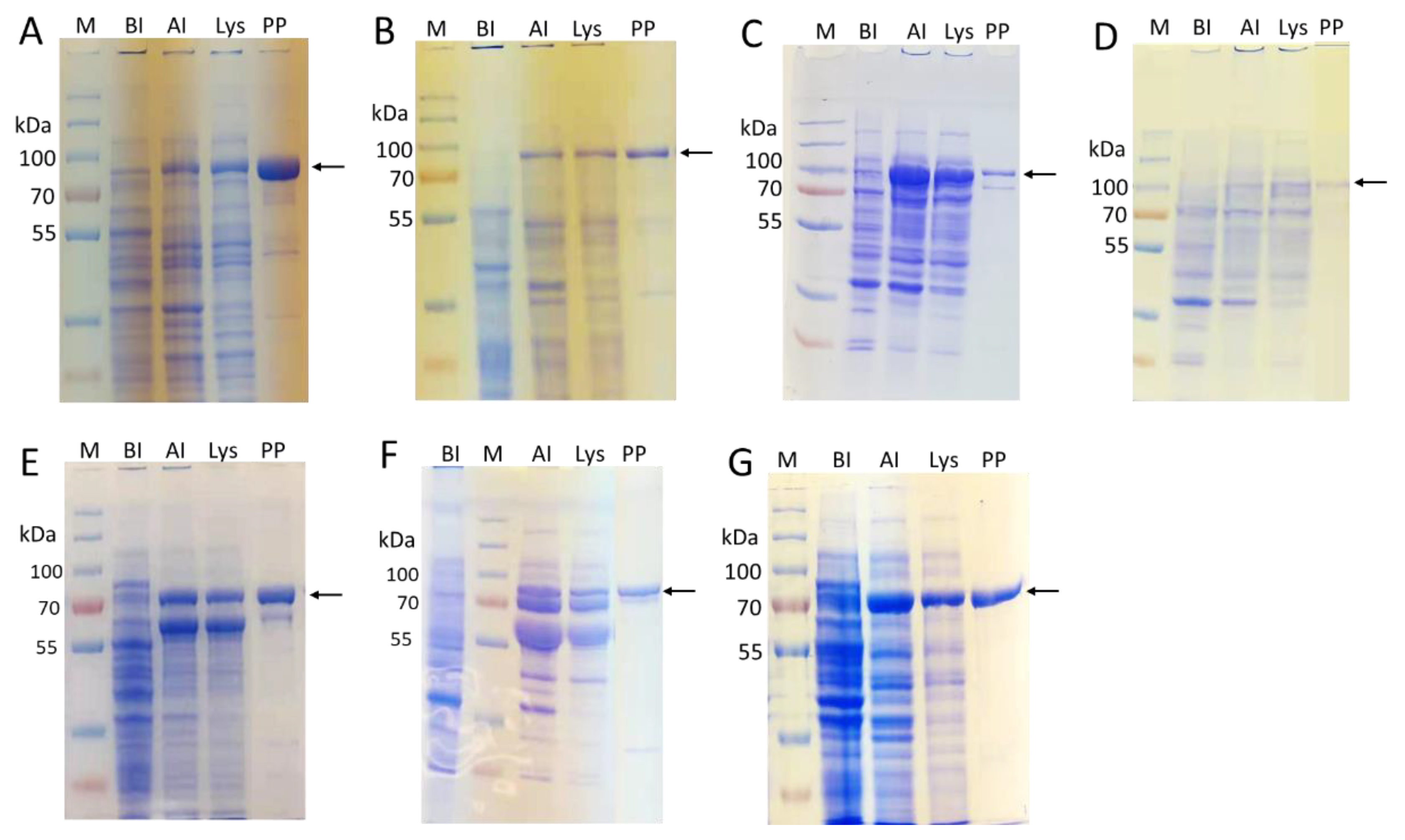
2.2. Enzyme Cloning and Expression
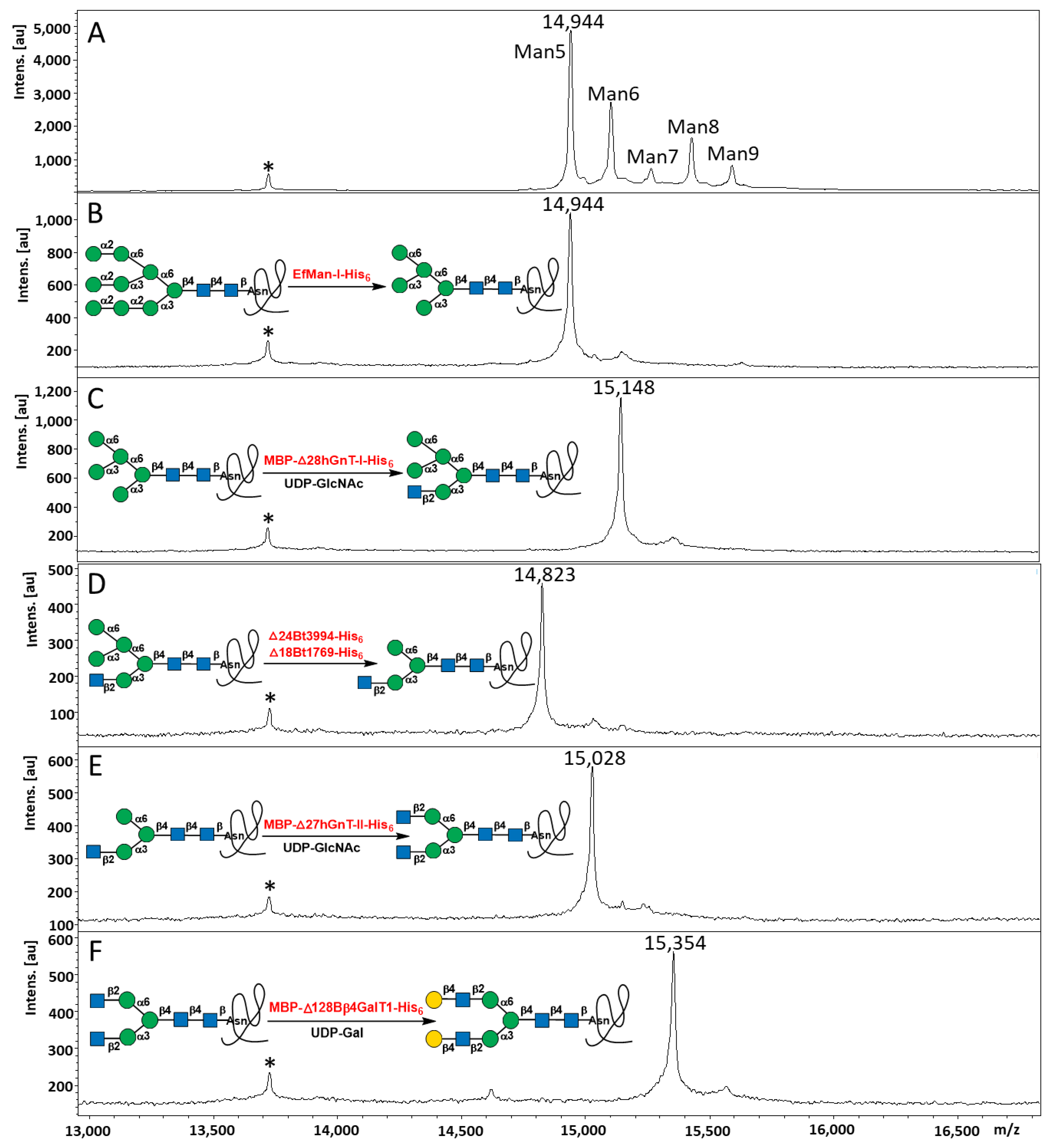
2.3. Step-Wise Reactions and Enzyme Activty Determination Using Glycoprotein Substrates
2.4. Multi-Step OPME N-Glycan Processing
3. Conclusions
4. Materials and Methods
4.1. Materials and Instruments
4.2. Cloning
4.3. Enzyme Expression and Purification
4.4. Stepwise Enzymatic Reaction Using RNase B as The Substrate
4.5. OPME Reactions for N-Glycan Processing of RNase B
4.6. MALDI-TOF MS Analyses of RNase B samples and the Released N-Glycans
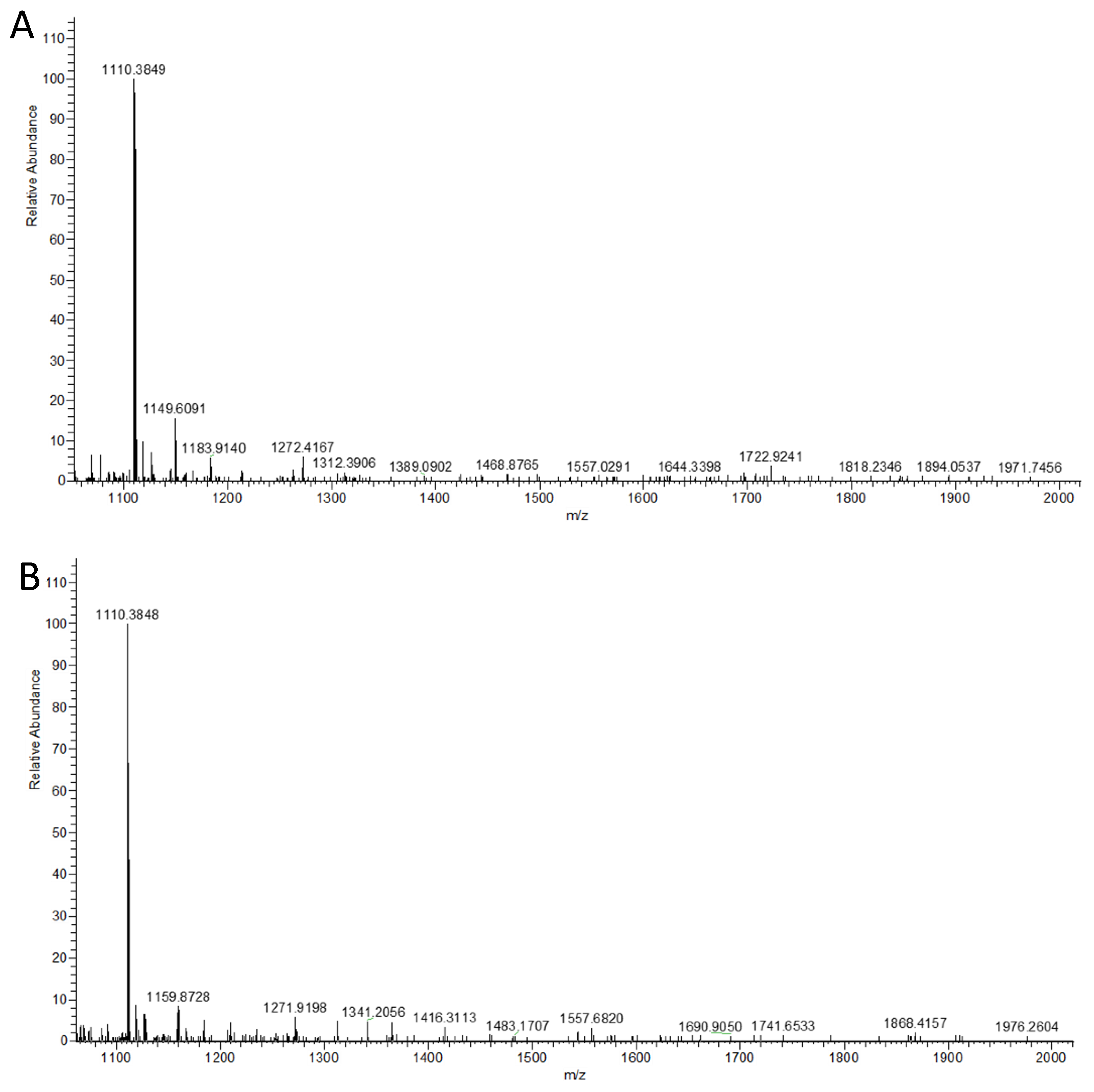
4.7. HRMS Analysis of Sialylated N-Glycans
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Li, X.; Chen, X. Glycoproteins: Chemical features and biological roles. In Carbohydrates Chemistry: State-of-the-Art and Challenges for Drug Development; Cipolla, L., Ed.; Imperial College Press: London, UK, 2015; pp. 3–33. [Google Scholar]
- Stanley, P.; Moremen, K.W.; Lewis, N.E.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2022; pp. 103–116. [Google Scholar]
- Borza, B.; Hajba, L.; Guttman, A. N-glycan analysis in molecular medicine: Innovator and biosimilar protein therapeutics. Curr. Mol. Med. 2020, 20, 828–839. [Google Scholar] [CrossRef]
- Wang, L.X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of antibodies for modulating functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Delobel, A. Glycosylation of therapeutic proteins: A critical quality attribute. Methods Mol. Biol. 2021, 2271, 1–21. [Google Scholar] [PubMed]
- Majewska, N.I.; Tejada, M.L.; Betenbaugh, M.J.; Agarwal, N. N-Glycosylation of IgG and IgG-like recombinant therapeutic proteins: Why is it important and how can we control it? Annu. Rev. Chem. Biomol. Eng. 2020, 11, 311–338. [Google Scholar] [CrossRef] [Green Version]
- Higel, F.; Seidl, A.; Sorgel, F.; Friess, W. N-Glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cymer, F.; Beck, H.; Rohde, A.; Reusch, D. Therapeutic monoclonal antibody N-glycosylation—Structure, function and therapeutic potential. Biologicals 2018, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Qiu, H. The mechanistic impact of N-glycosylation on stability, pharmacokinetics, and immunogenicity of therapeutic proteins. J. Pharm. Sci. 2019, 108, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Reusch, D.; Tejada, M.L. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology 2015, 25, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.; Krag, S.S.; Betenbaugh, M.J. Controlling N-linked glycan site occupancy. Biochim. Biophys. Acta 2005, 1726, 121–137. [Google Scholar] [CrossRef] [PubMed]
- de Haas, P.; Hendriks, W.; Lefeber, D.J.; Cambi, A. Biological and technical challenges in unraveling the role of N-glycans in immune receptor regulation. Front. Chem. 2020, 8, 55. [Google Scholar] [CrossRef]
- Čaval, T.; Heck, A.J.R.; Reiding, K.R. Meta-heterogeneity: Evaluating and describing the diversity in glycosylation between sites on the same glycoprotein. Mol. Cell. Proteom. 2021, 20, 100010. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.; Thomas-Oates, J.; Wood, A.J.; Ungar, D. The N-glycosylation processing potential of the mammalian golgi apparatus. Front. Cell Dev. Biol. 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, E.; Livanos, M.; Krueger, A.; Dell, A.; Haslam, S.M.; Mark Smales, C.; Bracewell, D.G. Strategies to control therapeutic antibody glycosylation during bioprocessing: Synthesis and separation. Biotechnol. Bioeng. 2022, 119, 1343–1358. [Google Scholar] [CrossRef]
- Rexer, T.; Laaf, D.; Gottschalk, J.; Frohnmeyer, H.; Rapp, E.; Elling, L. Enzymatic synthesis of glycans and glycoconjugates. Adv. Biochem. Eng. Biotechnol. 2021, 175, 231–280. [Google Scholar]
- Nomura, K.; Liu, Y.; Kajihara, Y. Synthesis of homogeneous glycoproteins with diverse N-glycans. Adv. Carbohydr. Chem. Biochem. 2022, 81, 57–93. [Google Scholar] [PubMed]
- Fairbanks, A.J. Chemoenzymatic synthesis of glycoproteins. Curr. Opin. Chem. Biol. 2019, 53, 9–15. [Google Scholar] [CrossRef]
- Li, C.; Wang, L.X. Chemoenzymatic methods for the synthesis of glycoproteins. Chem. Rev. 2018, 118, 8359–8413. [Google Scholar] [CrossRef]
- Mastrangeli, R.; Palinsky, W.; Bierau, H. Glycoengineered antibodies: Towards the next-generation of immunotherapeutics. Glycobiology 2018, 29, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, L.; Voglmeir, J. mAbs N-glycosylation: Implications for biotechnology and analytics. Carbohydr. Res. 2022, 514, 108541. [Google Scholar] [CrossRef]
- Mastrangeli, R.; Audino, M.C.; Palinsky, W.; Broly, H.; Bierau, H. The formidable challenge of controlling high mannose-type N-glycans in therapeutic mAbs. Trends Biotechnol. 2020, 38, 1154–1168. [Google Scholar] [CrossRef]
- Hsu, Y.P.; Verma, D.; Sun, S.; McGregor, C.; Mangion, I.; Mann, B.F. Successive remodeling of IgG glycans using a solid-phase enzymatic platform. Commun. Biol. 2022, 5, 328. [Google Scholar] [CrossRef] [PubMed]
- Warnock, D.; Bai, X.; Autote, K.; Gonzales, J.; Kinealy, K.; Yan, B.; Qian, J.; Stevenson, T.; Zopf, D.; Bayer, R.J. In vitro galactosylation of human IgG at 1 kg scale using recombinant galactosyltransferase. Biotechnol. Bioeng. 2005, 92, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, T.; Boons, G.J. Preparation of well-defined antibody-drug conjugates through glycan remodeling and strain-promoted azide-alkyne cycloadditions. Angew. Chem. Int. Ed. Engl. 2014, 53, 7179–7182. [Google Scholar] [CrossRef] [Green Version]
- Thomann, M.; Schlothauer, T.; Dashivets, T.; Malik, S.; Avenal, C.; Bulau, P.; Ruger, P.; Reusch, D. In vitro glycoengineering of IgG1 and its effect on Fc receptor binding and ADCC activity. PLoS ONE 2015, 10, e0134949. [Google Scholar] [CrossRef]
- Jaroentomeechai, T.; Kwon, Y.H.; Liu, Y.; Young, O.; Bhawal, R.; Wilson, J.D.; Li, M.; Chapla, D.G.; Moremen, K.W.; Jewett, M.C.; et al. A universal glycoenzyme biosynthesis pipeline that enables efficient cell-free remodeling of glycans. Nat. Commun. 2022, 13, 6325. [Google Scholar] [CrossRef]
- Moremen, K.W.; Ramiah, A.; Stuart, M.; Steel, J.; Meng, L.; Forouhar, F.; Moniz, H.A.; Gahlay, G.; Gao, Z.; Chapla, D.; et al. Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 2018, 14, 156–162. [Google Scholar] [CrossRef]
- Hidari, K.I.; Horie, N.; Murata, T.; Miyamoto, D.; Suzuki, T.; Usui, T.; Suzuki, Y. Purification and characterization of a soluble recombinant human ST6Gal I functionally expressed in Escherichia coli. Glycoconj. J. 2005, 22, 1–11. [Google Scholar] [CrossRef]
- Williams, R.L.; Greene, S.M.; McPherson, A. The crystal structure of ribonuclease B at 2.5-A resolution. J. Biol. Chem. 1987, 262, 16020–16031. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Joao, H.C.; Coghill, E.; Fiten, P.; Saunders, M.R.; Opdenakker, G.; Dwek, R.A. Glycoforms modify the dynamic stability and functional activity of an enzyme. Biochemistry 1994, 33, 17–22. [Google Scholar] [CrossRef]
- Li, Y.; Li, R.; Yu, H.; Sheng, X.; Wang, J.; Fisher, A.J.; Chen, X. Enterococcus faecalis alpha1-2-mannosidase (EfMan-I): An efficient catalyst for glycoprotein N-glycan modification. FEBS Lett. 2020, 594, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280 Pt 2, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Bairoch, A. Updating the sequence-based classification of glycosyl hydrolases. Biochem. J. 1996, 316 Pt 2, 695–696. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326 Pt 3, 929–939. [Google Scholar] [CrossRef]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Fujiyama, K.; Ido, Y.; Misaki, R.; Moran, D.G.; Yanagihara, I.; Honda, T.; Nishimura, S.; Yoshida, T.; Seki, T. Human N-acetylglucosaminyltransferase I. Expression in Escherichia coli as a soluble enzyme, and application as an immobilized enzyme for the chemoenzymatic synthesis of N-linked oligosaccharides. J. Biosci. Bioeng. 2001, 92, 569–574. [Google Scholar] [CrossRef]
- Zhu, Y.; Suits, M.D.; Thompson, A.J.; Chavan, S.; Dinev, Z.; Dumon, C.; Smith, N.; Moremen, K.W.; Xiang, Y.; Siriwardena, A.; et al. Mechanistic insights into a Ca2+-dependent family of alpha-mannosidases in a human gut symbiont. Nat. Chem. Biol. 2010, 6, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Kadirvelraj, R.; Yang, J.Y.; Sanders, J.H.; Liu, L.; Ramiah, A.; Prabhakar, P.K.; Boons, G.J.; Wood, Z.A.; Moremen, K.W. Human N-acetylglucosaminyltransferase II substrate recognition uses a modular architecture that includes a convergent exosite. Proc. Natl. Acad. Sci. USA 2018, 115, 4637–4642. [Google Scholar] [CrossRef] [Green Version]
- Boeggeman, E.E.; Balaji, P.V.; Sethi, N.; Masibay, A.S.; Qasba, P.K. Expression of deletion constructs of bovine beta-1,4-galactosyltransferase in Escherichia coli: Importance of Cys134 for its activity. Protein Eng. 1993, 6, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Gastinel, L.N.; Cambillau, C.; Bourne, Y. Crystal structures of the bovine beta4galactosyltransferase catalytic domain and its complex with uridine diphosphogalactose. EMBO J. 1999, 18, 3546–3557. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, B.; Boeggeman, E.; Ramasamy, V.; Qasba, P.K. Structure and catalytic cycle of beta-1,4-galactosyltransferase. Curr. Opin. Struct. Biol. 2004, 14, 593–600. [Google Scholar] [CrossRef]
- Li, Y.; Xue, M.; Sheng, X.; Yu, H.; Zeng, J.; Thon, V.; Chen, Y.; Muthana, M.M.; Wang, P.G.; Chen, X. Donor substrate promiscuity of bacterial beta1-3-N-acetylglucosaminyltransferases and acceptor substrate flexibility of beta1-4-galactosyltransferases. Bioorg. Med. Chem. 2016, 24, 1696–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Varki, A. Advances in the biology and chemistry of sialic acids. ACS Chem. Biol. 2010, 5, 163–176. [Google Scholar] [CrossRef]
- Yang, X.; Yu, H.; Yang, X.; Kooner, A.S.; Yuan, Y.; Luu, B.; Chen, X. One-pot multienzyme (OPME) chemoenzymatic synthesis of brain ganglioside glycans with human ST3GAL II expressed in E. coli. ChemCatChem 2022, 14, e202101498. [Google Scholar] [CrossRef]
- Zhang, Y.; Malinovskii, V.A.; Fiedler, T.J.; Brew, K. Role of a conserved acidic cluster in bovine beta1,4 galactosyltransferase-1 probed by mutagenesis of a bacterially expressed recombinant enzyme. Glycobiology 1999, 9, 815–822. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chokhawala, H.; Karpel, R.; Yu, H.; Wu, B.; Zhang, J.; Zhang, Y.; Jia, Q.; Chen, X. A multifunctional Pasteurella multocida sialyltransferase: A powerful tool for the synthesis of sialoside libraries. J. Am. Chem. Soc. 2005, 127, 17618–17619. [Google Scholar] [CrossRef]
- Sugiarto, G.; Lau, K.; Qu, J.; Li, Y.; Lim, S.; Mu, S.; Ames, J.B.; Fisher, A.J.; Chen, X. A sialyltransferase mutant with decreased donor hydrolysis and reduced sialidase activities for directly sialylating LewisX. ACS Chem. Biol. 2012, 7, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Thon, V.; Li, Y.; Yu, H.; Lau, K.; Chen, X. PmST3 from Pasteurella multocida encoded by Pm1174 gene is a monofunctional alpha2-3-sialyltransferase. Appl. Microbiol. Biotechnol. 2012, 94, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, S.; Chokhawala, H.; Sun, M.; Zheng, H.; Chen, X. Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural alpha-2,6-linked sialosides: A P. damsela alpha-2,6-sialyltransferase with extremely flexible donor-substrate specificity. Angew. Chem. Int. Ed. Engl. 2006, 45, 3938–3944. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Yang, X.; Yu, H.; Chen, X. Substrate and process engineering for biocatalytic synthesis and facile purification of human milk oligosaccharides. ChemSusChem 2022, 15, e202102539. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yu, H.; Lau, K.; Li, Y.; Muthana, S.; Wang, J.; Chen, X. Efficient chemoenzymatic synthesis of sialyl Tn-antigens and derivatives. Chem. Commun. 2011, 47, 8691–8693. [Google Scholar] [CrossRef]
- Ding, L.; Zhao, C.; Qu, J.; Li, Y.; Sugiarto, G.; Yu, H.; Wang, J.; Chen, X. A Photobacterium sp. alpha2-6-sialyltransferase (Psp2,6ST) mutant with an increased expression level and improved activities in sialylating Tn antigens. Carbohydr. Res. 2015, 408, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, X. Sialic acid metabolism and sialyltransferases: Natural functions and applications. Appl. Microbiol. Biotechnol. 2012, 94, 887–905. [Google Scholar] [CrossRef] [Green Version]
- Mbua, N.E.; Li, X.; Flanagan-Steet, H.R.; Meng, L.; Aoki, K.; Moremen, K.W.; Wolfert, M.A.; Steet, R.; Boons, G.J. Selective exo-enzymatic labeling of N-glycans on the surface of living cells by recombinant ST6Gal I. Angew. Chem. Int. Ed. Engl. 2013, 52, 13012–13015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.P.; Lairson, L.L.; Gilbert, M.; Wakarchuk, W.W.; Withers, S.G.; Strynadka, N.C. Structural analysis of the alpha-2,3-sialyltransferase Cst-I from Campylobacter jejuni in apo and substrate-analogue bound forms. Biochemistry 2007, 46, 7196–7204. [Google Scholar] [CrossRef]
- Geissner, A.; Baumann, L.; Morley, T.J.; Wong, A.K.O.; Sim, L.; Rich, J.R.; So, P.P.L.; Dullaghan, E.M.; Lessard, E.; Iqbal, U.; et al. 7-Fluorosialyl glycosides are hydrolysis resistant but readily assembled by sialyltransferases providing easy access to more metabolically stable glycoproteins. ACS Cent. Sci. 2021, 7, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.H.; Hemayatkar, M.; Deelder, A.M.; Wuhrer, M. Cotton HILIC SPE microtips for microscale purification and enrichment of glycans and glycopeptides. Anal. Chem. 2011, 83, 2492–2499. [Google Scholar] [CrossRef]
- Yu, H.; Yu, H.; Karpel, R.; Chen, X. Chemoenzymatic synthesis of CMP-sialic acid derivatives by a one-pot two-enzyme system: Comparison of substrate flexibility of three microbial CMP-sialic acid synthetases. Bioorg. Med. Chem. 2004, 12, 6427–6435. [Google Scholar] [CrossRef]
- Yu, H.; Chen, X. One-pot multienzyme (OPME) systems for chemoenzymatic synthesis of carbohydrates. Org. Biomol. Chem. 2016, 14, 2809–2818. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Thon, V.; Li, Y.; Yu, H.; Ding, L.; Lau, K.; Qu, J.; Hie, L.; Chen, X. One-pot three-enzyme synthesis of UDP-GlcNAc derivatives. Chem. Commun. 2011, 47, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Muthana, M.M.; Qu, J.; Li, Y.; Zhang, L.; Yu, H.; Ding, L.; Malekan, H.; Chen, X. Efficient one-pot multienzyme synthesis of UDP-sugars using a promiscuous UDP-sugar pyrophosphorylase from Bifidobacterium longum (BLUSP). Chem. Commun. 2012, 48, 2728–2730. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | CAZy Family | Source | Expression Host | Expression Level (mg/L LB) |
|---|---|---|---|---|
| EfMan-I-His6 | GH92 | Enterococcus faecalis V583 | BL21 (DE3) | 85 [a] |
| ∆24Bt3994-His6 | GH92 | Bacteroides thetaiotaomicron | BL21 (DE3) | 55 |
| ∆18Bt1769-His6 | GH92 | Bacteroides thetaiotaomicron | BL21 (DE3) | 55 |
| MBP-∆28hGnT-I-His6 | GT13 | Human | Origami B (DE3)/pGro7 | 5 |
| MBP-∆27hGnT-II-His6 | GT16 | Human | Origami B (DE3)/pGro7 | ~1 |
| MBP-∆128Bβ4GalT1-His6 | GT7 | Bovine | Origami B (DE3)/pGro7 | 60 |
| MBP-∆89ST6GAL-I-His6 | GT29 | Human | Origami B (DE3)/pGro7 | 30 |
| MBP-CjCst-I∆145-His6 | GT42 | Campylobacter jejuni | BL21 (DE3) | 60 |
| Plasmid | Ta (°C) | Primer Sequences (5′to 3′) | Restriction Enzyme | |
|---|---|---|---|---|
| pET15b-∆28hGnT-I | 52 | Forward | ATCCATATGTGGACCCGTCCAGCCCCAGGT | NdeⅠ |
| Reverse | CCGGGATCCTTAATTCCAGCTTGGATCATAACCTTC | BamHI | ||
| pMAL-c2X-∆28hGnT-I | 52 | Forward | GACCGAATTCTGGACCCGTCCAGCCCCAGGT | EcoRI |
| Reverse | CAGCAAGCTTTTAGTGGTGGTGATGATGATGATTCCAGCTTGGATCATAACCTTC | HindIII | ||
| pET22b(+)-Bt3994 | 55 | Forward | CTCCAGCATATGCTCCAGCATATGAAAACACCTATTTACCT | NdeI |
| Reverse | CTCCAGCTCGAGCTGGGTCTGTTCTTCAAATTCG | XhoI | ||
| pET22b(+)-∆24Bt3994 | 55 | Forward | CTCCAGCATATGCTCCAGCATATGAAAACACCTATTTACCT | NdeI |
| Reverse | CTCCAGCTCGAGCTGGGTCTGTTCTTCAAATTCG | XhoI | ||
| pET22b(+)-Bt1769 | 55 | Forward | CTCCAGCATATGAAACTTACACACATTTT | NdeI |
| Reverse | CTCCAGCTCGAGCTTGATTTCTAATGATGGAGG | XhoI | ||
| pET22b(+)-∆18Bt1769 | 55 | Forward | CTCCAGCATATGAAACTTACACACATTTT | NdeI |
| Reverse | CTCCAGCTCGAGCTTGATTTCTAATGATGGAGG | XhoI | ||
| pMAL-c4x-∆27hGnT-II | 56 | Forward | GATCGGATCCAACGGTCGTCAGCGTAAAAA | BamHI |
| Reverse | CGGAAGCTTTTAGTGGTGGTGGTGGTGGTGCTGCAGGCGGCGATAGG | HindIII | ||
| pMAL-c4X-∆128Bβ4GalT1 | 55 | Forward | GACCGAATTCCGCTCGCTGACCGCATG | EcoRI |
| Reverse | CAGCAAGCTTTCAATGGTGATGGTGATGGTGGCTCGGCGTCCCGATGTC | HindIII | ||
| pMAL-c2X-∆89hST6GAL-I | 50 | Forward | GACCGAATTCGAGGCCTCGTTTCAGGTTTG | EcoRI |
| Reverse | CAGCAAGCTTTTAGTGGTGGTGATGATGATGGCAGTGTATTGTTCGGAATC | HindIII | ||
| pMAL-c4x-CjCst-I∆145 | 56 | Forward | CTCTCTGAATTCATGACACGCACCAGAATGGAG | EcoRI |
| Reverse | CTCTCTGTCGACTTAATGATGATGATGATGATGATAAAAGTTCAGCATTAT | SalI | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Li, Y.; Li, R.; Yang, X.; Zheng, Z.; Fu, J.; Yu, H.; Chen, X. Glycoprotein In Vitro N-Glycan Processing Using Enzymes Expressed in E. coli. Molecules 2023, 28, 2753. https://doi.org/10.3390/molecules28062753
Zhang L, Li Y, Li R, Yang X, Zheng Z, Fu J, Yu H, Chen X. Glycoprotein In Vitro N-Glycan Processing Using Enzymes Expressed in E. coli. Molecules. 2023; 28(6):2753. https://doi.org/10.3390/molecules28062753
Chicago/Turabian StyleZhang, Libo, Yanhong Li, Riyao Li, Xiaohong Yang, Zimin Zheng, Jingxin Fu, Hai Yu, and Xi Chen. 2023. "Glycoprotein In Vitro N-Glycan Processing Using Enzymes Expressed in E. coli" Molecules 28, no. 6: 2753. https://doi.org/10.3390/molecules28062753
APA StyleZhang, L., Li, Y., Li, R., Yang, X., Zheng, Z., Fu, J., Yu, H., & Chen, X. (2023). Glycoprotein In Vitro N-Glycan Processing Using Enzymes Expressed in E. coli. Molecules, 28(6), 2753. https://doi.org/10.3390/molecules28062753