
Photogrammetry of Ultrafast Excited-State Intramolecular Proton Transfer Pathways in the Fungal Pigment Draconin Red

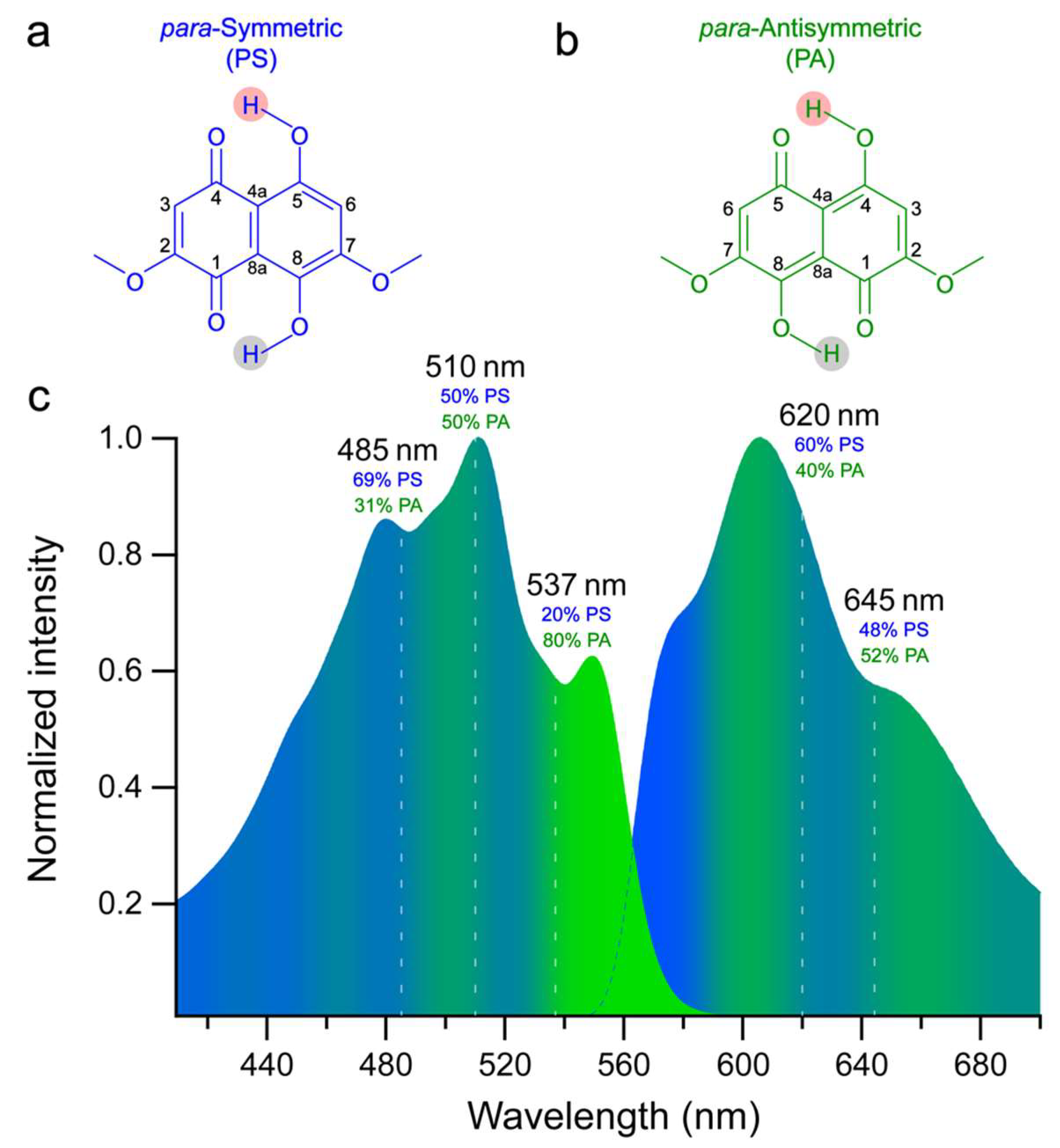{kind=link}
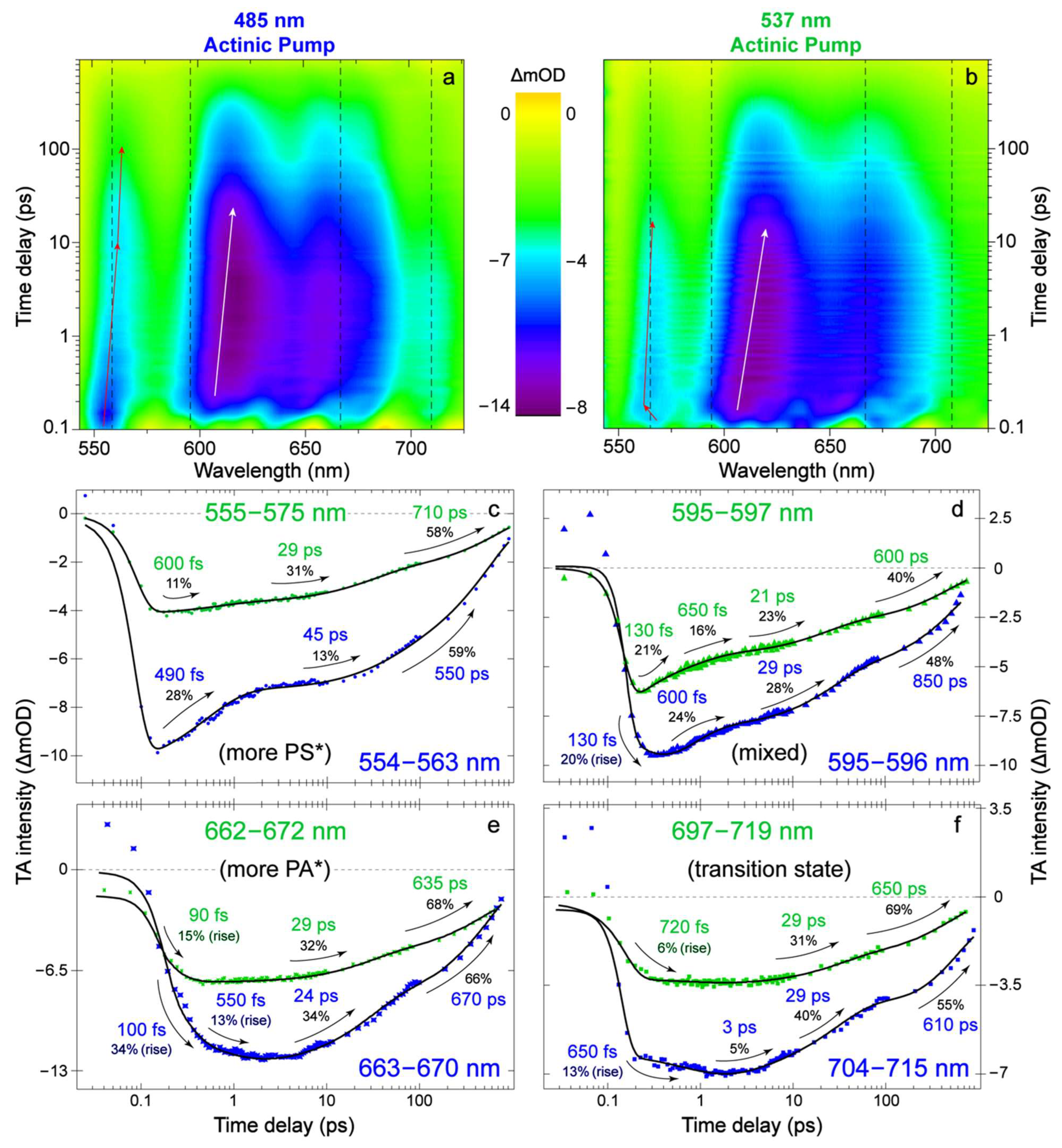{kind=link}
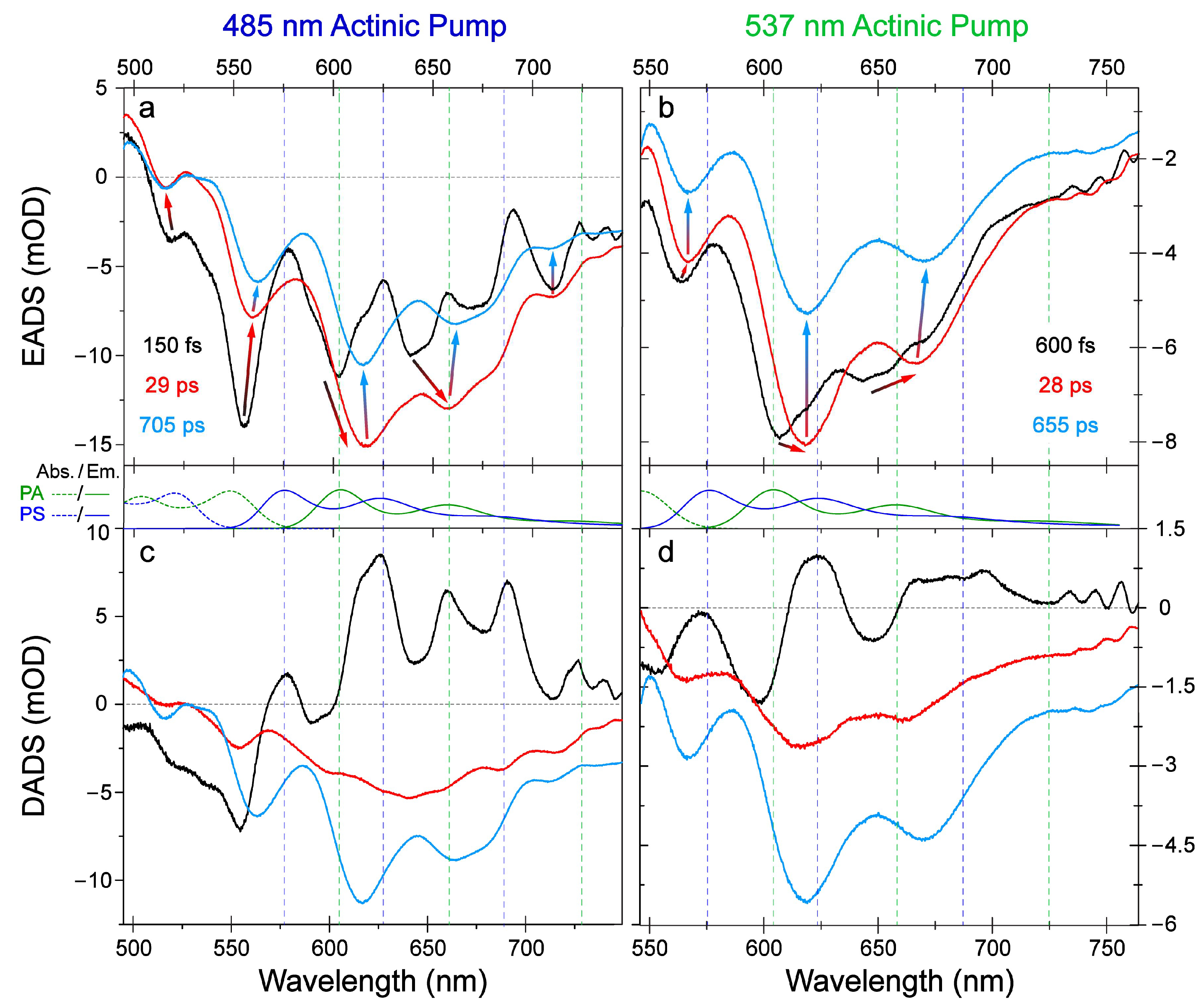{kind=link}
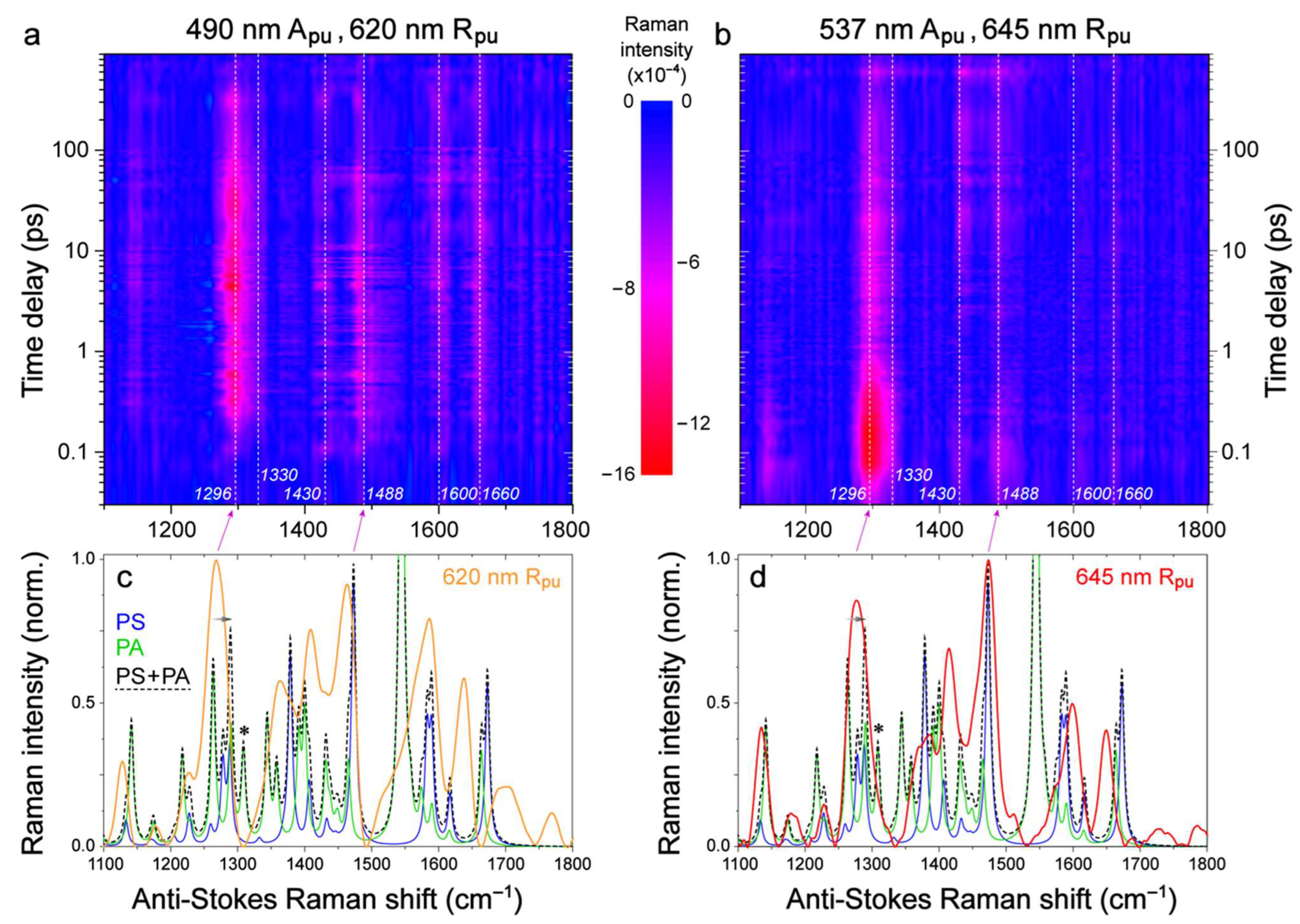{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Steady-State Electronic Spectroscopy and Femtosecond Transient Absorption Spectroscopy
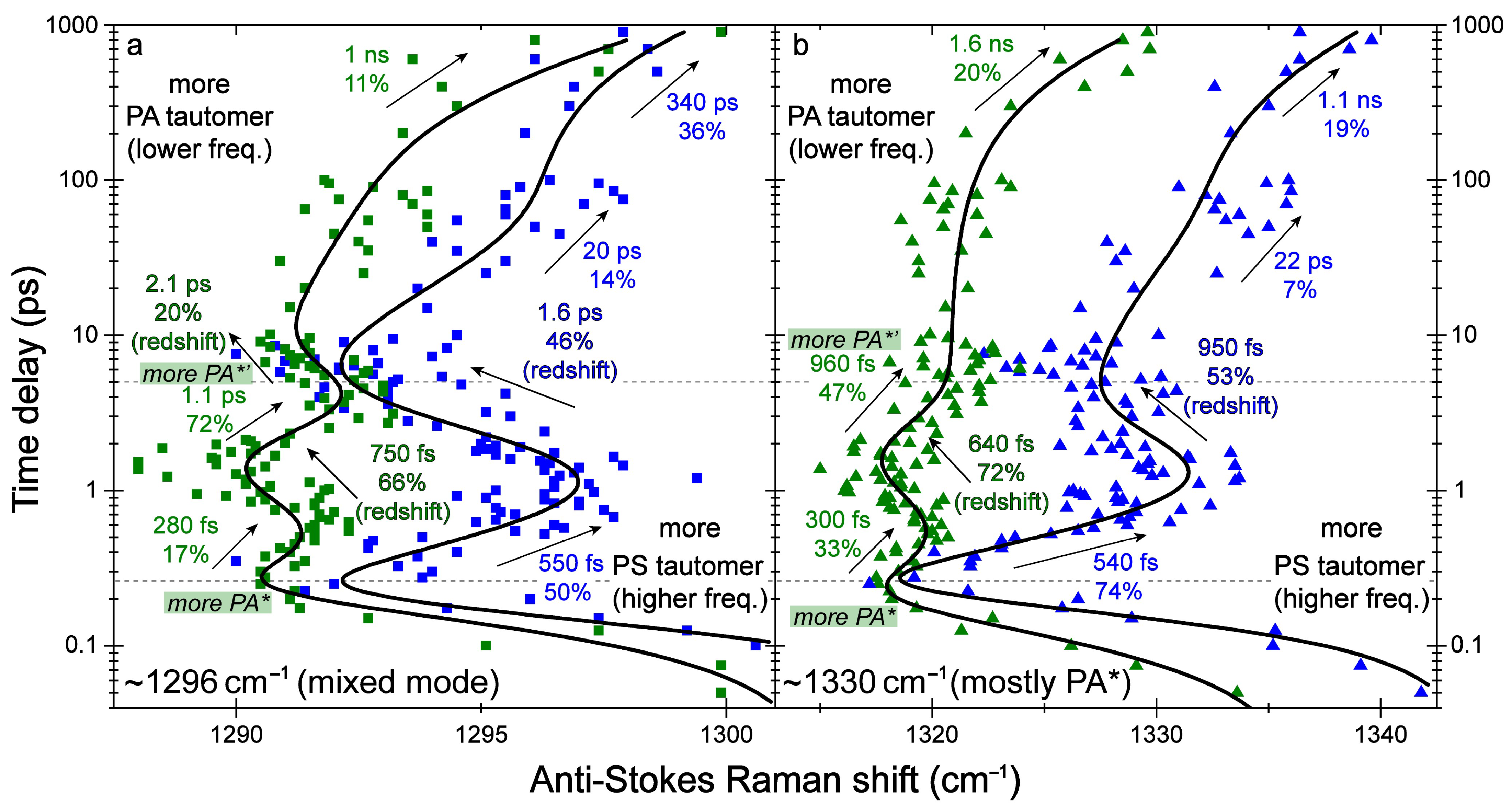
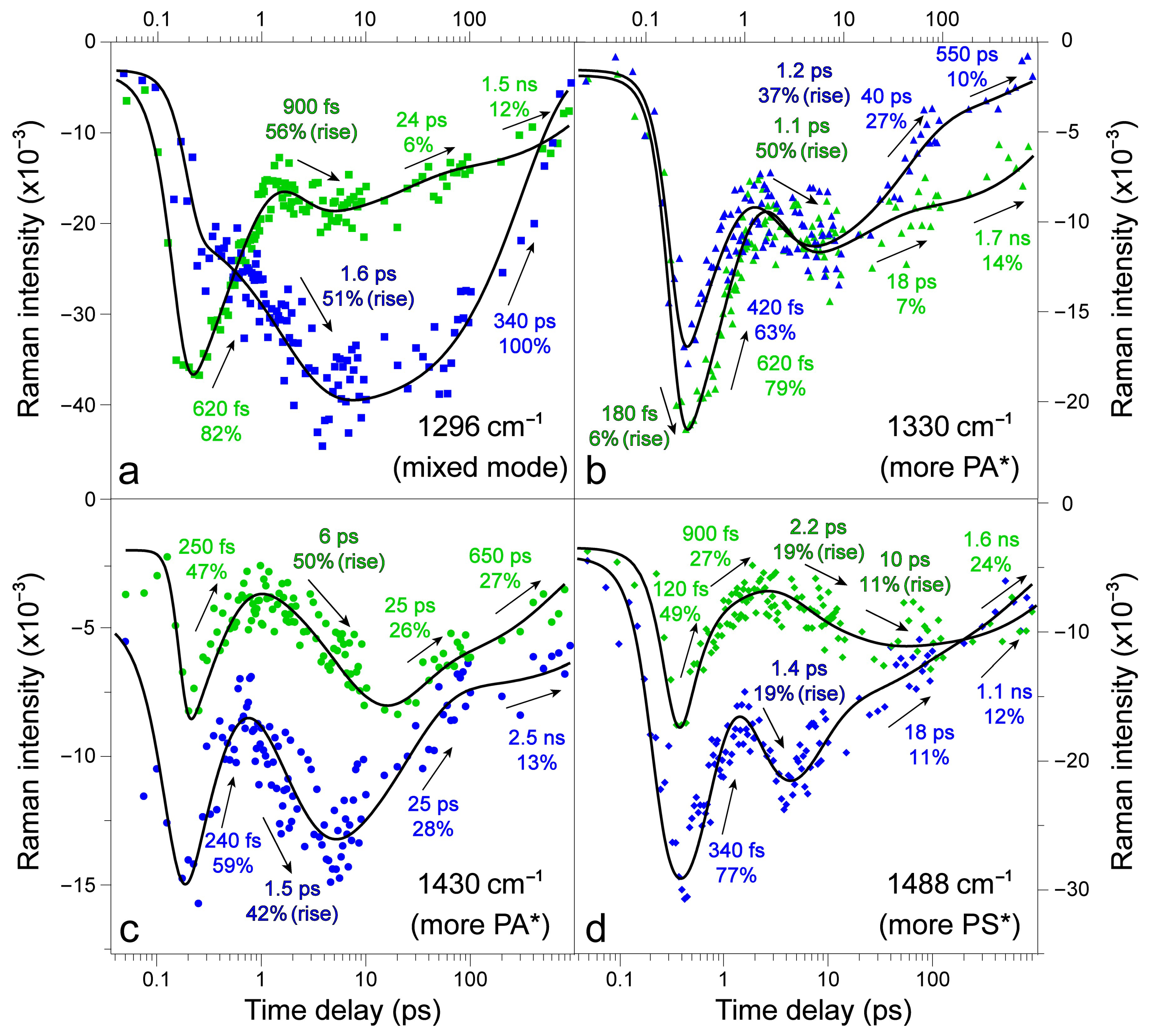
2.2. Femtosecond Stimulated Raman Spectroscopy (FSRS) of Draconin Red in Solution
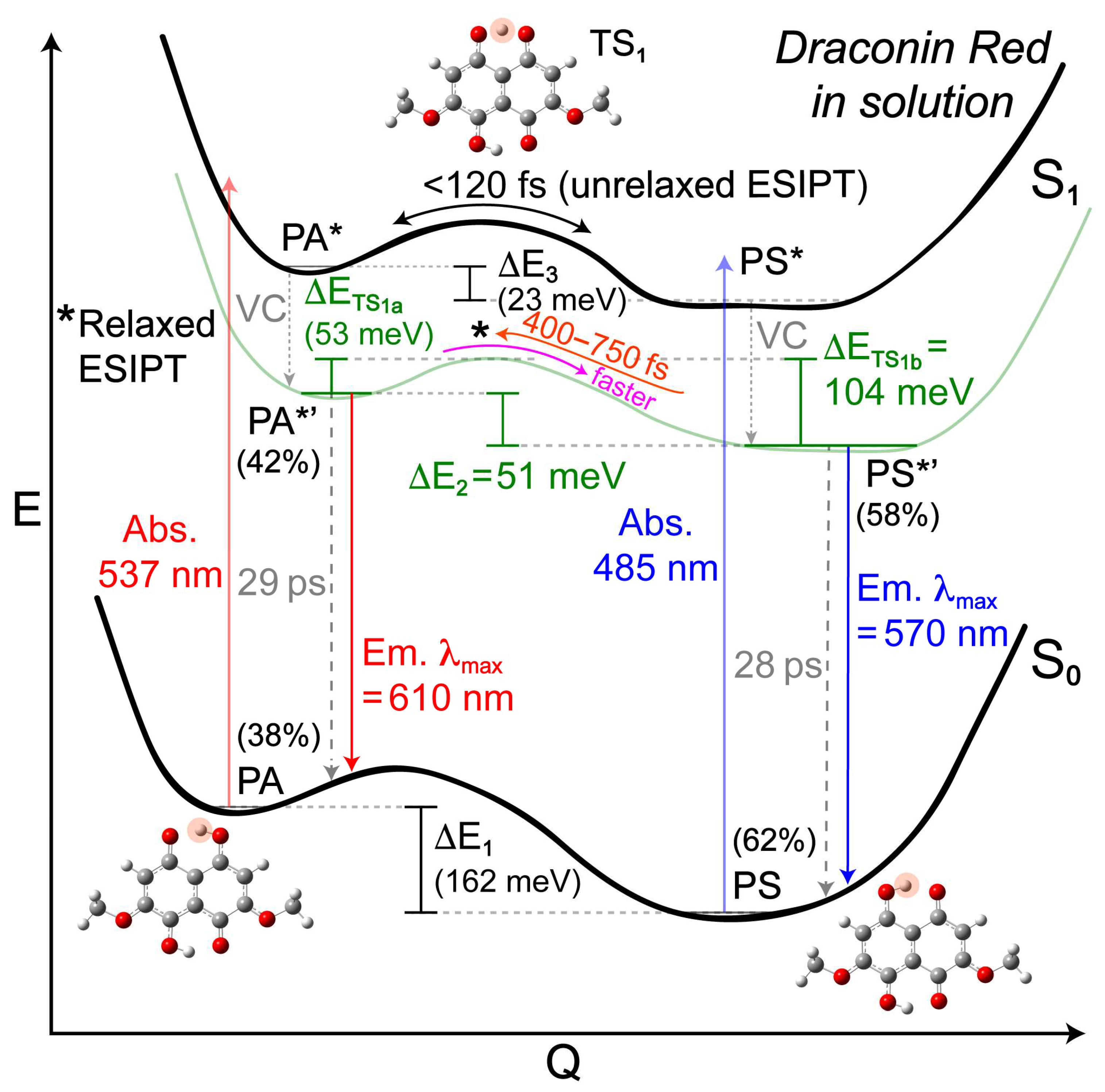
2.3. Revealing Bidirectional ESIPT during Excited-State Relaxation via Transient Raman Peak Dynamics
3. Perspectives and Conclusions
4. Materials and Methods
4.1. Sample Preparation
4.2. Steady-State Electronic Spectroscopy
4.3. Femtosecond Transient Absorption (fs-TA) and Femtosecond Stimulated Raman Spectroscopy (FSRS) from the Ground to Excited State
4.4. Quantum Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwon, J.E.; Park, S.Y. Advanced organic optoelectronic materials: Harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 2011, 23, 3615–3642. [Google Scholar] [CrossRef] [PubMed]
- Ostroverkhova, O. Organic optoelectronic materials: Mechanisms and applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef] [PubMed]
- Padalkar, V.S.; Seki, S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2016, 45, 169–202. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Fleming, G.R.; Chen, L.X.; Aspuru-Guzik, A.; Buchleitner, A.; Coker, D.F.; Engel, G.S.; van Grondelle, R.; Ishizaki, A.; Jonas, D.M.; et al. Using coherence to enhance function in chemical and biophysical systems. Nature 2017, 543, 647–656. [Google Scholar] [CrossRef]
- Mirkovic, T.; Ostroumov, E.E.; Anna, J.M.; van Grondelle, R.; Govindjee; Scholes, G.D. Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem. Rev. 2017, 117, 249–293. [Google Scholar] [CrossRef]
- Sedgwick, A.C.; Wu, L.; Han, H.-H.; Bull, S.D.; He, X.-P.; James, T.D.; Sessler, J.L.; Tang, B.Z.; Tian, H.; Yoon, J. Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842–8880. [Google Scholar] [CrossRef]
- Pradhan, S.; Brooks, A.K.; Yadavalli, V.K. Nature-derived materials for the fabrication of functional biodevices. Mater. Today Bio 2020, 7, 100065. [Google Scholar] [CrossRef]
- Bronstein, H.; Nielsen, C.B.; Schroeder, B.C.; McCulloch, I. The role of chemical design in the performance of organic semiconductors. Nat. Rev. Chem. 2020, 4, 66–77. [Google Scholar] [CrossRef]
- Krueger, T.D.; Fang, C. Elucidating inner workings of naturally sourced organic optoelectronic materials with ultrafast spectroscopy. Chem. Eur. J. 2021, 27, 17736–17750. [Google Scholar] [CrossRef]
- Di Mauro, E.; Rho, D.; Santato, C. Biodegradation of bio-sourced and synthetic organic electronic materials towards green organic electronics. Nat. Commun. 2021, 12, 3167. [Google Scholar] [CrossRef]
- Saikawa, Y.; Watanabe, T.; Hashimoto, K.; Nakata, M. Absolute configuration and tautomeric structure of xylindein, a blue–green pigment of Chlorociboria species. Phytochemistry 2000, 55, 237–240. [Google Scholar] [CrossRef]
- Robinson, S.C.; Tudor, D.; Snider, H.; Cooper, P.A. Stimulating growth and xylindein production of Chlorociboria aeruginascens in agar-based systems. AMB Express 2012, 2, 15. [Google Scholar] [CrossRef]
- Giesbers, G.; Krueger, T.; Van Schenck, J.; Van Court, R.; Morré, J.; Fang, C.; Robinson, S.; Ostroverkhova, O. Fungi-derived xylindein: Effect of purity on optical and electronic properties. MRS Adv. 2019, 4, 1769–1777. [Google Scholar] [CrossRef]
- Giesbers, G.; Van Schenck, J.; Quinn, A.; Van Court, R.; Vega Gutierrez, S.M.; Robinson, S.C.; Ostroverkhova, O. Xylindein: Naturally produced fungal compound for sustainable (opto)electronics. ACS Omega 2019, 4, 13309–13318. [Google Scholar] [CrossRef]
- Giesbers, G.; Krueger, T.D.; Van Schenck, J.D.B.; Kim, R.; Van Court, R.C.; Robinson, S.C.; Beaudry, C.M.; Fang, C.; Ostroverkhova, O. Role of hydroxyl groups in the photophysics, photostability, and (opto)electronic properties of the fungi-derived pigment xylindein. J. Phys. Chem. C 2021, 125, 6534–6545. [Google Scholar] [CrossRef]
- Krueger, T.D.; Giesbers, G.; Van Court, R.C.; Zhu, L.; Kim, R.; Beaudry, C.M.; Robinson, S.C.; Ostroverkhova, O.; Fang, C. Ultrafast dynamics and photoresponse of a fungi-derived pigment xylindein from solution to thin films. Chem. Eur. J. 2021, 27, 5627–5631. [Google Scholar] [CrossRef]
- Le Person, A.; Cornard, J.-P.; Say-Liang-Fat, S. Studies of the tautomeric forms of alizarin in the ground state by electronic spectroscopy combined with quantum chemical calculations. Chem. Phys. Lett. 2011, 517, 41–45. [Google Scholar] [CrossRef]
- Mech, J.; Grela, M.A.; Szaciłowski, K. Ground and excited state properties of alizarin and its isomers. Dye. Pigment. 2014, 103, 202–213. [Google Scholar] [CrossRef]
- Jen, M.; Lee, S.; Jeon, K.; Hussain, S.; Pang, Y. Ultrafast intramolecular proton transfer of alizarin investigated by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2017, 121, 4129–4136. [Google Scholar] [CrossRef]
- Flom, S.R.; Barbara, P.F. Proton transfer and hydrogen bonding in the internal conversion of S1 anthraquinones. J. Phys. Chem. 1985, 89, 4489–4494. [Google Scholar] [CrossRef]
- Gai, F.; Fehr, M.J.; Petrich, J.W. Observation of excited-state tautomerization in the antiviral agent hypericin and identification of its fluorescent species. J. Phys. Chem. 1994, 98, 5784–5795. [Google Scholar] [CrossRef]
- Lochbrunner, S.; Wurzer, A.J.; Riedle, E. Ultrafast excited-state proton transfer and subsequent coherent skeletal motion of 2-(2′-hydroxyphenyl)benzothiazole. J. Chem. Phys. 2000, 112, 10699–10702. [Google Scholar] [CrossRef]
- Ameer-Beg, S.; Ormson, S.M.; Brown, R.G.; Matousek, P.; Towrie, M.; Nibbering, E.T.J.; Foggi, P.; Neuwahl, F.V.R. Ultrafast measurements of excited state intramolecular proton transfer (ESIPT) in room temperature solutions of 3-hydroxyflavone and derivatives. J. Phys. Chem. A 2001, 105, 3709–3718. [Google Scholar] [CrossRef]
- Lochbrunner, S.; Wurzer, A.J.; Riedle, E. Microscopic mechanism of ultrafast excited-state intramolecular proton transfer: A 30-fs study of 2-(2‘-hydroxyphenyl)benzothiazole. J. Phys. Chem. A 2003, 107, 10580–10590. [Google Scholar] [CrossRef]
- Chen, K.-Y.; Cheng, Y.-M.; Lai, C.-H.; Hsu, C.-C.; Ho, M.-L.; Lee, G.-H.; Chou, P.-T. Ortho green fluorescence protein synthetic chromophore; excited-state intramolecular proton transfer via a seven-membered-ring hydrogen-bonding system. J. Am. Chem. Soc. 2007, 129, 4534–4535. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, J.; Yang, P.; Huang, D.; Zhang, C.; Li, Q. New excited state intramolecular proton transfer (ESIPT) dyes based on naphthalimide and observation of long-lived triplet excited states. Chem. Commun. 2012, 48, 9720–9722. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, H.; Zheng, Y. Theoretical insights into the excited state double proton transfer mechanism of deep red pigment alkannin. J. Phys. Chem. A 2018, 122, 1200–1208. [Google Scholar] [CrossRef]
- Berenbeim, J.A.; Boldissar, S.; Owens, S.; Haggmark, M.R.; Gate, G.; Siouri, F.M.; Cohen, T.; Rode, M.F.; Patterson, C.S.; de Vries, M.S. Excited state intramolecular proton transfer in hydroxyanthraquinones: Toward predicting fading of organic red colorants in art. Sci. Adv. 2019, 5, eaaw5227. [Google Scholar] [CrossRef]
- Yoneda, Y.; Sotome, H.; Mathew, R.; Lakshmanna, Y.A.; Miyasaka, H. Non-condon effect on ultrafast excited-state intramolecular proton transfer. J. Phys. Chem. A 2020, 124, 265–271. [Google Scholar] [CrossRef]
- Qin, C.; Liu, H.; Sun, S.; Zhou, Z.; Liu, Y. Ultrafast investigation of intramolecular proton transfer dynamics and vibration relaxation in 1,8-dihydroxyanthraquinone. J. Mol. Struct. 2021, 1229, 129502. [Google Scholar] [CrossRef]
- Nag, P.; Anand, N.; Vennapusa, S.R. Ultrafast nonadiabatic excited-state intramolecular proton transfer in 3-hydroxychromone: A surface hopping approach. J. Chem. Phys. 2021, 155, 094301. [Google Scholar] [CrossRef]
- Boulanger, S.A.; Chen, C.; Myasnyanko, I.N.; Baranov, M.S.; Fang, C. Fluorescence modulation of ortho-green fluorescent protein chromophores following ultrafast proton transfer in solution. J. Phys. Chem. B 2022, 126, 5081–5093. [Google Scholar] [CrossRef]
- Zhang, X.; Schwarz, K.N.; Zhang, L.; Fassioli, F.; Fu, B.; Nguyen, L.Q.; Knowles, R.R.; Scholes, G.D. Interference of nuclear wavepackets in a pair of proton transfer reactions. Proc. Natl. Acad. Sci. USA 2022, 119, e2212114119. [Google Scholar] [CrossRef]
- Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Excited state intramolecular proton transfer (ESIPT): From principal photophysics to the development of new chromophores and applications in fluorescent molecular probes and luminescent materials. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. [Google Scholar] [CrossRef]
- Giesbers, G.; Van Schenck, J.; Vega Gutierrez, S.; Robinson, S.; Ostroverkhova, O. Fungi-derived pigments for sustainable organic (opto)electronics. MRS Adv. 2018, 3, 3459–3464. [Google Scholar] [CrossRef]
- Wu, J.-J.; Gao, H.; Lai, R.; Zhuo, M.-P.; Feng, J.; Wang, X.-D.; Wu, Y.; Liao, L.-S.; Jiang, L. Near-infrared organic single-crystal nanolaser arrays activated by excited-state intramolecular proton transfer. Matter 2020, 2, 1233–1243. [Google Scholar] [CrossRef]
- Weber, G.; Chen, H.-L.; Hinsch, E.; Freitas, S.; Robinson, S. Pigments extracted from the wood-staining fungi Chlorociboria aeruginosa, Scytalidium cuboideum, and S. ganodermophthorum show potential for use as textile dyes. Color. Technol. 2014, 130, 445–452. [Google Scholar] [CrossRef]
- Vega Gutierrez, S.M.; Hazell, K.K.; Simonsen, J.; Robinson, S.C. Description of a naphthoquinonic crystal produced by the fungus Scytalidium cuboideum. Molecules 2018, 23, 1905. [Google Scholar] [CrossRef]
- Vega Gutierrez, S.M.; He, Y.; Cao, Y.; Stone, D.; Walsh, Z.; Malhotra, R.; Chen, H.-L.; Chang, C.-H.; Robinson, S.C. Feasibility and surface evaluation of the pigment from Scytalidium cuboideum for inkjet printing on textiles. Coatings 2019, 9, 266. [Google Scholar] [CrossRef]
- Chang, C.W.J.; Moore, R.E.; Scheuer, P.J. The structure of Spinochrome M. J. Am. Chem. Soc. 1964, 86, 2959–2961. [Google Scholar] [CrossRef]
- Gerber, N.N.; Wieclawek, B. The structures of two naphthoquinone pigments from an actinomycete. J. Org. Chem. 1966, 31, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- McGovern, E.P.; Bentley, R. Biosynthesis of flaviolin and 5,8-dihydroxy-2,7-dimethoxy-1,4-naphthoquinone. Biochemistry 1975, 14, 3138–3143. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Rentzepis, P.M. On the picosecond kinetics and photostability of indigo and 6,6’-dimethoxyindigo. J. Chem. Phys. 1979, 70, 886–892. [Google Scholar] [CrossRef]
- Klimovich, I.V.; Leshanskaya, L.I.; Troyanov, S.I.; Anokhin, D.V.; Novikov, D.V.; Piryazev, A.A.; Ivanov, D.A.; Dremova, N.N.; Troshin, P.A. Design of indigo derivatives as environment-friendly organic semiconductors for sustainable organic electronics. J. Mater. Chem. C 2014, 2, 7621–7631. [Google Scholar] [CrossRef]
- He, X.; Yang, F.; Li, S.; He, X.; Yu, A.; Chen, J.; Xu, J.; Wang, J. Ultrafast excited-state intermolecular proton transfer in indigo oligomer. J. Phys. Chem. A 2019, 123, 6463–6471. [Google Scholar] [CrossRef]
- Krueger, T.D.; Solaris, J.; Tang, L.; Zhu, L.; Webber, C.; Van Court, R.C.; Robinson, S.C.; Ostroverkhova, O.; Fang, C. Illuminating excited-state intramolecular proton transfer of a fungi-derived red pigment for sustainable functional materials. J. Phys. Chem. C 2022, 126, 459–477. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L.; Oscar, B.G.; Chen, C. Capturing structural snapshots during photochemical reactions with ultrafast Raman spectroscopy: From materials transformation to biosensor responses. J. Phys. Chem. Lett. 2018, 9, 3253–3263. [Google Scholar] [CrossRef]
- Bravaya, K.B.; Grigorenko, B.L.; Nemukhin, A.V.; Krylov, A.I. Quantum chemistry behind bioimaging: Insights from ab initio studies of fluorescent proteins and their chromophores. Acc. Chem. Res. 2012, 45, 265–275. [Google Scholar] [CrossRef]
- Gozem, S.; Melaccio, F.; Luk, H.L.; Rinaldi, S.; Olivucci, M. Learning from photobiology how to design molecular devices using a computer. Chem. Soc. Rev. 2014, 43, 4019–4036. [Google Scholar] [CrossRef]
- Donati, G.; Petrone, A.; Caruso, P.; Rega, N. The mechanism of a green fluorescent protein proton shuttle unveiled in the time-resolved frequency domain by excited state ab initio dynamics. Chem. Sci. 2018, 9, 1126–1135. [Google Scholar] [CrossRef]
- Zhou, P.; Han, K. Unraveling the detailed mechanism of excited-state proton transfer. Acc. Chem. Res. 2018, 51, 1681–1690. [Google Scholar] [CrossRef]
- Fedorov, D.A.; Seritan, S.; Fales, B.S.; Martínez, T.J.; Levine, B.G. PySpawn: Software for nonadiabatic quantum molecular dynamics. J. Chem. Theory Comput. 2020, 16, 5485–5498. [Google Scholar] [CrossRef]
- Coppola, F.; Cimino, P.; Raucci, U.; Chiariello, M.G.; Petrone, A.; Rega, N. Exploring the Franck–Condon region of a photoexcited charge transfer complex in solution to interpret femtosecond stimulated Raman spectroscopy: Excited state electronic structure methods to unveil non-radiative pathways. Chem. Sci. 2021, 12, 8058–8072. [Google Scholar] [CrossRef]
- Jones, C.M.; List, N.H.; Martínez, T.J. Resolving the ultrafast dynamics of the anionic green fluorescent protein chromophore in water. Chem. Sci. 2021, 12, 11347–11363. [Google Scholar] [CrossRef]
- Loe, C.M.; Liekhus-Schmaltz, C.; Govind, N.; Khalil, M. Spectral signatures of ultrafast excited-state intramolecular proton transfer from computational multi-edge transient X-ray absorption spectroscopy. J. Phys. Chem. Lett. 2021, 12, 9840–9847. [Google Scholar] [CrossRef]
- Xu, X.; Chen, Z.; Yang, Y. Molecular dynamics with constrained nuclear electronic orbital density functional theory: Accurate vibrational spectra from efficient incorporation of nuclear quantum effects. J. Am. Chem. Soc. 2022, 144, 4039–4046. [Google Scholar] [CrossRef]
- Liu, W.; Han, F.; Smith, C.; Fang, C. Ultrafast conformational dynamics of pyranine during excited state proton transfer in aqueous solution revealed by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 10535–10550. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, W.; Fang, C. A versatile femtosecond stimulated Raman spectroscopy setup with tunable pulses in the visible to near infrared. Appl. Phys. Lett. 2014, 105, 041106. [Google Scholar]
- Dietze, D.R.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. ChemPhysChem 2016, 17, 1224–1251. [Google Scholar] [CrossRef]
- Liu, W.; Tang, L.; Oscar, B.G.; Wang, Y.; Chen, C.; Fang, C. Tracking ultrafast vibrational cooling during excited state proton transfer reaction with anti-Stokes and Stokes femtosecond stimulated Raman spectroscopy. J. Phys. Chem. Lett. 2017, 8, 997–1003. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L.; Chen, C. Unveiling coupled electronic and vibrational motions of chromophores in condensed phases. J. Chem. Phys. 2019, 151, 200901. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhu, L.; Taylor, M.A.; Wang, Y.; Remington, S.J.; Fang, C. Excited state structural evolution of a GFP single-site mutant tracked by tunable femtosecond-stimulated Raman spectroscopy. Molecules 2018, 23, 2226. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhu, L.; Fang, C. Femtosecond stimulated Raman line shapes: Dependence on resonance conditions of pump and probe pulses. Chin. J. Chem. Phys. 2018, 31, 492–502. [Google Scholar] [CrossRef]
- Fang, C.; Tang, L. Mapping structural dynamics of proteins with femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2020, 71, 239–265. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.; Chen, C.; Zhu, R.; Jiang, J.; Weng, T.-C.; Ji, Q.; Huang, Y.; Fang, C.; Liu, W. Mapping the complete photocycle that powers a large Stokes shift red fluorescent protein. Angew. Chem. Int. Ed. 2023, 62, e202212209. [Google Scholar]
- Provencher, F.; Bérubé, N.; Parker, A.W.; Greetham, G.M.; Towrie, M.; Hellmann, C.; Côté, M.; Stingelin, N.; Silva, C.; Hayes, S.C. Direct observation of ultrafast long-range charge separation at polymer-fullerene heterojunctions. Nat. Commun. 2014, 5, 4288. [Google Scholar] [CrossRef]
- Tang, L.; Liu, W.; Wang, Y.; Zhao, Y.; Oscar, B.G.; Campbell, R.E.; Fang, C. Unraveling ultrafast photoinduced proton transfer dynamics in a fluorescent protein biosensor for Ca2+ imaging. Chem. Eur. J. 2015, 21, 6481–6490. [Google Scholar] [CrossRef]
- Zhou, J.; Yu, W.; Bragg, A.E. Structural relaxation of photoexcited quaterthiophenes probed with vibrational specificity. J. Phys. Chem. Lett. 2015, 6, 3496–3502. [Google Scholar] [CrossRef]
- Batignani, G.; Pontecorvo, E.; Ferrante, C.; Aschi, M.; Elles, C.G.; Scopigno, T. Visualizing excited-state dynamics of a diaryl thiophene: Femtosecond stimulated Raman scattering as a probe of conjugated molecules. J. Phys. Chem. Lett. 2016, 7, 2981–2988. [Google Scholar] [CrossRef]
- Hall, C.R.; Conyard, J.; Heisler, I.A.; Jones, G.; Frost, J.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast dynamics in light-driven molecular rotary motors probed by femtosecond stimulated Raman spectroscopy. J. Am. Chem. Soc. 2017, 139, 7408–7414. [Google Scholar] [CrossRef]
- Barclay, M.S.; Caricato, M.; Elles, C.G. Femtosecond stimulated Raman scattering from triplet electronic states: Experimental and theoretical study of resonance enhancements. J. Phys. Chem. A 2019, 123, 7720–7732. [Google Scholar] [CrossRef]
- Ferrante, C.; Batignani, G.; Pontecorvo, E.; Montemiglio, L.C.; Vos, M.H.; Scopigno, T. Ultrafast dynamics and vibrational relaxation in six-coordinate heme proteins revealed by femtosecond stimulated Raman spectroscopy. J. Am. Chem. Soc. 2020, 142, 2285–2292. [Google Scholar] [CrossRef]
- Batignani, G.; Ferrante, C.; Scopigno, T. Accessing excited state molecular vibrations by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. Lett. 2020, 11, 7805–7813. [Google Scholar] [CrossRef]
- Bailey-Darland, S.; Krueger, T.D.; Fang, C. Ultrafast spectroscopies of nitrophenols and nitrophenolates in solution: From electronic dynamics and vibrational structures to photochemical and environmental implications. Molecules 2023, 28, 601. [Google Scholar] [CrossRef]
- Roy, P.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast motion in a third generation photomolecular motor. Nat. Commun. 2023, 14, 1253. [Google Scholar] [CrossRef]
- Mukamel, S.; Biggs, J.D. Communication: Comment on the effective temporal and spectral resolution of impulsive stimulated Raman signals. J. Chem. Phys. 2011, 134, 161101. [Google Scholar] [CrossRef]
- Fumero, G.; Batignani, G.; Dorfman, K.E.; Mukamel, S.; Scopigno, T. On the resolution limit of femtosecond stimulated Raman spectroscopy: Modelling fifth-order signals with overlapping pulses. ChemPhysChem 2015, 16, 3438–3443. [Google Scholar] [CrossRef]
- Liebel, M.; Schnedermann, C.; Wende, T.; Kukura, P. Principles and applications of broadband impulsive vibrational spectroscopy. J. Phys. Chem. A 2015, 119, 9506–9517. [Google Scholar] [CrossRef]
- Kuramochi, H.; Takeuchi, S.; Tahara, T. Femtosecond time-resolved impulsive stimulated Raman spectroscopy using sub-7-fs pulses: Apparatus and applications. Rev. Sci. Instrum. 2016, 87, 043107. [Google Scholar]
- Kuramochi, H.; Takeuchi, S.; Yonezawa, K.; Kamikubo, H.; Kataoka, M.; Tahara, T. Probing the early stages of photoreception in photoactive yellow protein with ultrafast time-domain Raman spectroscopy. Nat. Chem. 2017, 9, 660–666. [Google Scholar] [CrossRef]
- Batignani, G.; Sansone, C.; Ferrante, C.; Fumero, G.; Mukamel, S.; Scopigno, T. Excited-state energy surfaces in molecules revealed by impulsive stimulated Raman excitation profiles. J. Phys. Chem. Lett. 2021, 12, 9239–9247. [Google Scholar] [CrossRef] [PubMed]
- Dierksen, M.; Grimme, S. Density functional calculations of the vibronic structure of electronic absorption spectra. J. Chem. Phys. 2004, 120, 3544–3554. [Google Scholar] [CrossRef] [PubMed]
- Bloino, J.; Biczysko, M.; Santoro, F.; Barone, V. General approach to compute vibrationally resolved one-photon electronic spectra. J. Chem. Theory Comput. 2010, 6, 1256–1274. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with time-dependent density functional theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Chen, F.; Zhao, X.; Liang, W. One- and two-photon absorption spectra of the yellow fluorescent protein citrine: Effects of intramolecular electron-vibrational coupling and intermolecular interactions. Mol. Phys. 2018, 116, 885–897. [Google Scholar] [CrossRef]
- Benkyi, I.; Tapavicza, E.; Fliegl, H.; Sundholm, D. Calculation of vibrationally resolved absorption spectra of acenes and pyrene. Phys. Chem. Chem. Phys. 2019, 21, 21094–21103. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Montoya-Castillo, A.; Napoli, J.A.; Markland, T.E.; Isborn, C.M. Optical spectra in the condensed phase: Capturing anharmonic and vibronic features using dynamic and static approaches. J. Chem. Phys. 2019, 151, 074111. [Google Scholar] [CrossRef]
- Zuehlsdorff, T.J.; Shedge, S.V.; Lu, S.-Y.; Hong, H.; Aguirre, V.P.; Shi, L.; Isborn, C.M. Vibronic and environmental effects in simulations of optical spectroscopy. Annu. Rev. Phys. Chem. 2021, 72, 165–188. [Google Scholar] [CrossRef]
- Bader, A.N.; Pivovarenko, V.G.; Demchenko, A.P.; Ariese, F.; Gooijer, C. Excited state and ground state proton transfer rates of 3-hydroxyflavone and its derivatives studied by Shpol’skii spectroscopy: The influence of redistribution of electron density. J. Phys. Chem. B 2004, 108, 10589–10595. [Google Scholar] [CrossRef]
- Rodriguez, A.A.; Schwartz, M. Raman study of peak frequencies and linewidths of the νi mode of trichloroethane in solution. Spectrochim. Acta A 1988, 44, 43–46. [Google Scholar] [CrossRef]
- Horng, M.L.; Gardecki, J.A.; Papazyan, A.; Maroncelli, M. Subpicosecond measurements of polar solvation dynamics: Coumarin 153 revisited. J. Phys. Chem. 1995, 99, 17311–17337. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.P.; Seger, R.; Mullen, K.M.; van Stokkum, I.H.M. Glotaran: A Java-based graphical user interface for the R-package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Kim, P.W.; Rockwell, N.C.; Martin, S.S.; Lagarias, J.C.; Larsen, D.S. Dynamic inhomogeneity in the photodynamics of cyanobacterial phytochrome Cph1. Biochemistry 2014, 53, 2818–2826. [Google Scholar] [CrossRef]
- Tang, L.; Liu, W.; Wang, Y.; Zhu, L.; Han, F.; Fang, C. Ultrafast structural evolution and chromophore inhomogeneity inside a green-fluorescent-protein-based Ca2+ biosensor. J. Phys. Chem. Lett. 2016, 7, 1225–1230. [Google Scholar] [CrossRef]
- Fang, C.; Hochstrasser, R.M. Two-dimensional infrared spectra of the 13c=18o isotopomers of alanine residues in an α-helix. J. Phys. Chem. B 2005, 109, 18652–18663. [Google Scholar] [CrossRef]
- Laptenok, S.P.; Gil, A.A.; Hall, C.R.; Lukacs, A.; Iuliano, J.N.; Jones, G.A.; Greetham, G.M.; Donaldson, P.; Miyawaki, A.; Tonge, P.J.; et al. Infrared spectroscopy reveals multi-step multi-timescale photoactivation in the photoconvertible protein archetype Dronpa. Nat. Chem. 2018, 10, 845–852. [Google Scholar] [CrossRef]
- Harbola, U.; Umapathy, S.; Mukamel, S. Loss and gain signals in broadband stimulated-Raman spectra: Theoretical analysis. Phys. Rev. A 2013, 88, 011801. [Google Scholar] [CrossRef]
- Nakamura, R.; Hamada, N.; Abe, K.; Yoshizawa, M. Ultrafast hydrogen-bonding dynamics in the electronic excited state of photoactive yellow protein revealed by femtosecond stimulated Raman spectroscopy. J. Phys. Chem. B 2012, 116, 14768–14775. [Google Scholar] [CrossRef]
- Batignani, G.; Fumero, G.; Mukamel, S.; Scopigno, T. Energy flow between spectral components in 2D broadband stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2015, 17, 10454–10461. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Yoon, S.; Wandschneider, D.B.; Mathies, R.A. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science 2005, 310, 1006–1009. [Google Scholar] [CrossRef]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Batignani, G.; Pontecorvo, E.; Giovannetti, G.; Ferrante, C.; Fumero, G.; Scopigno, T. Electronic resonances in broadband stimulated Raman spectroscopy. Sci. Rep. 2016, 6, 18445. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.; Pontecorvo, E.; Cerullo, G.; Vos, M.H.; Scopigno, T. Direct observation of subpicosecond vibrational dynamics in photoexcited myoglobin. Nat. Chem. 2016, 8, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Oscar, B.G.; Chen, C.; Liu, W.; Zhu, L.; Fang, C. Dynamic Raman line shapes on an evolving excited-state landscape: Insights from tunable femtosecond stimulated Raman spectroscopy. J. Phys. Chem. A 2017, 121, 5428–5441. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Tang, L.; Oscar, B.G.; Zhu, L.; Fang, C. Panoramic portrait of primary molecular events preceding excited state proton transfer in water. Chem. Sci. 2016, 7, 5484–5494. [Google Scholar] [CrossRef]
- Hamm, P.; Ohline, S.M.; Zinth, W. Vibrational cooling after ultrafast photoisomerization of azobenzene measured by femtosecond infrared spectroscopy. J. Chem. Phys. 1997, 106, 519–529. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Schanz, R.; Hennig, H.; Ernsting, N.P. Cooling dynamics of an optically excited molecular probe in solution from femtosecond broadband transient absorption spectroscopy. J. Chem. Phys. 2001, 115, 3256–3273. [Google Scholar] [CrossRef]
- Kumpulainen, T.; Lang, B.; Rosspeintner, A.; Vauthey, E. Ultrafast elementary photochemical processes of organic molecules in liquid solution. Chem. Rev. 2017, 117, 10826–10939. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Y.; Zhu, L.; Lee, C.; Fang, C. Correlated molecular structural motions for photoprotection after deep-UV irradiation. J. Phys. Chem. Lett. 2018, 9, 2311–2319. [Google Scholar] [CrossRef]
- Baumler, S.M.; Mutchler, J.M.; Blanchard, G.J. Comparing rotational and translational diffusion to evaluate heterogeneity in binary solvent systems. J. Phys. Chem. B 2019, 123, 216–224. [Google Scholar] [CrossRef]
- Chen, C.; Zhu, L.; Baranov, M.S.; Tang, L.; Baleeva, N.S.; Smirnov, A.Y.; Yampolsky, I.V.; Solntsev, K.M.; Fang, C. Photoinduced proton transfer of GFP-inspired fluorescent superphotoacids: Principles and design. J. Phys. Chem. B 2019, 123, 3804–3821. [Google Scholar] [CrossRef]
- Krueger, T.D.; Tang, L.; Fang, C. Delineating ultrafast structural dynamics of a green-red fluorescent protein for calcium sensing. Biosensors 2023, 13, 218. [Google Scholar] [CrossRef]
- Hoffman, D.P.; Mathies, R.A. Femtosecond stimulated Raman exposes the role of vibrational coherence in condensed-phase photoreactivity. Acc. Chem. Res. 2016, 49, 616–625. [Google Scholar] [CrossRef]
- Nandy, R.; Sankararaman, S. Donor-acceptor substituted phenylethynyltriphenylenes—Excited state intramolecular charge transfer, solvatochromic absorption and fluorescence emission. Beilstein J. Org. Chem. 2010, 6, 992–1001. [Google Scholar] [CrossRef]
- Gilbert, M.; Albinsson, B. Photoinduced charge and energy transfer in molecular wires. Chem. Soc. Rev. 2015, 44, 845–862. [Google Scholar] [CrossRef]
- Chen, C.; Fang, C. Devising efficient red-shifting strategies for bioimaging: A generalizable donor-acceptor fluorophore prototype. Chem. Asian J. 2020, 15, 1514–1523. [Google Scholar] [CrossRef]
- Vega Gutierrez, S.M.; Van Court, R.C.; Stone, D.W.; Konkler, M.J.; Groth, E.N.; Robinson, S.C. Relationship between molarity and color in the crystal (‘Dramada’) produced by Scytalidium cuboideum, in two solvents. Molecules 2018, 23, 2581. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Shim, S.; Mathies, R.A. Origin of negative and dispersive features in anti-Stokes and resonance femtosecond stimulated Raman spectroscopy. J. Chem. Phys. 2008, 129, 064507. [Google Scholar] [CrossRef]
- Weigel, A.; Dobryakov, A.; Klaumünzer, B.; Sajadi, M.; Saalfrank, P.; Ernsting, N.P. Femtosecond stimulated Raman spectroscopy of flavin after optical excitation. J. Phys. Chem. B 2011, 115, 3656–3680. [Google Scholar] [CrossRef]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar] [CrossRef]
- Kashinski, D.O.; Chase, G.M.; Nelson, R.G.; Di Nallo, O.E.; Scales, A.N.; VanderLey, D.L.; Byrd, E.F.C. Harmonic vibrational frequencies: Approximate global scaling factors for TPSS, M06, and M11 functional families using several common basis sets. J. Phys. Chem. A 2017, 121, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kayal, S.; Ariese, F.; Beeby, A.; Umapathy, S. Mode specific excited state dynamics study of bis(phenylethynyl)benzene from ultrafast Raman loss spectroscopy. J. Chem. Phys. 2017, 146, 064303. [Google Scholar] [CrossRef] [PubMed]
- Berera, R.; van Grondelle, R.; Kennis, J.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.; Schatz, G.C. Resonance Raman scattering of rhodamine 6G as calculated using time-dependent density functional theory. J. Phys. Chem. A 2006, 110, 5973–5977. [Google Scholar] [CrossRef]
- Quincy, T.J.; Barclay, M.S.; Caricato, M.; Elles, C.G. Probing dynamics in higher-lying electronic states with resonance-enhanced femtosecond stimulated Raman spectroscopy. J. Phys. Chem. A 2018, 122, 8308–8319. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solaris, J.; Krueger, T.D.; Chen, C.; Fang, C. Photogrammetry of Ultrafast Excited-State Intramolecular Proton Transfer Pathways in the Fungal Pigment Draconin Red. Molecules 2023, 28, 3506. https://doi.org/10.3390/molecules28083506
Solaris J, Krueger TD, Chen C, Fang C. Photogrammetry of Ultrafast Excited-State Intramolecular Proton Transfer Pathways in the Fungal Pigment Draconin Red. Molecules. 2023; 28(8):3506. https://doi.org/10.3390/molecules28083506
Chicago/Turabian StyleSolaris, Janak, Taylor D. Krueger, Cheng Chen, and Chong Fang. 2023. "Photogrammetry of Ultrafast Excited-State Intramolecular Proton Transfer Pathways in the Fungal Pigment Draconin Red" Molecules 28, no. 8: 3506. https://doi.org/10.3390/molecules28083506
APA StyleSolaris, J., Krueger, T. D., Chen, C., & Fang, C. (2023). Photogrammetry of Ultrafast Excited-State Intramolecular Proton Transfer Pathways in the Fungal Pigment Draconin Red. Molecules, 28(8), 3506. https://doi.org/10.3390/molecules28083506