
Photoinduced Dynamics of 13,13′-Diphenylpropyl-β-carotene

 , , and
, , and
Abstract
:
1. Introduction
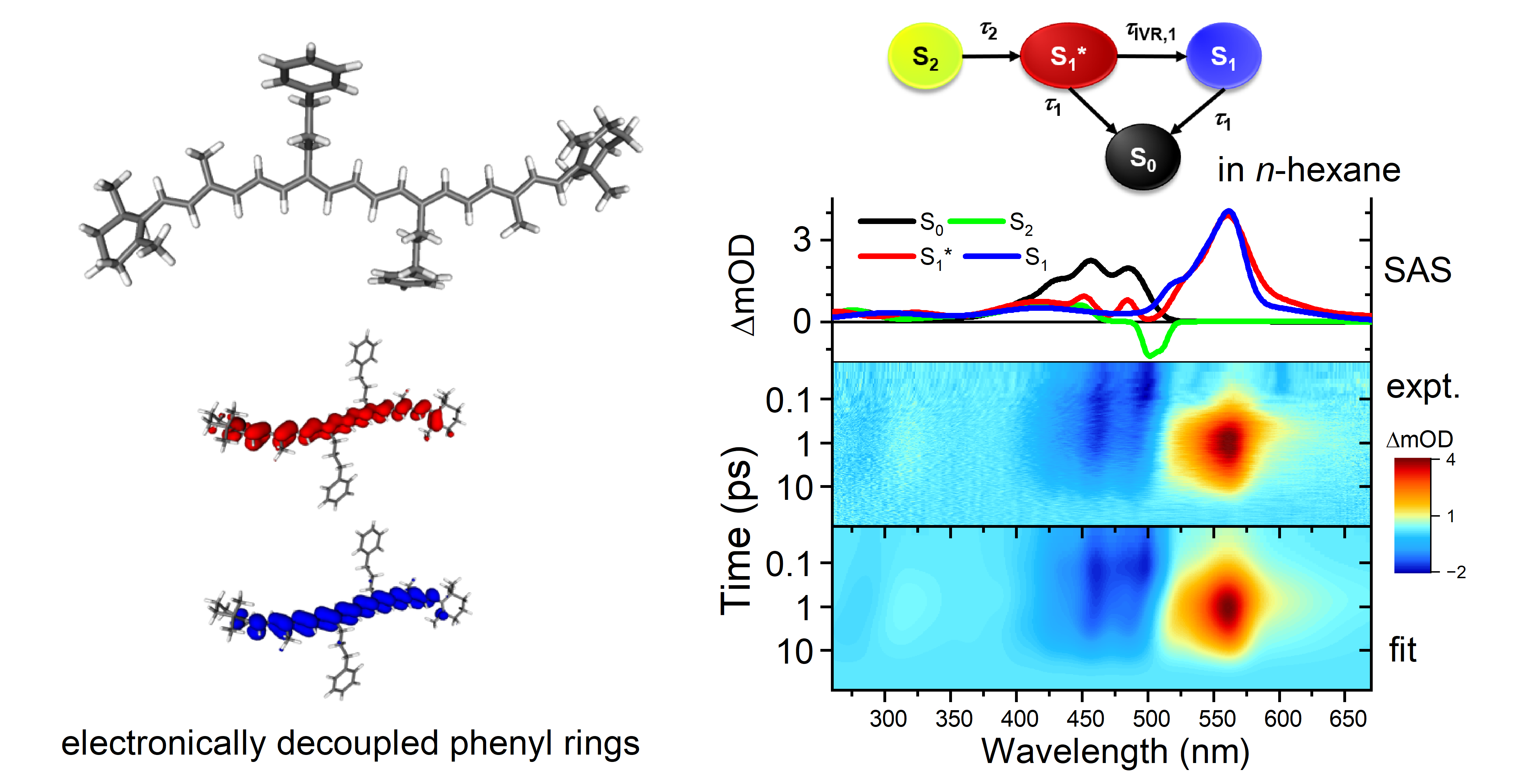
2. Results and Discussion
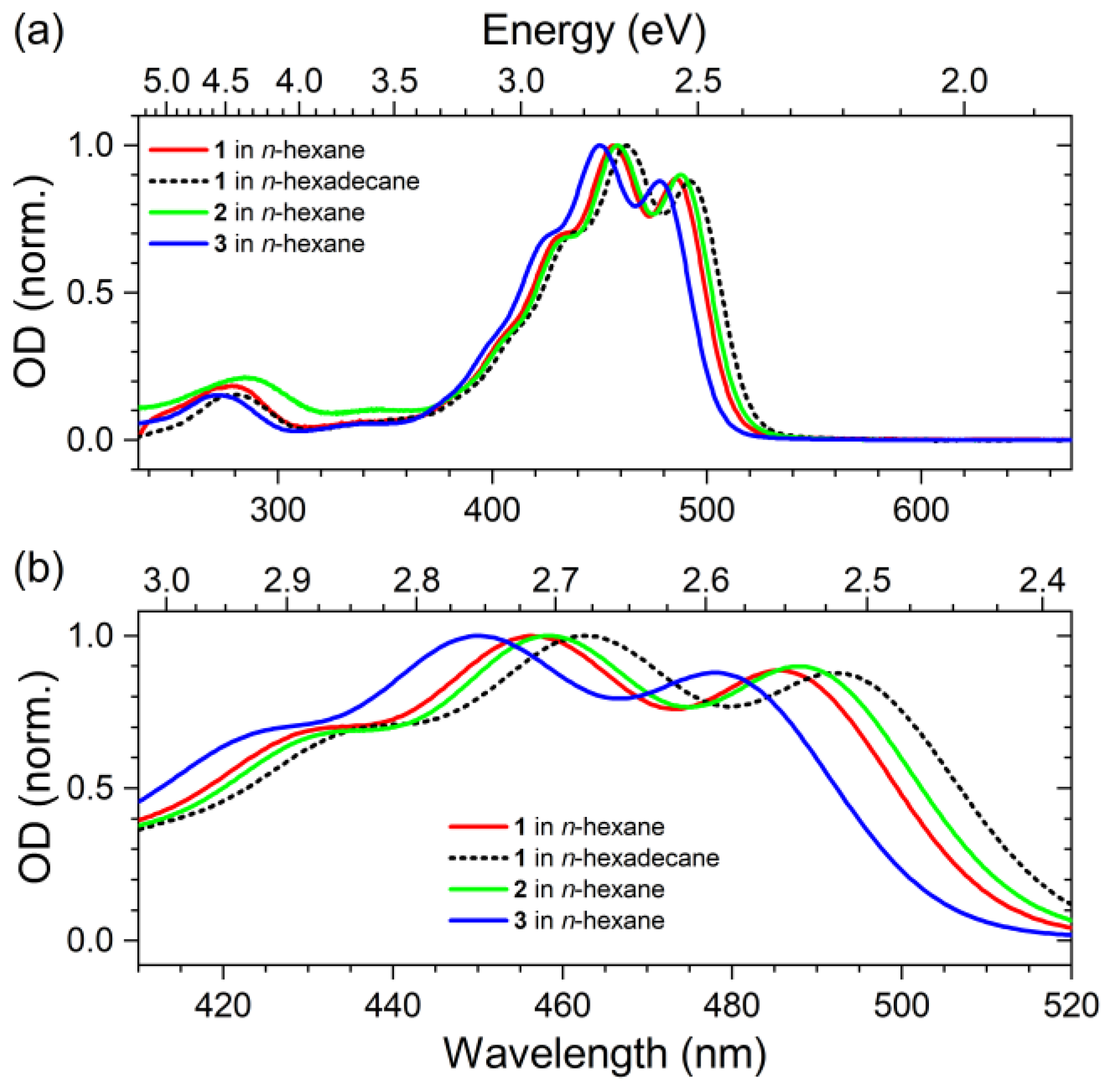
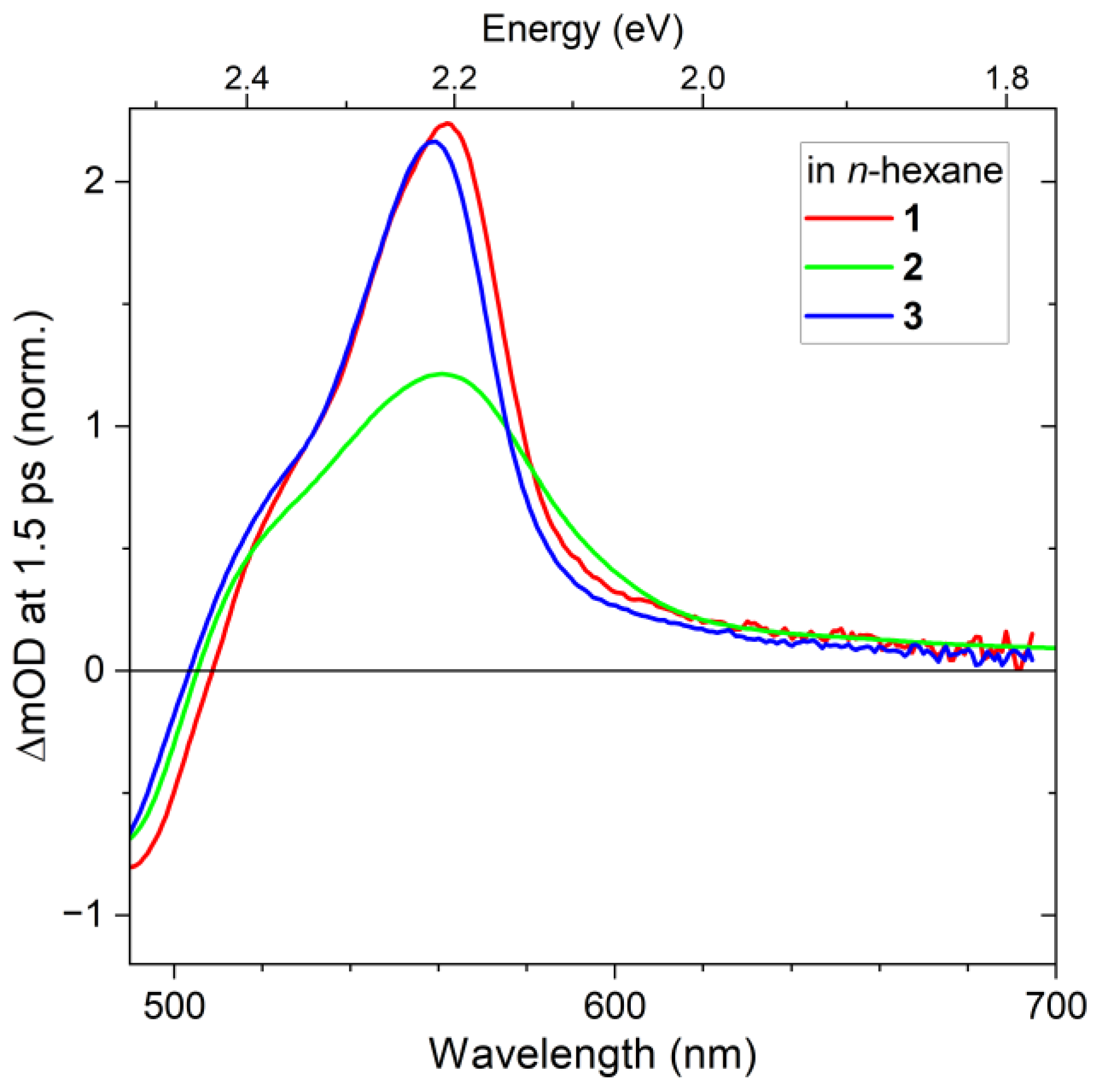
2.1. Steady-State Absorption Spectra
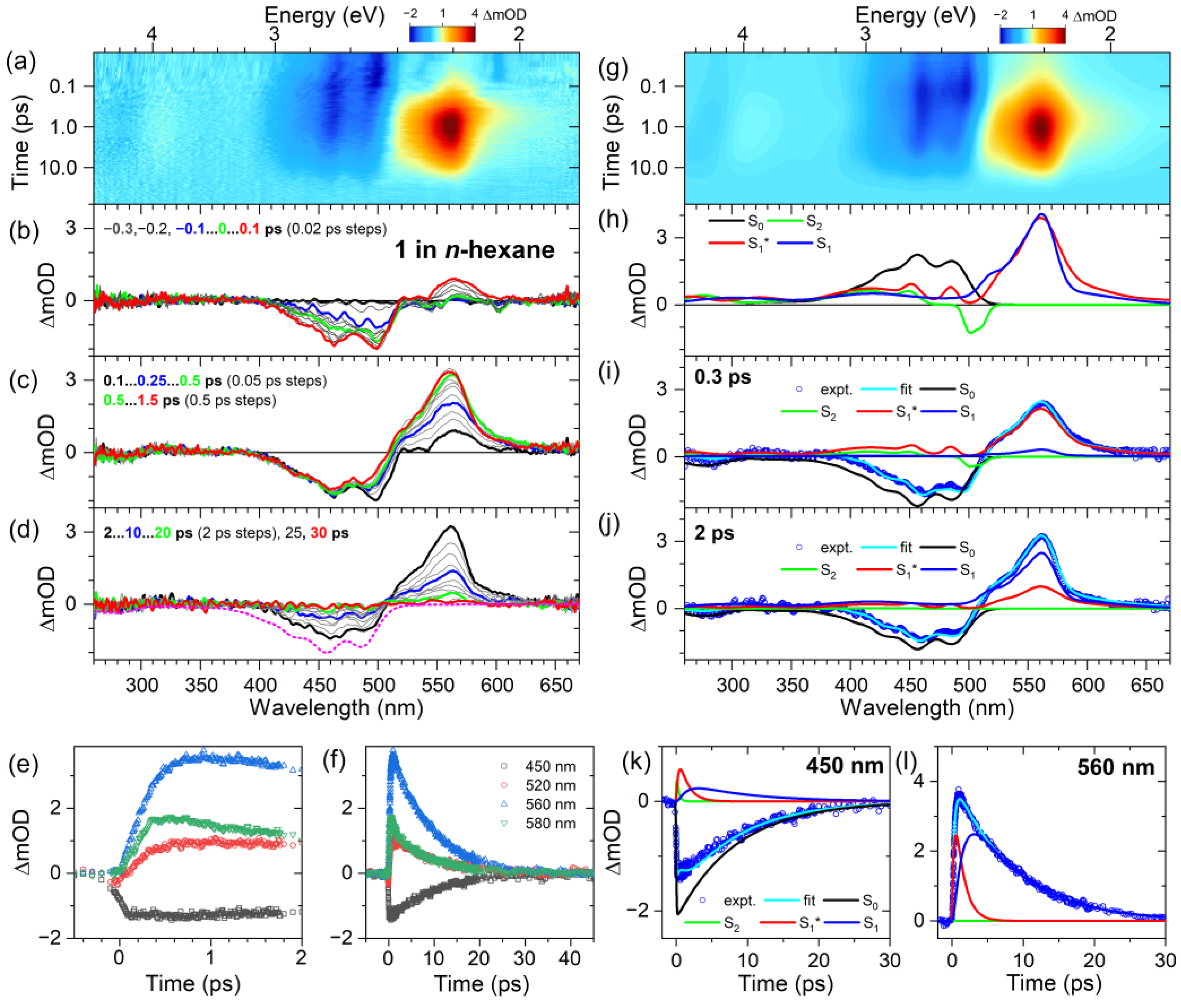
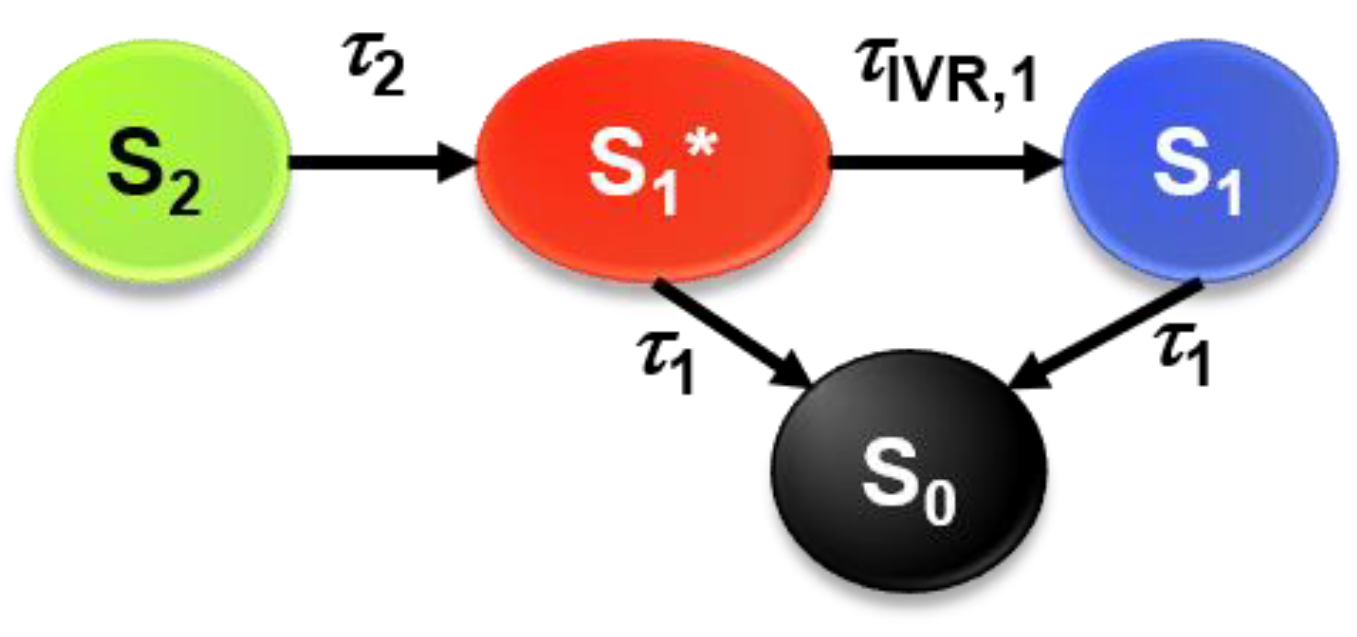
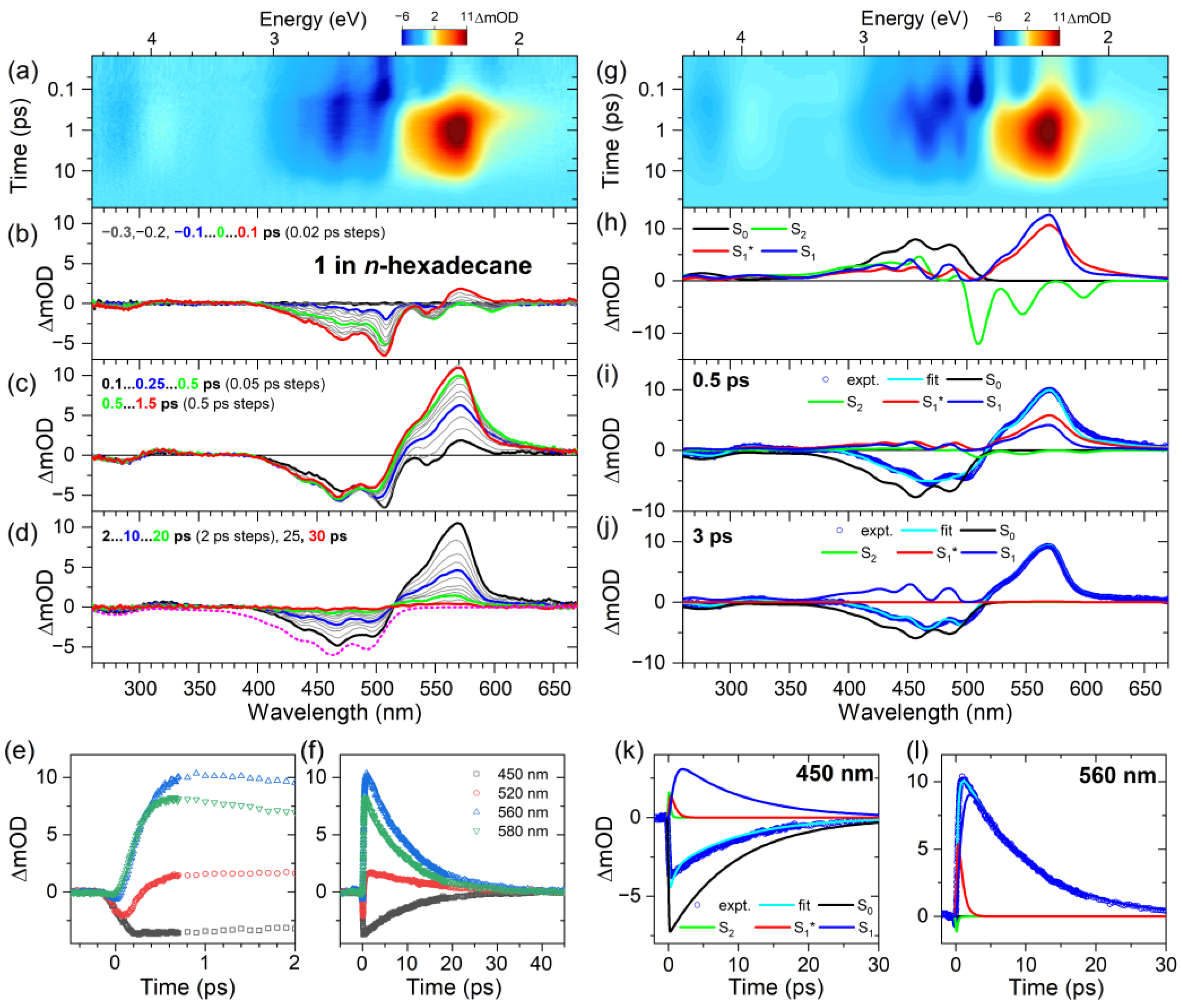
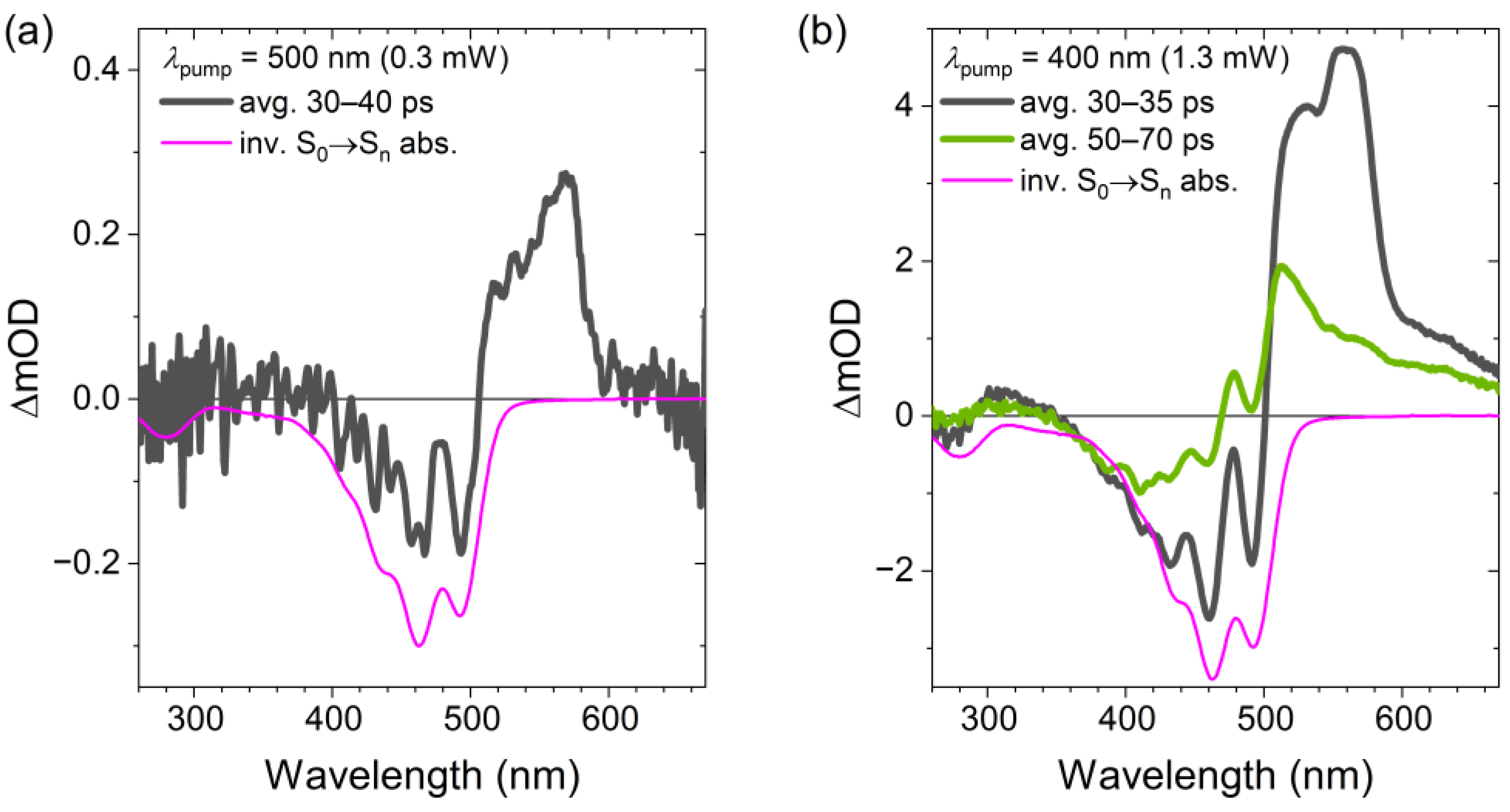
2.2. Transient Absorption Spectra and Their Global Kinetic Analysis
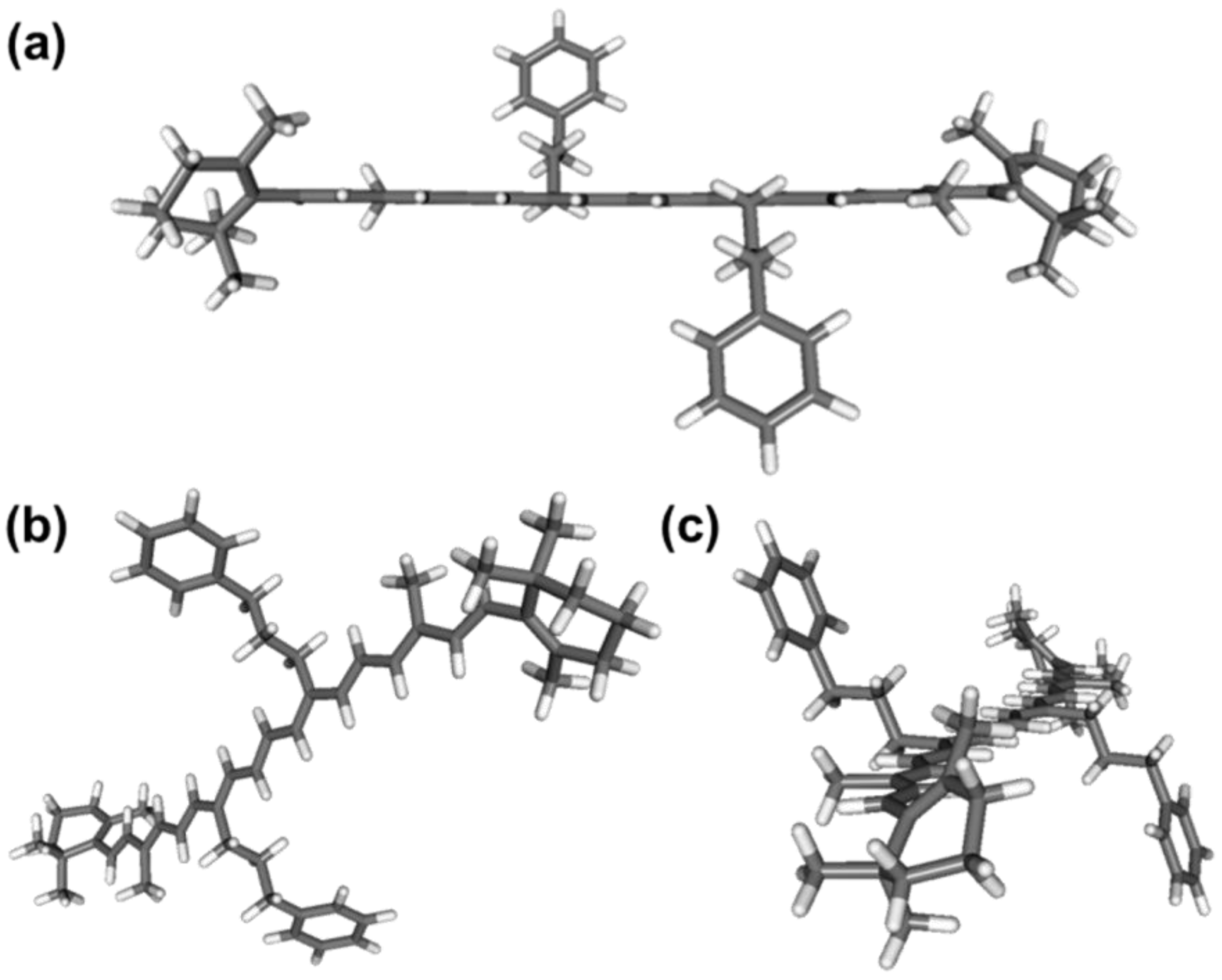
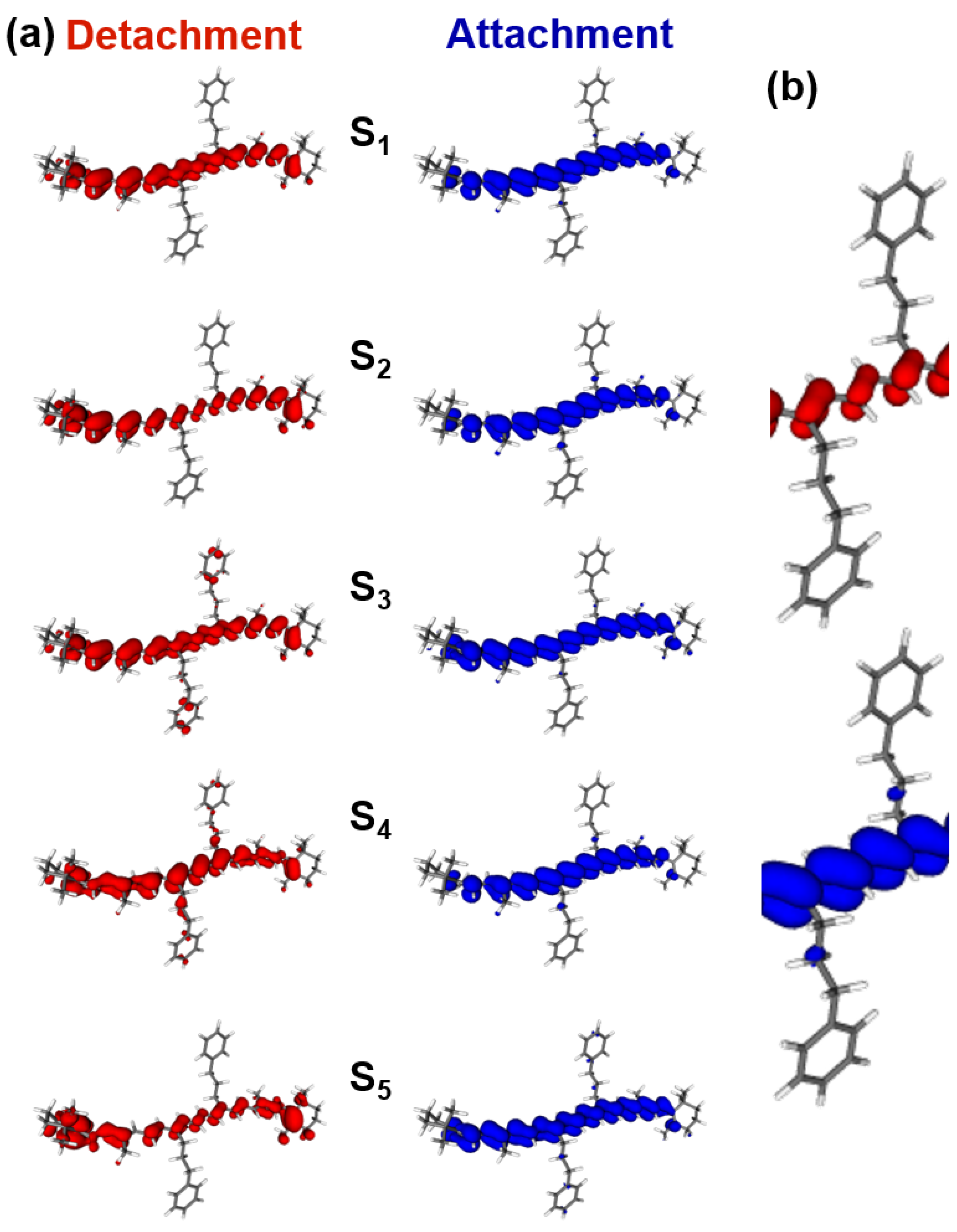
2.3. DFT/TDDFT Calculations
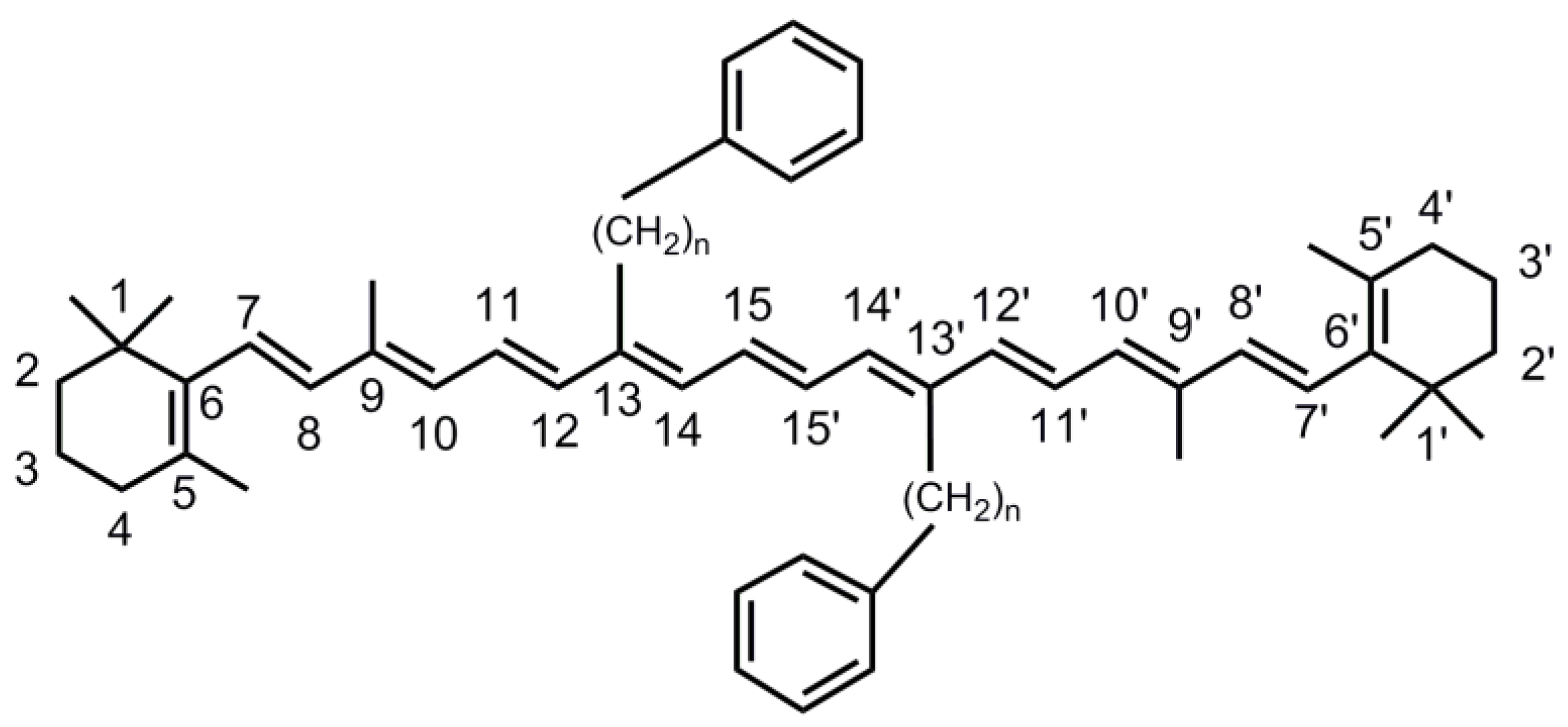
3. Materials and Methods
3.1. Synthesis of the Carotenoid and Sample Treatment
3.2. Ultrafast Transient Absorption Spectroscopy
3.3. Global Kinetic Analysis of the Transient Spectra
3.4. DFT/TDDFT Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Frank, H.A.; Young, A.J.; Britton, G.; Cogdell, R.J. The Photochemistry of Carotenoids; Kluwer: Dordrecht, The Netherlands, 1999; Volume 8, p. 399. [Google Scholar]
- Polívka, T.; Sundström, V. Ultrafast Dynamics of Carotenoid Excited States—From Solution to Natural and Artificial Systems. Chem. Rev. 2004, 104, 2021–2071. [Google Scholar] [CrossRef] [PubMed]
- Frank, H.A.; Cua, A.; Chynwat, V.; Young, A.; Gosztola, D.; Wasielewski, M.R. Photophysics of the carotenoids associated with the xanthophyll cycle in photosynthesis. Photosynth. Res. 1994, 41, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, F.; Scholz, M.; Lenzer, T.; Oum, K. Excited State Dynamics of Selected All-trans C40 Xanthophyll Carotenoids. Z. Phys. Chem. 2015, 229, 1815–1830. [Google Scholar] [CrossRef]
- Scholz, M.; Flender, O.; Lenzer, T.; Oum, K. Ultrafast Excited-State Dynamics of all-trans-Capsanthin in Organic Solvents. J. Phys. Chem. A 2017, 121, 8380–8388. [Google Scholar] [CrossRef]
- Niedzwiedzki, D.M.; Chatterjee, N.; Enriquez, M.M.; Kajikawa, T.; Hasegawa, S.; Katsumura, S.; Frank, H.A. Spectroscopic Investigation of Peridinin Analogues Having Different π-Electron Conjugated Chain Lengths: Exploring the Nature of the Intramolecular Charge Transfer State. J. Phys. Chem. B 2009, 113, 13604–13612. [Google Scholar] [CrossRef]
- Kosumi, D.; Kajikawa, T.; Okumura, S.; Sugisaki, M.; Sakaguchi, K.; Katsumura, S.; Hashimoto, H. Elucidation and Control of an Intramolecular Charge Transfer Property of Fucoxanthin by a Modification of Its Polyene Chain Length. J. Phys. Chem. Lett. 2014, 5, 792–797. [Google Scholar] [CrossRef]
- Ilagan, R.P.; Koscielecki, J.F.; Hiller, R.G.; Sharples, F.P.; Gibson, G.N.; Birge, R.R.; Frank, H.A. Femtosecond Time-Resolved Absorption Spectroscopy of Main-Form and High-Salt Peridinin-Chlorophyll a-Proteins at Low Temperatures. Biochemistry 2006, 45, 14052–14063. [Google Scholar] [CrossRef]
- Gelzinis, A.; Butkus, V.; Songaila, E.; Auguli, R.; Gal, A.; Büchel, C.; Robert, B.; Abramavicius, D.; Zigmantas, D.; Valkunas, L. Mapping energy transfer channels in fucoxanthin-chlorophyll protein complex. Biochim. Biophys. Acta 2015, 1847, 241–247. [Google Scholar] [CrossRef]
- Macernis, M.; Galzerano, D.; Sulskus, J.; Kish, E.; Kim, Y.-H.; Koo, S.; Valkunas, L.; Robert, B. Resonance Raman Spectra of Carotenoid Molecules: Influence of Methyl Substitutions. J. Phys. Chem. A 2015, 119, 56–66. [Google Scholar] [CrossRef]
- Golibrzuch, K.; Ehlers, F.; Scholz, M.; Oswald, R.; Lenzer, T.; Oum, K.; Kim, H.; Koo, S. Ultrafast excited state dynamics and spectroscopy of 13,13’-diphenyl-β-carotene. Phys. Chem. Chem. Phys. 2011, 13, 6340–6351. [Google Scholar] [CrossRef]
- Maeng, J.; Kim, S.B.; Lee, N.J.; Choi, E.; Jung, S.-Y.; Hong, I.; Bae, S.-H.; Oh, J.T.; Lim, B.; Kim, J.W.; et al. Conductance Control in Stabilized Carotenoid Wires. Chem. Eur. J. 2010, 16, 7395–7399. [Google Scholar] [CrossRef]
- Zhao, Y.; Lindsay, S.; Jeon, S.; Kim, H.-J.; Su, L.; Lim, B.; Koo, S. Combined Effect of Polar Substituents on the Electronic Flows in the Carotenoid Molecular Wires. Chem. Eur. J. 2013, 19, 10832–10835. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, A.C.; Darwish, N.; Im, J.; Lim, B.; Choi, J.; Koo, S.; Díez-Pérez, I. Fine-Tuning of Single-Molecule Conductance by Tweaking Both Electronic Structure and Conformation of Side Substituents. Chem. Eur. J. 2015, 21, 7716–7720. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Oh, E.-T.; Im, J.; Lee, K.S.; Jung, H.; Kim, M.; Kim, D.; Oh, J.T.; Bae, S.-H.; Chung, W.-J.; et al. Systematic Synthesis of Diphenyl-Substituted Carotenoids as Molecular Wires. Eur. J. Org. Chem. 2017, 6390–6400. [Google Scholar] [CrossRef]
- Lim, B.; Kim, Y.-H.; Kim, H.; Park, M.; Yeo, H.; Koo, S. 13,13’-Diphenalkyl β-carotenes as pi-stacking models of chlorophylls and carotenoids in photosynthesis. Bull. Korean Chem. Soc. 2022, 43, 1037–1039. [Google Scholar] [CrossRef]
- Lenzer, T.; Ehlers, F.; Scholz, M.; Oswald, R.; Oum, K. Assignment of carotene S* state features to the vibrationally hot ground electronic state. Phys. Chem. Chem. Phys. 2010, 12, 8832–8839. [Google Scholar] [CrossRef]
- Andersson, P.O.; Gillbro, T.; Ferguson, L.; Cogdell, R.J. Absorption spectral shifts of carotenoids related to medium polarizability. Photochem. Photobiol. 1991, 54, 353–360. [Google Scholar] [CrossRef]
- Chen, Z.; Lee, C.; Lenzer, T.; Oum, K. Solvent Effects on the S0(11Ag−) → S2(11Bu+) Transition of β-Carotene, Echinenone, Canthaxanthin, and Astaxanthin in Supercritical CO2 and CF3H. J. Phys. Chem. A 2006, 110, 11291–11297. [Google Scholar] [CrossRef] [PubMed]
- Christensen, R.L.; Goyette, M.; Gallagher, L.; Duncan, J.; DeCoster, B.; Lugtenburg, J.; Jansen, F.J.; van der Hoef, I. S1 and S2 States of Apo- and Diapocarotenes. J. Phys. Chem. A 1999, 103, 2399–2407. [Google Scholar] [CrossRef]
- Kopczynski, M.; Lenzer, T.; Oum, K.; Seehusen, J.; Seidel, M.T.; Ushakov, V.G. Ultrafast transient lens spectroscopy of various C40 carotenoids: Lycopene, β-carotene, (3R,3’R)-zeaxanthin, (3R,3’R,6’R)-lutein, echinenone, canthaxanthin, and astaxanthin. Phys. Chem. Chem. Phys. 2005, 7, 2793–2803. [Google Scholar] [CrossRef]
- Ehlers, F.; Scholz, M.; Schimpfhauser, J.; Bienert, J.; Oum, K.; Lenzer, T. Collisional relaxation of apocarotenals: Identifying the S* state with vibrationally excited molecules in the ground electronic state S0*. Phys. Chem. Chem. Phys. 2015, 17, 10478–10488. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.O.; Gillbro, T. Photophysics and dynamics of the lowest excited singlet state in long substituted polyenes with implications to the very long-chain limit. J. Chem. Phys. 1995, 103, 2509–2519. [Google Scholar] [CrossRef]
- Corsetti, S.; Rabl, T.; McGloin, D.; Kiefer, J. Intermediate phases during solid to liquid transitions in long-chain n-alkanes. Phys. Chem. Chem. Phys. 2017, 19, 13941–13950. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Dobryakov, A.L.; Ruthmann, J.; Ernsting, N.P. Femtosecond spectroscopy of condensed phases with chirped supercontinuum probing. Phys. Rev. A 1999, 59, 2369–2384. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Grana, A.M.; Maurice, D.; White, C.A. Analysis of Electronic Transitions as the Difference of Electron Attachment and Detachment Densities. J. Phys. Chem. 1995, 99, 14261–14270. [Google Scholar] [CrossRef]
- Dreuw, A. Influence of Geometry Relaxation on the Energies of the S1 and S2 States of Violaxanthin, Zeaxanthin, and Lutein. J. Phys. Chem. A 2006, 110, 4592–4599. [Google Scholar] [CrossRef]
- Starcke, J.H.; Wormit, M.; Schirmer, J.; Dreuw, A. How much double excitation character do the lowest excited states of linear polyenes have? Chem. Phys. 2006, 329, 39–49. [Google Scholar] [CrossRef]
- Oum, K.; Lenzer, T.; Scholz, M.; Jung, D.Y.; Sul, O.; Cho, B.J.; Lange, J.; Müller, A. Observation of Ultrafast Carrier Dynamics and Phonon Relaxation of Graphene from the Deep-Ultraviolet to the Visible Region. J. Phys. Chem. C 2014, 118, 6454–6461. [Google Scholar] [CrossRef]
- Ehlers, F.; Lenzer, T.; Oum, K. Excited-State Dynamics of 12’-Apo-β-caroten-12’-al and 8’-Apo-β-caroten-8’-al in Supercritical CO2, N2O, and CF3H. J. Phys. Chem. B 2008, 112, 16690–16700. [Google Scholar] [CrossRef]
- Dobryakov, A.L.; Kovalenko, S.A.; Weigel, A.; Pérez Lustres, J.L.; Lange, J.; Müller, A.; Ernsting, N.P. Femtosecond pump/supercontinuum-probe spectroscopy: Optimized setup and signal analysis for single-shot spectral referencing. Rev. Sci. Instrum. 2010, 81, 113106. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C. Quantum Theory of Molecules and Solids, Vol. 4: The Self-Consistent Field for Molecules and Solids, 1st ed.; McGraw-Hill: New York, NY, USA, 1974. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]
- Wild, D.A.; Winkler, K.; Stalke, S.; Oum, K.; Lenzer, T. Extremely strong solvent dependence of the S1 → S0 internal conversion lifetime of 12’-apo-β-caroten-12’-al. Phys. Chem. Chem. Phys. 2006, 8, 2499–2505. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | τ2 (fs) | τIVR,1 (ps) | τ1 (ps) |
|---|---|---|---|
| n-Hexane | 300 | 1.42 | 8.3 |
| n-Hexadecane | 190 | 0.66 | 9.5 |
| State | E (eV) | λ (nm) | f | ΔE(2-1) (eV) | ΔE(3-1) (eV) |
|---|---|---|---|---|---|
| B3LYP/G-31+G(d) | |||||
| S1 | 2.37 | 523 | 5.2 | −0.03 | +0.02 |
| S2 | 2.63 | 472 | 0.0 | −0.04 | 0.00 |
| S3 | 3.27 | 379 | 0.3 | −0.06 | 0.00 |
| S4 | 3.44 | 360 | 0.0 | −0.05 | +0.01 |
| S5 | 3.71 | 335 | 0.2 | −0.19 | −0.02 |
| BLYP/G-31+G(d) | |||||
| S1 | 2.01 | 617 | 0.0 | −0.03 | −0.01 |
| S2 | 2.11 | 587 | 4.3 | −0.02 | 0.00 |
| S3 | 2.62 | 473 | 1.5 | −0.05 | +0.01 |
| S4 | 2.75 | 450 | 0.0 | +0.01 | +0.02 |
| S5 | 2.90 | 427 | 0.5 | −0.09 | 0.00 |
| SVWN/G-31+G(d) | |||||
| S1 | 1.99 | 623 | 0.0 | −0.03 | 0.00 |
| S2 | 2.09 | 594 | 4.1 | −0.02 | 0.00 |
| S3 | 2.60 | 478 | 1.8 | −0.04 | +0.01 |
| S4 | 2.69 | 460 | 0.0 | 0.00 | +0.03 |
| S5 | 2.90 | 429 | 0.5 | −0.10 | −0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, S.; Kim, Y.H.; Flender, O.; Scholz, M.; Oum, K.; Lenzer, T. Photoinduced Dynamics of 13,13′-Diphenylpropyl-β-carotene. Molecules 2023, 28, 3505. https://doi.org/10.3390/molecules28083505
Koo S, Kim YH, Flender O, Scholz M, Oum K, Lenzer T. Photoinduced Dynamics of 13,13′-Diphenylpropyl-β-carotene. Molecules. 2023; 28(8):3505. https://doi.org/10.3390/molecules28083505
Chicago/Turabian StyleKoo, Sangho, Yeong Hun Kim, Oliver Flender, Mirko Scholz, Kawon Oum, and Thomas Lenzer. 2023. "Photoinduced Dynamics of 13,13′-Diphenylpropyl-β-carotene" Molecules 28, no. 8: 3505. https://doi.org/10.3390/molecules28083505
APA StyleKoo, S., Kim, Y. H., Flender, O., Scholz, M., Oum, K., & Lenzer, T. (2023). Photoinduced Dynamics of 13,13′-Diphenylpropyl-β-carotene. Molecules, 28(8), 3505. https://doi.org/10.3390/molecules28083505