
Designing Electron-Deficient Diketone Unit Based Non-Fused Ring Acceptors with Amplified Optoelectronic Features for Highly Efficient Organic Solar Cells: A DFT Study

 , ,
, ,
Abstract
:1. Introduction
2. Result and Discussion
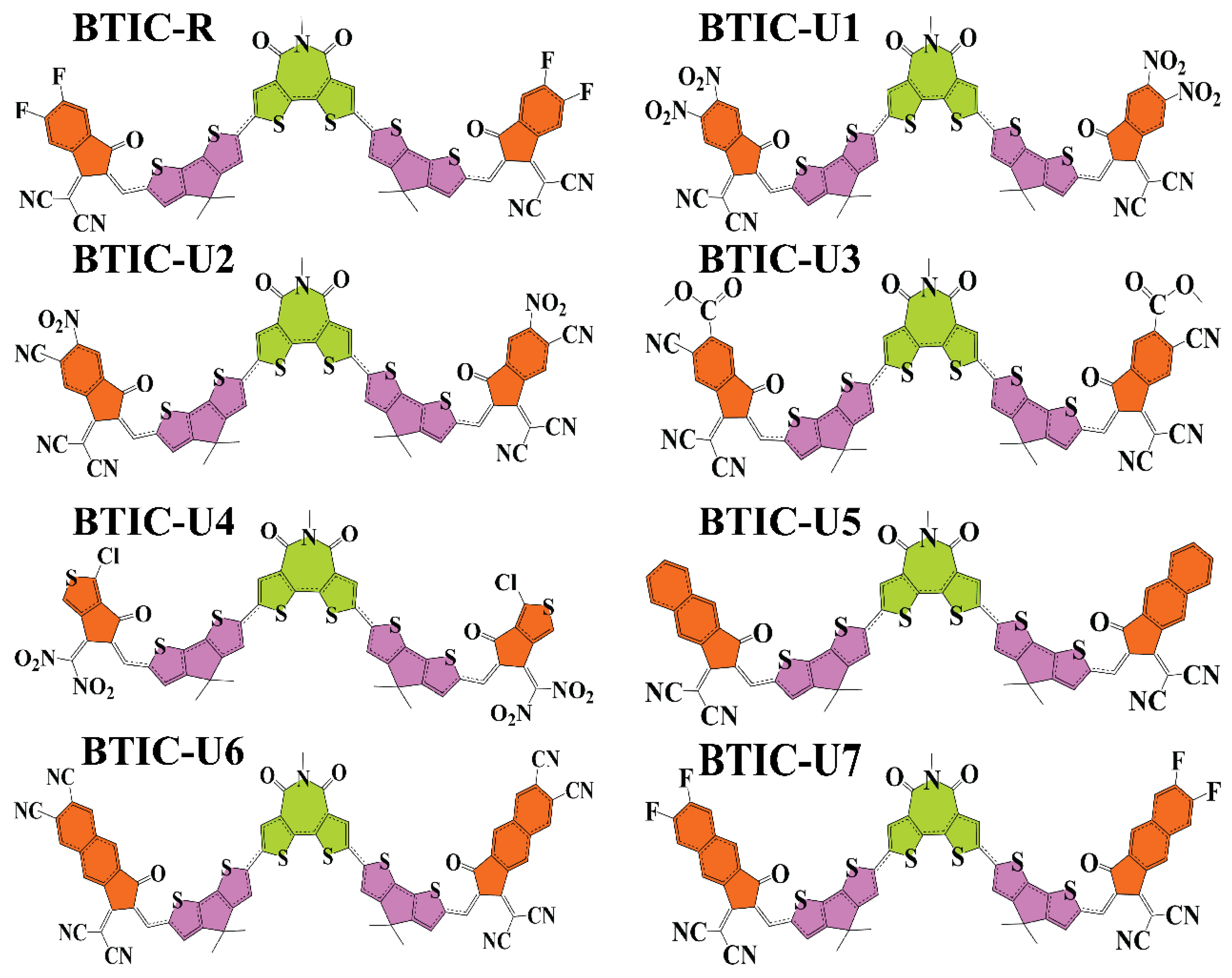
2.1. Chemistry of Molecules
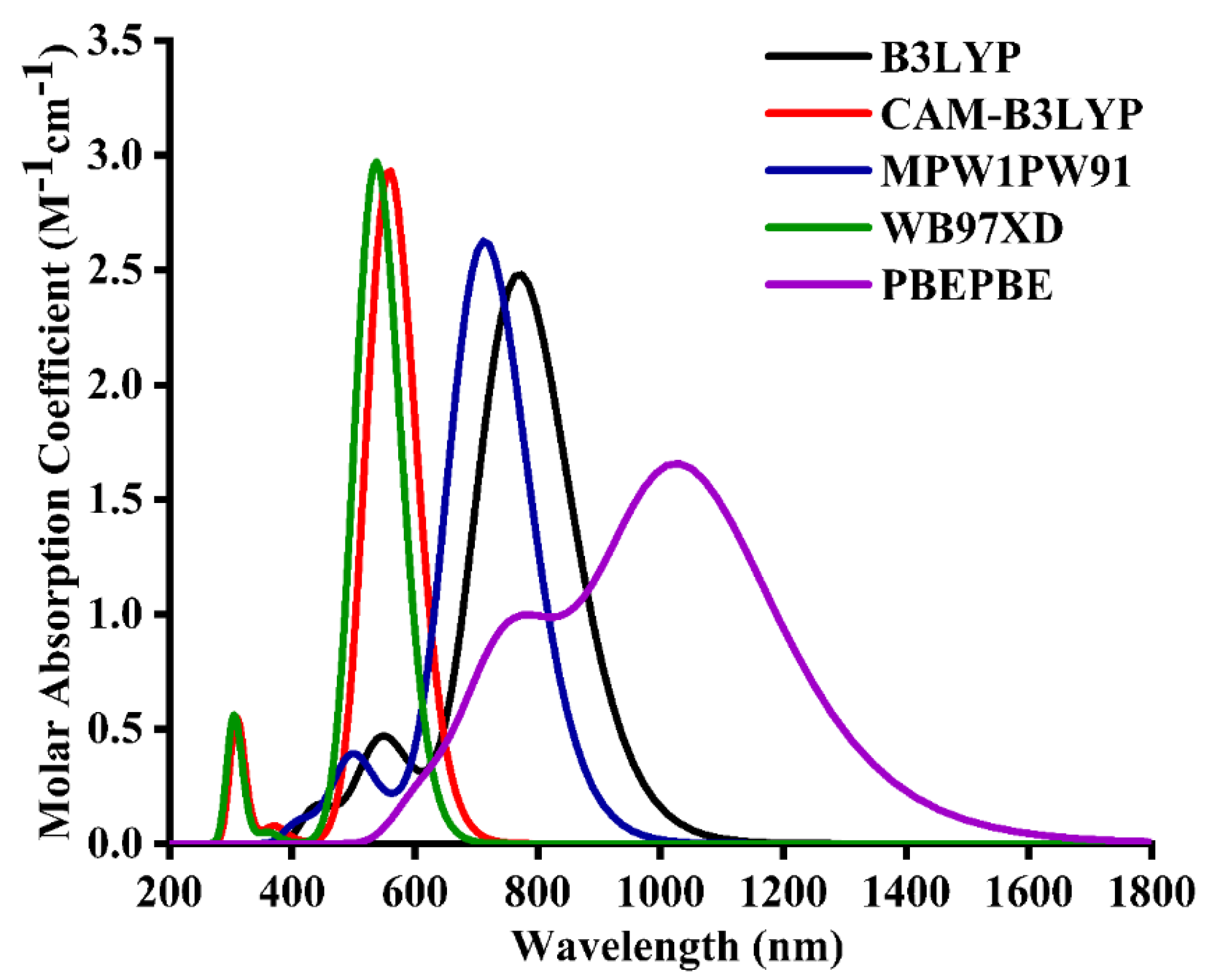
2.2. Method Selection and Optimized Geometries
2.3. Quantum Mechanical Descriptors
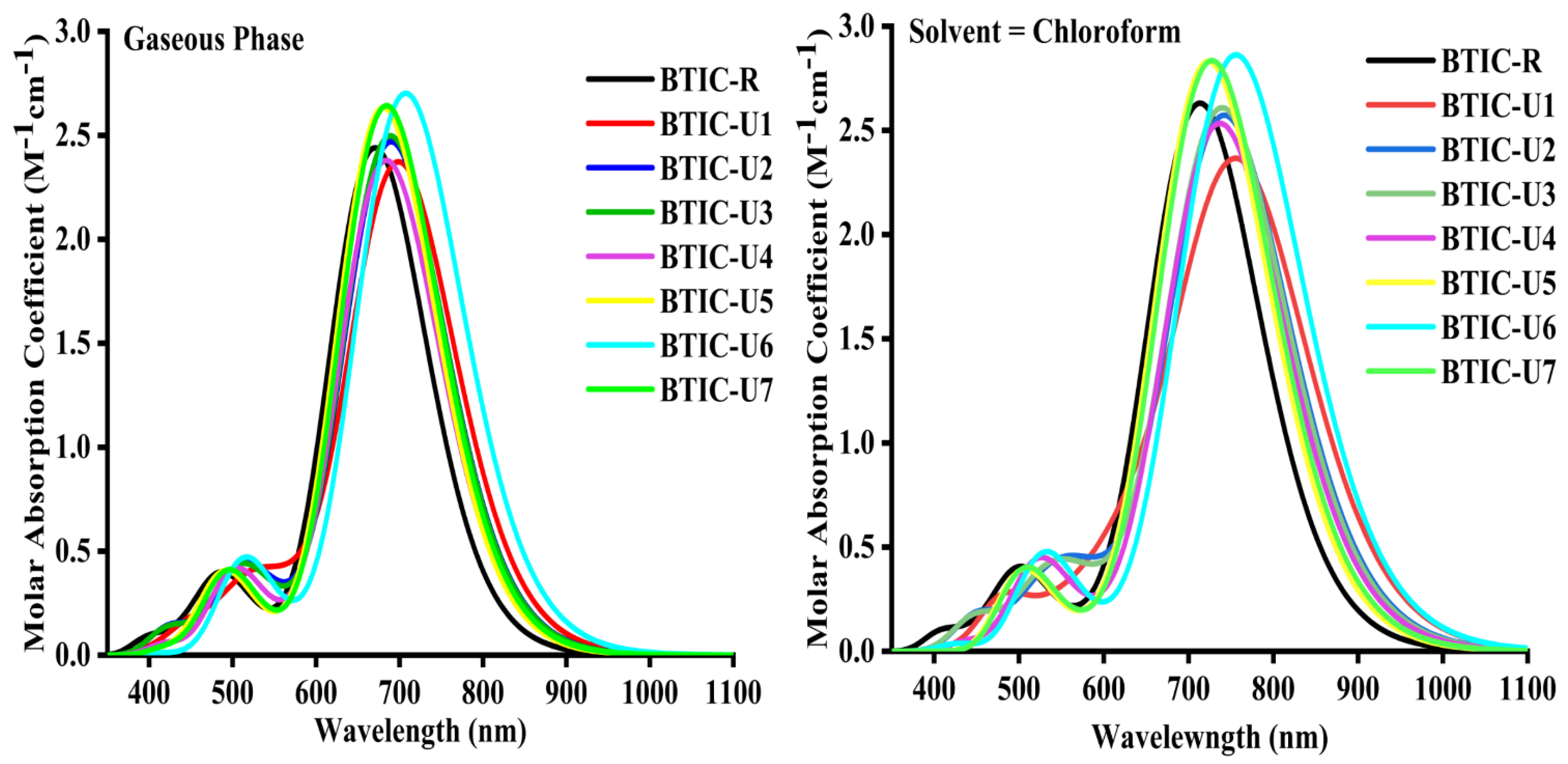
2.4. Absorption Spectrum
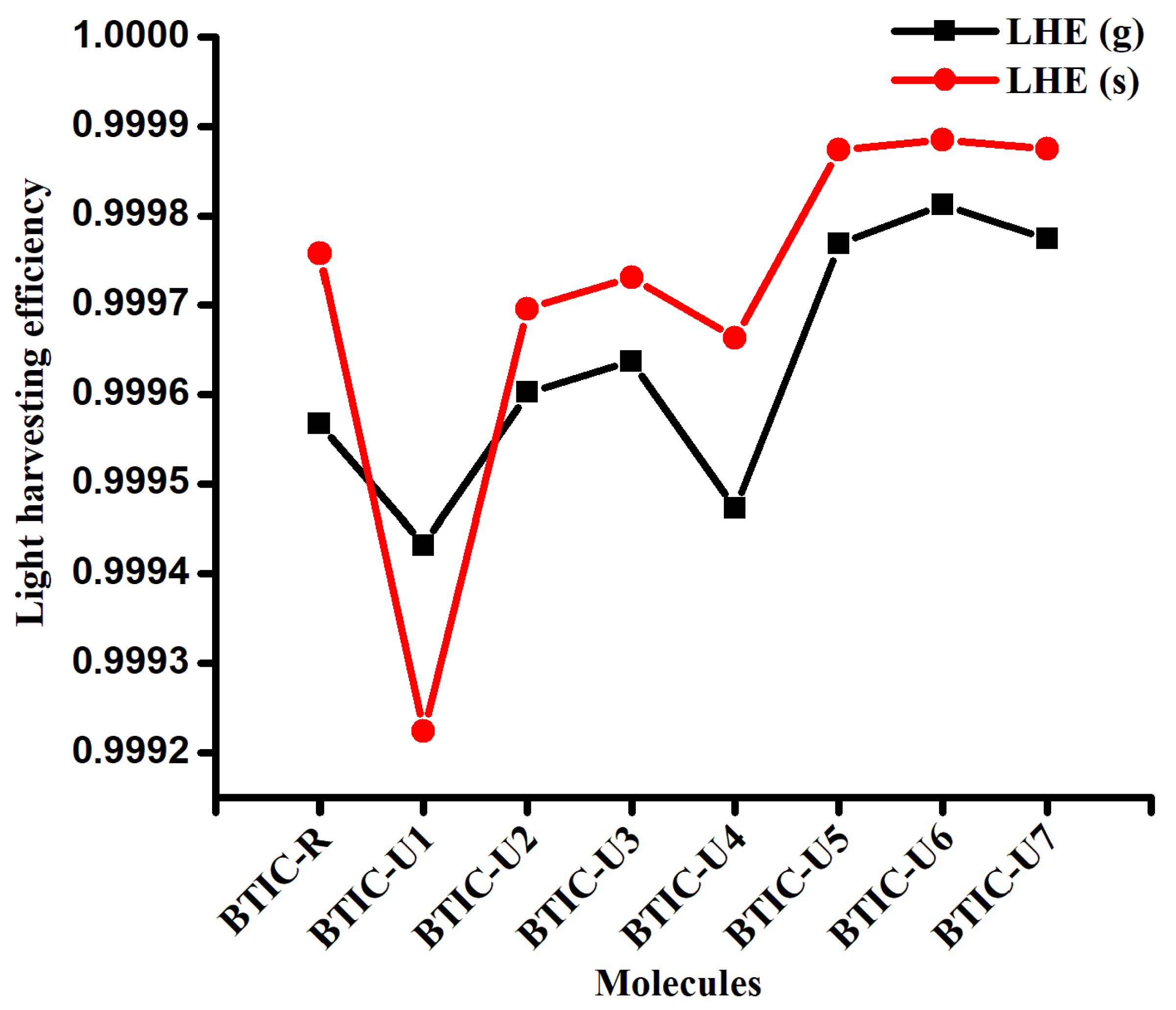
2.5. Light Harvesting Efficiency (LHE)
2.6. Dipole Moment
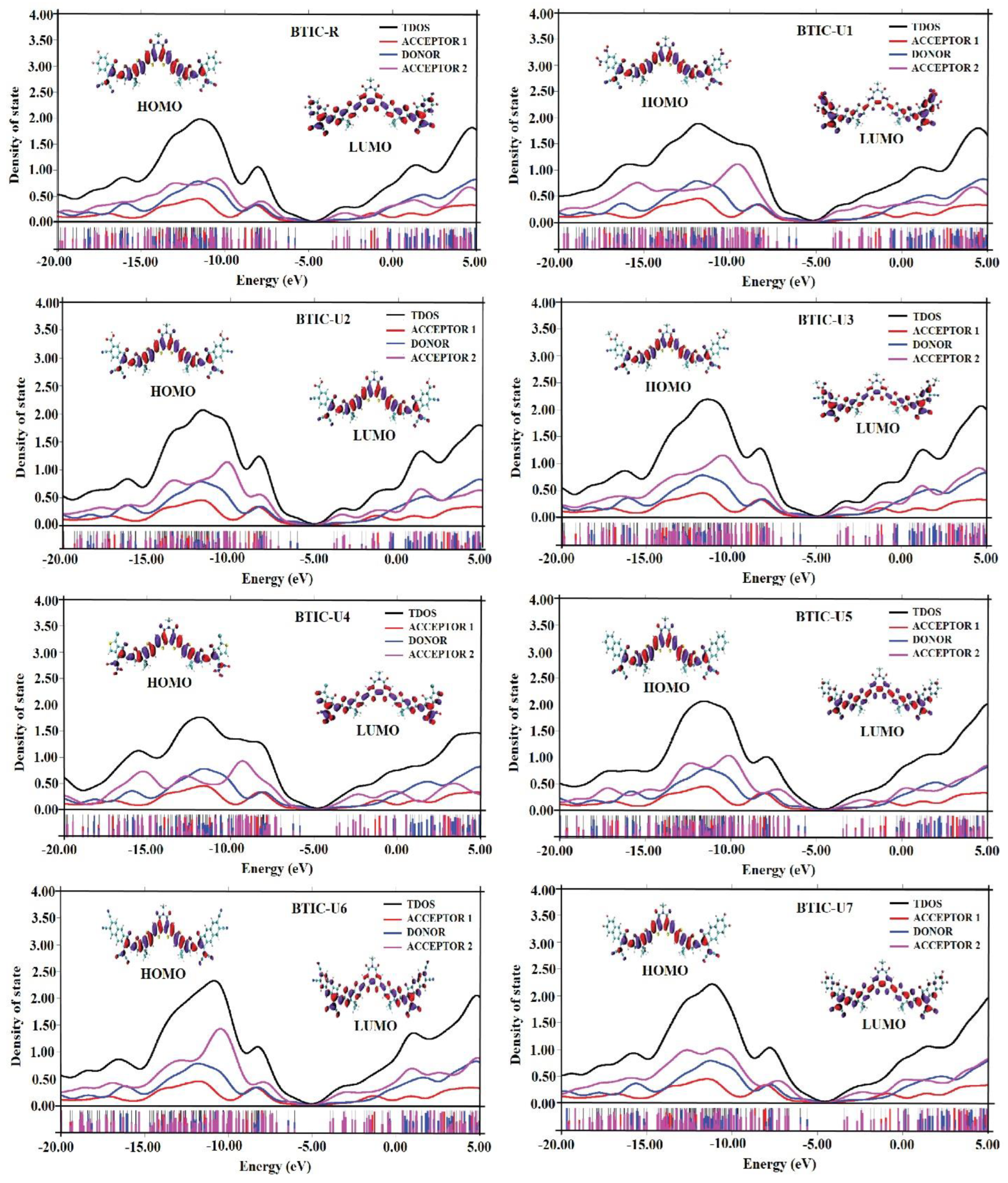
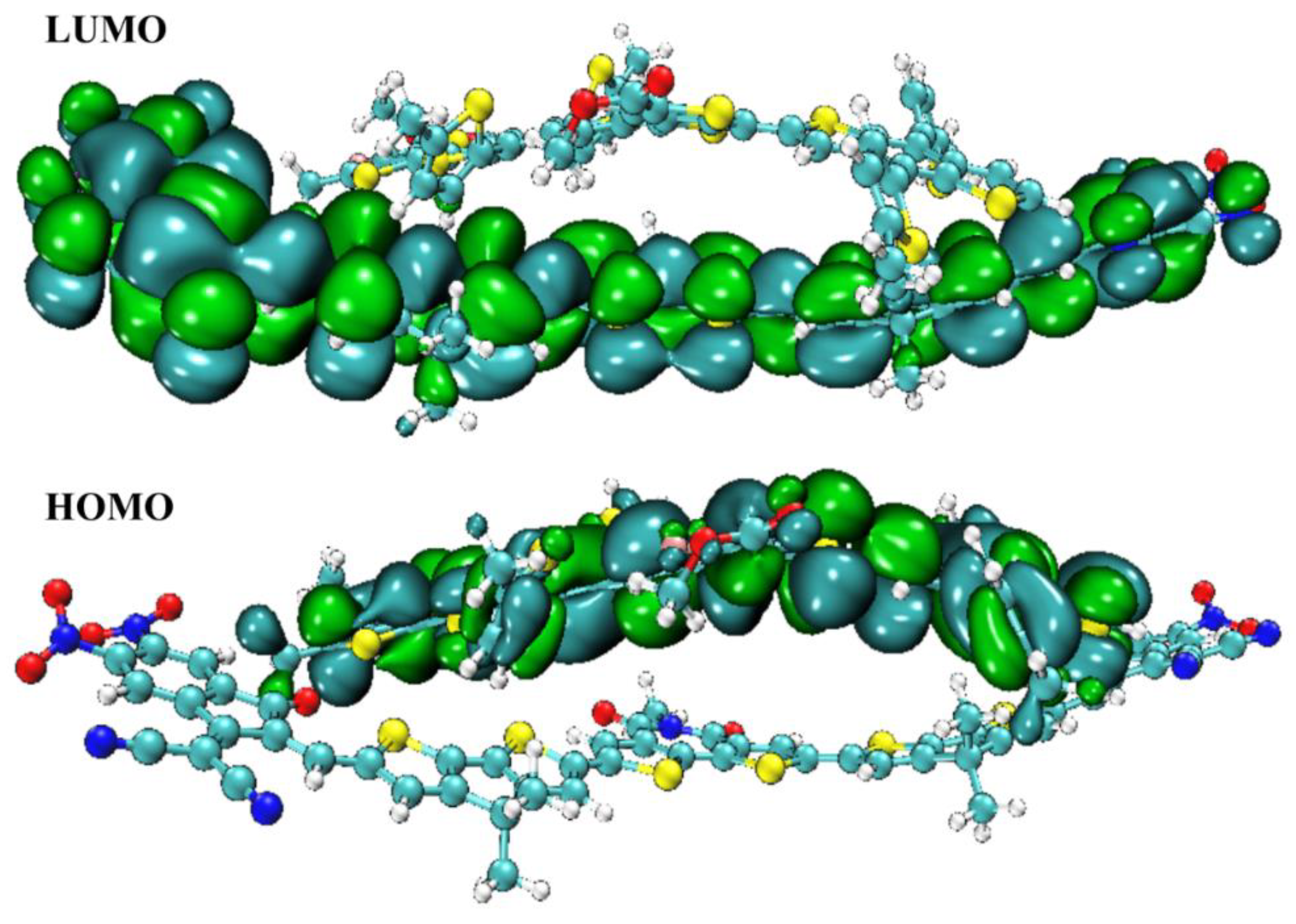
2.7. Density of State
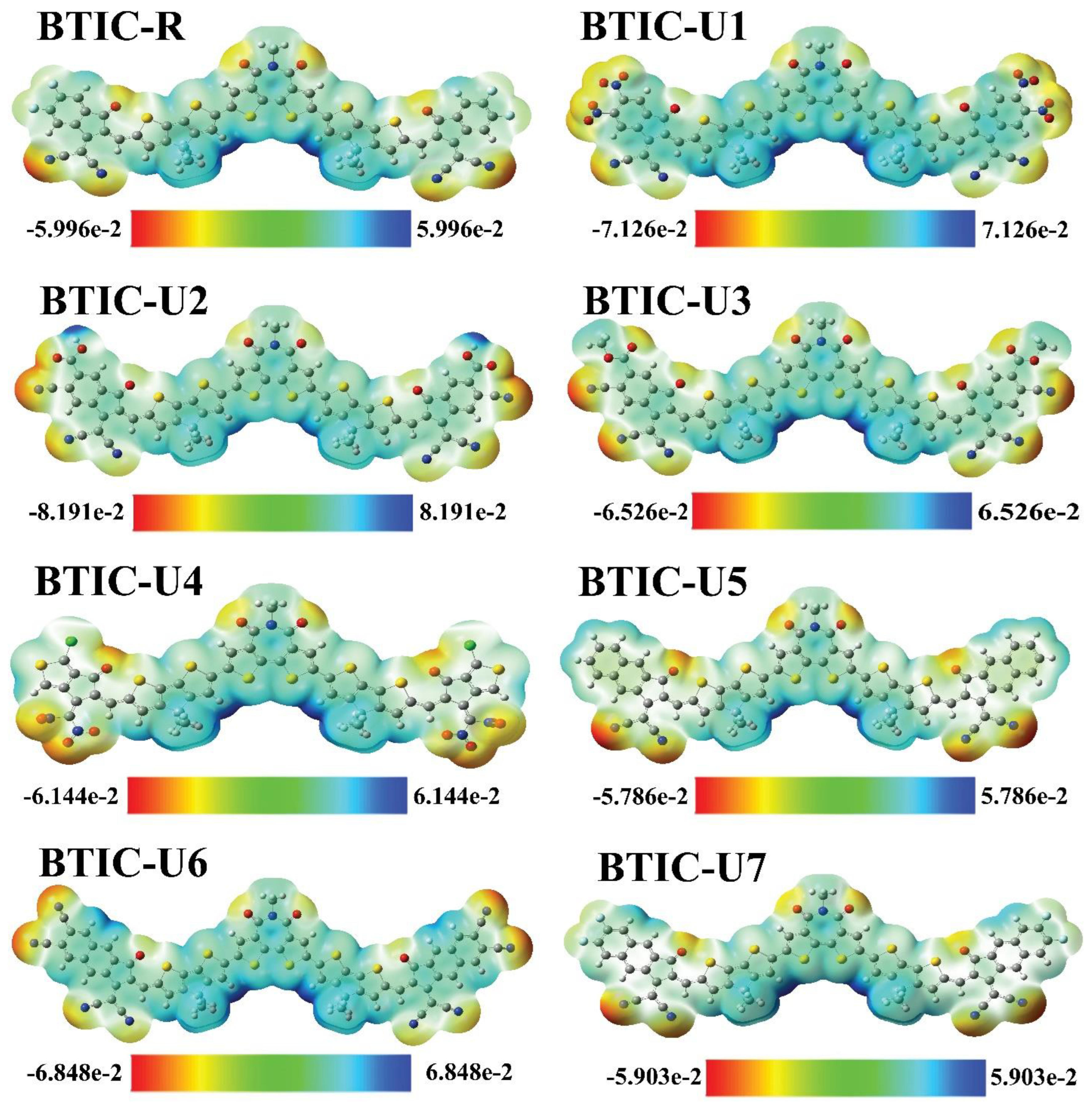
2.8. Electrostatic Potential (ESP)
2.9. Analysis of Charge Mobility
2.10. Chemical Reactivity Parameters
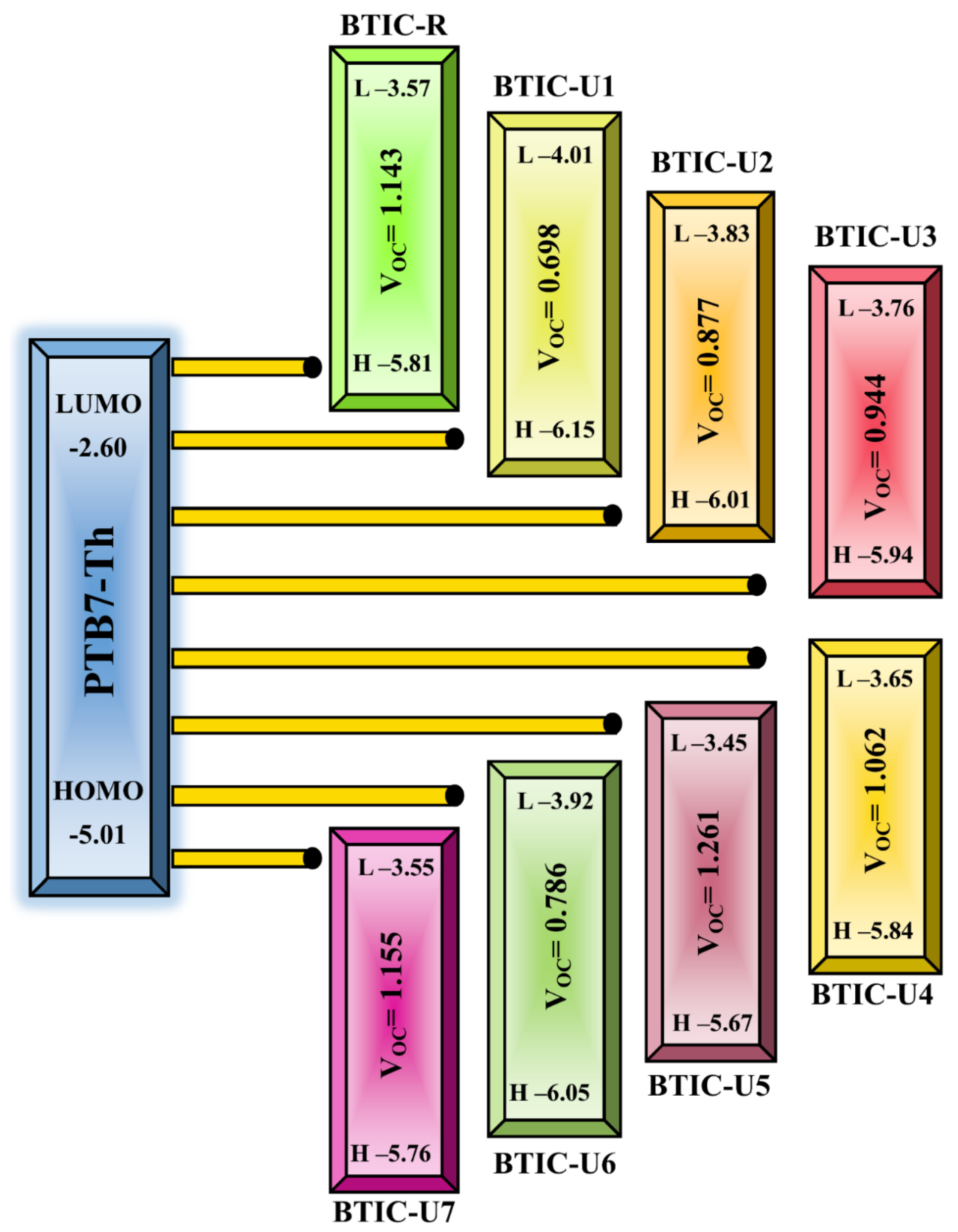
2.11. Device Performance
2.12. Transition Density Matrix (TDM) and Binding Energy (Eb)
2.13. Charge Transfer Analysis
3. Computational Methodology
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Polman, A.; Knight, M.; Garnett, E.C.; Ehrler, B.; Sinke, W.C. Photovoltaic materials: Present efficiencies and future challenges. Science 2016, 352, aad4424. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Guo, Y.; Han, N.; Jiang, X.; Zhang, J.; Ahuja, R.; Su, Y.; Zhao, J. 2D lateral heterostructures of group-III monochalcogenide: Potential photovoltaic applications. Appl. Phys. Lett. 2018, 112, 143902. [Google Scholar] [CrossRef]
- Cheng, P.; Li, G.; Zhan, X.; Yang, Y. Next-generation organic photovoltaics based on non-fullerene acceptors. Nat. Photonics 2018, 12, 131–142. [Google Scholar] [CrossRef]
- Zhang, J.; Tan, H.S.; Guo, X.; Facchetti, A.; Yan, H. Material insights and challenges for non-fullerene organic solar cells based on small molecular acceptors. Nat. Energy 2018, 3, 720–731. [Google Scholar] [CrossRef]
- Inganäs, O. Organic photovoltaics over three decades. Adv. Mater. 2018, 30, 1800388. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Jiang, K.; Lin, F.; Wu, Z.; Zhang, H.; Gao, W.; Li, Y.; Cai, Z.; Woo, H.Y.; Zhu, Z. Over 17% efficiency binary organic solar cells with photoresponses reaching 1000 nm enabled by selenophene-fused nonfullerene acceptors. ACS Energy Lett. 2020, 6, 9–15. [Google Scholar] [CrossRef]
- Li, D.; Zhu, L.; Liu, X.; Xiao, W.; Yang, J.; Ma, R.; Ding, L.; Liu, F.; Duan, C.; Fahlman, M. Enhanced and balanced charge transport boosting ternary solar cells over 17% efficiency. Adv. Mater. 2020, 32, 2002344. [Google Scholar] [CrossRef]
- Zhu, C.; Yuan, J.; Cai, F.; Meng, L.; Zhang, H.; Chen, H.; Li, J.; Qiu, B.; Peng, H.; Chen, S. Tuning the electron-deficient core of a non-fullerene acceptor to achieve over 17% efficiency in a single-junction organic solar cell. Energy Environ. Sci. 2020, 13, 2459–2466. [Google Scholar] [CrossRef]
- Ma, X.; Wang, J.; Gao, J.; Hu, Z.; Xu, C.; Zhang, X.; Zhang, F. Achieving 17.4% efficiency of ternary organic photovoltaics with two well-compatible nonfullerene acceptors for minimizing energy loss. Adv. Energy Mater. 2020, 10, 2001404. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, L.; Zhou, G.; Hao, T.; Qiu, C.; Zhao, Z.; Hu, Q.; Larson, B.W.; Zhu, H.; Ma, Z. Single-layered organic photovoltaics with double cascading charge transport pathways: 18% efficiencies. Nat. Commun. 2021, 12, 309. [Google Scholar] [CrossRef]
- Lin, Y.; Wang, J.; Zhang, Z.G.; Bai, H.; Li, Y.; Zhu, D.; Zhan, X. An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater. 2015, 27, 1170–1174. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, Y.; Zhou, L.; Zhang, G.; Yip, H.-L.; Lau, T.-K.; Lu, X.; Zhu, C.; Peng, H.; Johnson, P.A. Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule 2019, 3, 1140–1151. [Google Scholar] [CrossRef]
- Xiao, Z.; Jia, X.; Li, D.; Wang, S.; Geng, X.; Liu, F.; Chen, J.; Yang, S.; Russell, T.P.; Ding, L. 26 mA cm−2 Jsc from organic solar cells with a low-bandgap nonfullerene acceptor. Sci. Bull. 2017, 62, 1494–1496. [Google Scholar] [CrossRef]
- Cui, Y.; Yao, H.; Zhang, J.; Zhang, T.; Wang, Y.; Hong, L.; Xian, K.; Xu, B.; Zhang, S.; Peng, J. Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages. Nat. Commun. 2019, 10, 2515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, S.; Yao, H.; Zhang, S.; Zhang, Y.; Yang, B.; Hou, J. Molecular optimization enables over 13% efficiency in organic solar cells. J. Am. Chem. Soc. 2017, 139, 7148–7151. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ma, X.; Zhang, Z.; Yu, J.; Zhou, J.; Yin, X.; Yang, L.; Geng, R.; Zhu, R.; Zhang, F. Dithieno [3, 2-b: 2′, 3′-d] pyrrol fused nonfullerene acceptors enabling over 13% efficiency for organic solar cells. Adv. Mater. 2018, 30, 1707150. [Google Scholar] [CrossRef]
- Yao, H.; Cui, Y.; Yu, R.; Gao, B.; Zhang, H.; Hou, J. Design, synthesis, and photovoltaic characterization of a small molecular acceptor with an ultra-narrow band gap. Angew. Chem. Int. Ed. 2017, 56, 3045–3049. [Google Scholar] [CrossRef]
- Zhang, G.; Zhao, J.; Chow, P.C.; Jiang, K.; Zhang, J.; Zhu, Z.; Zhang, J.; Huang, F.; Yan, H. Nonfullerene acceptor molecules for bulk heterojunction organic solar cells. Chem. Rev. 2018, 118, 3447–3507. [Google Scholar] [CrossRef]
- Li, X.; Pan, F.; Sun, C.; Zhang, M.; Wang, Z.; Du, J.; Wang, J.; Xiao, M.; Xue, L.; Zhang, Z.-G. Simplified synthetic routes for low cost and high photovoltaic performance n-type organic semiconductor acceptors. Nat. Commun. 2019, 10, 519. [Google Scholar] [CrossRef]
- Min, J.; Luponosov, Y.N.; Cui, C.; Kan, B.; Chen, H.; Wan, X.; Chen, Y.; Ponomarenko, S.A.; Li, Y.; Brabec, C.J. Evaluation of electron donor materials for solution-processed organic solar cells via a novel figure of merit. Adv. Energy Mater. 2017, 7, 1700465. [Google Scholar] [CrossRef]
- Sun, C.; Pan, F.; Bin, H.; Zhang, J.; Xue, L.; Qiu, B.; Wei, Z.; Zhang, Z.-G.; Li, Y. A low cost and high performance polymer donor material for polymer solar cells. Nat. Commun. 2018, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Li, M.; Ma, X.; Huang, H.; Lu, H.; Jia, Q.; Liu, Y.; Xu, X.; Li, H.-B.; Bo, Z. Noncovalently Fused-Ring Electron Acceptors with C 2 v Symmetry for Regulating the Morphology of Organic Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 46220–46230. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Qin, J.; Xiao, Z.; Zhang, J.; Chen, Z.; Zhang, L.; Cheng, M.; Jin, Z.; Yuan, Y.; Wu, W.-Q. Multiple conformation locks gift polymer donor high efficiency. Nano Energy 2020, 77, 105161. [Google Scholar] [CrossRef]
- Huang, H.; Guo, Q.; Feng, S.; Zhang, C.; Bi, Z.; Xue, W.; Yang, J.; Song, J.; Li, C.; Xu, X. Noncovalently fused-ring electron acceptors with near-infrared absorption for high-performance organic solar cells. Nat. Commun. 2019, 10, 3038. [Google Scholar] [CrossRef]
- Li, S.; Zhan, L.; Liu, F.; Ren, J.; Shi, M.; Li, C.Z.; Russell, T.P.; Chen, H. An unfused-core-based nonfullerene acceptor enables high-efficiency organic solar cells with excellent morphological stability at high temperatures. Adv. Mater. 2018, 30, 1705208. [Google Scholar] [CrossRef]
- Liu, X.; Wei, Y.; Zhang, X.; Qin, L.; Wei, Z.; Huang, H. An ADA′-DA type unfused nonfullerene acceptor for organic solar cells with approaching 14% efficiency. Sci. China Chem. 2021, 64, 228–231. [Google Scholar] [CrossRef]
- Chang, M.; Meng, L.; Wang, Y.; Ke, X.; Yi, Y.-Q.-Q.; Zheng, N.; Zheng, W.; Xie, Z.; Zhang, M.; Yi, Y. Achieving an efficient and stable morphology in organic solar cells via fine-tuning the side chains of small-molecule acceptors. Chem. Mater. 2020, 32, 2593–2604. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, M.; Shen, S.; Wang, J.; Zheng, R.; Lu, H.; Liu, Y.; Ma, Z.; Song, J.; Bo, Z. Hybrid nonfused-ring electron acceptors with fullerene pendant for high-efficiency organic solar cells. ACS Appl. Mater. Interfaces 2020, 13, 1603–1611. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, L.; Yu, J.; Li, Y.; Wei, Y.; Liu, X.; Lu, X.; Gao, F.; Huang, H. High-Performance Noncovalently Fused-Ring Electron Acceptors for Organic Solar Cells Enabled by Noncovalent Intramolecular Interactions and End-Group Engineering. Angew. Chem. 2021, 133, 12583–12589. [Google Scholar] [CrossRef]
- Luo, D.; Lai, X.; Zheng, N.; Duan, C.; Wang, Z.; Wang, K.; Kyaw, A.K.K. High-performance and low-energy loss organic solar cells with non-fused ring acceptor by alkyl chain engineering. Chem. Eng. J. 2021, 420, 129768. [Google Scholar] [CrossRef]
- Li, S.; Zhan, L.; Sun, C.; Zhu, H.; Zhou, G.; Yang, W.; Shi, M.; Li, C.-Z.; Hou, J.; Li, Y. Highly efficient fullerene-free organic solar cells operate at near zero highest occupied molecular orbital offsets. J. Am. Chem. Soc. 2019, 141, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.-Z.; Yang, W.-T.; Gao, J.; Li, S.-X.; Shi, M.-M.; Lau, T.-K.; Lu, X.-H.; Li, C.-Z.; Chen, H.-Z. Non-fullerene acceptors with a thieno [3, 4-c] pyrrole-4, 6-dione (TPD) core for efficient organic solar cells. Chin. J. Polym. Sci. 2019, 37, 1005–1014. [Google Scholar] [CrossRef]
- Letizia, J.A.; Salata, M.R.; Tribout, C.M.; Facchetti, A.; Ratner, M.A.; Marks, T.J. n-Channel polymers by design: Optimizing the interplay of solubilizing substituents, crystal packing, and field-effect transistor characteristics in polymeric bithiophene-imide semiconductors. J. Am. Chem. Soc. 2008, 130, 9679–9694. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, N.; Lou, S.J.; Smith, J.; Tice, D.B.; Hennek, J.W.; Ortiz, R.P.; Navarrete, J.T.L.; Li, S.; Strzalka, J. Polymer solar cells with enhanced fill factors. Nat. Photonics 2013, 7, 825–833. [Google Scholar] [CrossRef]
- Shi, Y.; Guo, H.; Huang, J.; Zhang, X.; Wu, Z.; Yang, K.; Zhang, Y.; Feng, K.; Woo, H.Y.; Ortiz, R.P. Distannylated Bithiophene Imide: Enabling High-Performance n-Type Polymer Semiconductors with an Acceptor–Acceptor Backbone. Angew. Chem. 2020, 132, 14557–14565. [Google Scholar] [CrossRef]
- Shi, Y.; Guo, H.; Qin, M.; Zhao, J.; Wang, Y.; Wang, H.; Wang, Y.; Facchetti, A.; Lu, X.; Guo, X. Thiazole imide-based all-acceptor homopolymer: Achieving high-performance unipolar electron transport in organic thin-film transistors. Adv. Mater. 2018, 30, 1705745. [Google Scholar] [CrossRef]
- Sun, H.; Yu, H.; Shi, Y.; Yu, J.; Peng, Z.; Zhang, X.; Liu, B.; Wang, J.; Singh, R.; Lee, J. A narrow-bandgap n-type polymer with an acceptor–acceptor backbone enabling efficient all-polymer solar cells. Adv. Mater. 2020, 32, 2004183. [Google Scholar] [CrossRef]
- Luo, D.; Li, L.; Shi, Y.; Zhang, J.; Wang, K.; Guo, X.; Kyaw, A.K.K. Electron-deficient diketone unit engineering for non-fused ring acceptors enabling over 13% efficiency in organic solar cells. J. Mater. Chem. A 2021, 9, 14948–14957. [Google Scholar] [CrossRef]
- Rafiq, A.; Hussain, R.; Khan, M.U.; Mehboob, M.Y.; Khalid, M.; Alam, M.M.; Imran, M.; Ayub, K. Novel Star-Shaped Benzotriindole-Based Nonfullerene Donor Materials: Toward the Development of Promising Photovoltaic Compounds for High-Performance Organic Solar Cells. Energy Technol. 2022, 10, 2100751. [Google Scholar] [CrossRef]
- Khalid, M.; Khan, M.U.; Ahmed, S.; Shafiq, Z.; Alam, M.M.; Imran, M.; Braga, A.A.C.; Akram, M.S. Exploration of promising optical and electronic properties of (non-polymer) small donor molecules for organic solar cells. Sci. Rep. 2021, 11, 21540. [Google Scholar] [CrossRef]
- Khalid, M.; Khan, M.U.; Razia, E.-T.; Shafiq, Z.; Alam, M.M.; Imran, M.; Akram, M.S. Exploration of efficient electron acceptors for organic solar cells: Rational design of indacenodithiophene based non-fullerene compounds. Sci. Rep. 2021, 11, 19931. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.U.; Khalid, M.; Hussain, R.; Umar, A.; Mehboob, M.Y.; Shafiq, Z.; Imran, M.; Irfan, A. Novel W-shaped oxygen heterocycle-fused fluorene-based non-fullerene acceptors: First theoretical framework for designing environment-friendly organic solar cells. Energy Fuels 2021, 35, 12436–12450. [Google Scholar] [CrossRef]
- Khalid, M.; Imran, M.; Braga, A.A.C.; Akram, M.S. Molecular engineering of indenoindene-3-ethylrodanine acceptors with A2-A1-D-A1-A2 architecture for promising fullerene-free organic solar cells. Sci. Rep. 2021, 11, 20320. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Ullah, M.A.; Adeel, M.; Khan, M.U.; Tahir, M.N.; Braga, A.A.C. Synthesis, crystal structure analysis, spectral IR, UV–Vis, NMR assessments, electronic and nonlinear optical properties of potent quinoline based derivatives: Interplay of experimental and DFT study. J. Saudi Chem. Soc. 2019, 23, 546–560. [Google Scholar] [CrossRef]
- Khalid, M.; Ali, A.; Jawaria, R.; Asghar, M.A.; Asim, S.; Khan, M.U.; Hussain, R.; ur Rehman, M.F.; Ennis, C.J.; Akram, M.S. First principles study of electronic and nonlinear optical properties of A–D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 2020, 10, 22273–22283. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Lodhi, H.M.; Khan, M.U.; Imran, M. Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: A DFT approach. RSC Adv. 2021, 11, 14237–14250. [Google Scholar] [CrossRef]
- Khalid, M.; Hussain, R.; Hussain, A.; Ali, B.; Jaleel, F.; Imran, M.; Assiri, M.A.; Usman Khan, M.; Ahmed, S.; Abid, S. Electron donor and acceptor influence on the nonlinear optical response of diacetylene-functionalized organic materials (DFOMs): Density functional theory calculations. Molecules 2019, 24, 2096. [Google Scholar] [CrossRef]
- Khalid, M.; Khan, M.U.; Shafiq, I.; Hussain, R.; Ali, A.; Imran, M.; Braga, A.A.; Fayyaz ur Rehman, M.; Akram, M.S. Structural modulation of π-conjugated linkers in D–π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties. R. Soc. Open Sci. 2021, 8, 210570. [Google Scholar] [CrossRef]
- Khalid, M.; Ali, A.; Rehman, M.F.U.; Mustaqeem, M.; Ali, S.; Khan, M.U.; Asim, S.; Ahmad, N.; Saleem, M. Exploration of noncovalent interactions, chemical reactivity, and nonlinear optical properties of piperidone derivatives: A concise theoretical approach. ACS Omega 2020, 5, 13236–13249. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.; Shafiq, I.; Zhu, M.; Khan, M.U.; Shafiq, Z.; Iqbal, J.; Alam, M.M.; Braga, A.A.C.; Imran, M. Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation. J. Saudi Chem. Soc. 2021, 25, 101305. [Google Scholar] [CrossRef]
- Janjua, M.R.S.A.; Khan, M.U.; Khalid, M.; Ullah, N.; Kalgaonkar, R.; Alnoaimi, K.; Baqader, N.; Jamil, S. Theoretical and conceptual framework to design efficient dye-sensitized solar cells (DSSCs): Molecular engineering by DFT method. J. Clust. Sci. 2021, 32, 243–253. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. 0109, Revision D. 01; Gaussian. Inc.: Wallingford, UK, 2009. [Google Scholar]
- Dennington, R.D.; Keith, T.A.; Millam, J.M. GaussView 5.0.8; Gaussian. Inc.: Wallingford, UK, 2008. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Civalleri, B.; Zicovich-Wilson, C.M.; Valenzano, L.; Ugliengo, P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, A.; Xu, X.; Yan, Y. OPBE: A promising density functional for the calculation of nuclear shielding constants. Chem. Phys. Lett. 2006, 421, 383–388. [Google Scholar] [CrossRef]
- O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 1993, 65, 599. [Google Scholar] [CrossRef]
- Tang, S.; Zhang, J. Design of donors with broad absorption regions and suitable frontier molecular orbitals to match typical acceptors via substitution on oligo (thienylenevinylene) toward solar cells. J. Comput. Chem. 2012, 33, 1353–1363. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | EHOMO (eV) | ELUMO (eV) | Energy Gap (eV) |
|---|---|---|---|
| BTIC-R | −5.81 | −3.57 | 2.24 |
| BTIC-U1 | −6.15 | −4.01 | 2.14 |
| BTIC-U2 | −6.01 | −3.83 | 2.17 |
| BTIC-U3 | −5.94 | −3.76 | 2.18 |
| BTIC-U4 | −5.84 | −3.65 | 2.19 |
| BTIC-U5 | −5.67 | −3.45 | 2.22 |
| BTIC-U6 | −6.05 | −3.92 | 2.12 |
| BTIC-U7 | −5.76 | −3.55 | 2.21 |
| Molecules | Cal. λmax (nm) | Exp. λmax (nm) | Electronic Excitation Energy (eV) | Oscillator Strength (f) | Major Transition |
|---|---|---|---|---|---|
| BTIC-R | 670 | 696 | 1.8499 | 3.3638 | HOMO→LUMO (95%) |
| BTIC-U1 | 699 | 696 | 1.7727 | 3.2453 | HOMO→LUMO (94%) |
| BTIC-U2 | 689 | 696 | 1.7975 | 3.3996 | HOMO→LUMO (94%) |
| BTIC-U3 | 688 | 696 | 2.3108 | 3.4397 | HOMO→LUMO (94%) |
| BTIC-U4 | 683 | 696 | 1.8131 | 3.2777 | HOMO→LUMO (94%) |
| BTIC-U5 | 680 | 696 | 1.8230 | 3.6336 | HOMO→LUMO (93%) |
| BTIC-U6 | 707 | 696 | 1.7535 | 3.7263 | HOMO→LUMO (92%) |
| BTIC-U7 | 684 | 696 | 1.8126 | 3.645 | HOMO→LUMO (93%) |
| Molecules | Cal. λmax (nm) | Exp. λmax (nm) | Electronic Excitation Energy (eV) | Oscillator Strength (f) | Major Transition |
|---|---|---|---|---|---|
| BTIC-R | 713 | 696 | 1.7376 | 3.6158 | HOMO→LUMO (93%) |
| BTIC-U1 | 761 | 696 | 1.6284 | 3.1103 | HOMO→LUMO (90%) |
| BTIC-U2 | 742 | 696 | 1.6692 | 3.5172 | HOMO→LUMO (92%) |
| BTIC-U3 | 740 | 696 | 1.6751 | 3.5707 | HOMO→LUMO (92%) |
| BTIC-U4 | 737 | 696 | 1.6811 | 3.4724 | HOMO→LUMO (92%) |
| BTIC-U5 | 723 | 696 | 1.7145 | 3.8991 | HOMO→LUMO (93%) |
| BTIC-U6 | 755 | 696 | 1.6404 | 3.9393 | HOMO→LUMO (92%) |
| BTIC-U7 | 727 | 696 | 1.7046 | 3.9032 | HOMO→LUMO (93%) |
| Molecules | LHE (Gas) | LHE (Solvent) |
|---|---|---|
| BTIC-R | 0.999567 | 0.999758 |
| BTIC-U1 | 0.999432 | 0.999224 |
| BTIC-U2 | 0.999602 | 0.999696 |
| BTIC-U3 | 0.999637 | 0.999731 |
| BTIC-U4 | 0.999472 | 0.999663 |
| BTIC-U5 | 0.999768 | 0.999874 |
| BTIC-U6 | 0.999812 | 0.999885 |
| BTIC-U7 | 0.999774 | 0.999875 |
| Molecules | μ (Gaseous Phase) | μ (Solvent Phase) |
|---|---|---|
| BTIC-R | 1.159 | 0.899 |
| BTIC-U1 | 5.582 | 7.164 |
| BTIC-U2 | 2.433 | 2.170 |
| BTIC-U3 | 1.532 | 0.866 |
| BTIC-U4 | 2.479 | 2.772 |
| BTIC-U5 | 5.132 | 5.602 |
| BTIC-U6 | 5.552 | 7.307 |
| BTIC-U7 | 2.351 | 2.394 |
| Molecules | Acceptor1 (%) | Acceptor2 (%) | Donor (%) | |
|---|---|---|---|---|
| BTIC-R | HOMO | 30.5 | 18.1 | 51.4 |
| LUMO | 24.0 | 39.0 | 37.0 | |
| BTIC-U1 | HOMO | 31.8 | 19.1 | 49.1 |
| LUMO | 15.5 | 52.3 | 32.2 | |
| BTIC-U2 | HOMO | 31.0 | 19.0 | 50.0 |
| LUMO | 18.7 | 46.7 | 34.6 | |
| BTIC-U3 | HOMO | 30.9 | 19.0 | 50.1 |
| LUMO | 19.2 | 45.8 | 34.9 | |
| BTIC-U4 | HOMO | 30.4 | 19.3 | 50.3 |
| LUMO | 19.0 | 48.1 | 32.9 | |
| BTIC-U5 | HOMO | 29.1 | 19.5 | 51.4 |
| LUMO | 24.6 | 39.0 | 36.4 | |
| BTIC-U6 | HOMO | 30.6 | 20.0 | 49.4 |
| LUMO | 17.4 | 48.2 | 34.5 | |
| BTIC-U7 | HOMO | 29.5 | 19.5 | 51.0 |
| LUMO | 23.0 | 40.8 | 36.2 |
| Molecules | λe | λh |
|---|---|---|
| BTIC−R | 0.1914 | 0.2108 |
| BTIC−U1 | 0.1309 | 0.2129 |
| BTIC−U2 | 0.1467 | 0.2081 |
| BTIC−U3 | 0.1522 | 0.2104 |
| BTIC−U4 | 0.1915 | 0.2277 |
| BTIC−U5 | 0.1772 | 0.1989 |
| BTIC−U6 | 0.1217 | 0.2022 |
| BTIC−U7 | 0.1732 | 0.2033 |
| Molecules | η (eV) | S (eV−1) | μ (eV) | ω (eV) |
|---|---|---|---|---|
| BTIC-R | 1.122 | 0.899 | −4.689 | 9.794 |
| BTIC-U1 | 1.068 | 0.936 | −5.080 | 12.083 |
| BTIC-U2 | 1.087 | 0.919 | −4.920 | 11.132 |
| BTIC-U3 | 1.090 | 0.917 | −4.856 | 10.814 |
| BTIC-U4 | 1.096 | 0.912 | −4.744 | 10.262 |
| BTIC-U5 | 1.112 | 0.899 | −4.561 | 9.355 |
| BTIC-U6 | 1.064 | 0.939 | −4.988 | 11.689 |
| BTIC-U7 | 1.104 | 0.905 | −4.659 | 9.829 |
| Molecules | Voc (eV) | Normalized Voc | FF | PCE |
|---|---|---|---|---|
| BTIC−R | 1.143 | 44.146 | 0.893 | 20.94 |
| BTIC−U1 | 0.698 | 26.936 | 0.845 | 12.09 |
| BTIC−U2 | 0.877 | 33.860 | 0.869 | 15.63 |
| BTIC−U3 | 0.944 | 36.465 | 0.877 | 16.98 |
| BTIC−U4 | 1.062 | 41.025 | 0.887 | 19.33 |
| BTIC−U5 | 1.261 | 48.674 | 0.901 | 23.29 |
| BTIC−U6 | 0.786 | 30.351 | 0.858 | 13.83 |
| BTIC−U7 | 1.155 | 44.597 | 0.894 | 21.18 |
| Molecules | Egap (eV) | Ex (eV) | Eb (eV) Solvent |
|---|---|---|---|
| BTIC-R | 2.2449 | 1.7376 | 0.5073 |
| BTIC-U1 | 2.1361 | 1.6284 | 0.5077 |
| BTIC-U2 | 2.1747 | 1.6692 | 0.5055 |
| BTIC-U3 | 2.1802 | 1.6751 | 0.5051 |
| BTIC-U4 | 2.1929 | 1.6811 | 0.5118 |
| BTIC-U5 | 2.224 | 1.7145 | 0.5095 |
| BTIC-U6 | 2.1285 | 1.6404 | 0.4881 |
| BTIC-U7 | 2.2088 | 1.7046 | 0.5042 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.U.; Shafiq, F.; Al Abbad, S.S.; Yaqoob, J.; Hussain, R.; Alsunaidi, Z.H.A.; Mustafa, G.; Hussain, S. Designing Electron-Deficient Diketone Unit Based Non-Fused Ring Acceptors with Amplified Optoelectronic Features for Highly Efficient Organic Solar Cells: A DFT Study. Molecules 2023, 28, 3625. https://doi.org/10.3390/molecules28083625
Khan MU, Shafiq F, Al Abbad SS, Yaqoob J, Hussain R, Alsunaidi ZHA, Mustafa G, Hussain S. Designing Electron-Deficient Diketone Unit Based Non-Fused Ring Acceptors with Amplified Optoelectronic Features for Highly Efficient Organic Solar Cells: A DFT Study. Molecules. 2023; 28(8):3625. https://doi.org/10.3390/molecules28083625
Chicago/Turabian StyleKhan, Muhammad Usman, Faiza Shafiq, Sanaa S. Al Abbad, Junaid Yaqoob, Riaz Hussain, Zainab H. A. Alsunaidi, Ghulam Mustafa, and Shabbir Hussain. 2023. "Designing Electron-Deficient Diketone Unit Based Non-Fused Ring Acceptors with Amplified Optoelectronic Features for Highly Efficient Organic Solar Cells: A DFT Study" Molecules 28, no. 8: 3625. https://doi.org/10.3390/molecules28083625
APA StyleKhan, M. U., Shafiq, F., Al Abbad, S. S., Yaqoob, J., Hussain, R., Alsunaidi, Z. H. A., Mustafa, G., & Hussain, S. (2023). Designing Electron-Deficient Diketone Unit Based Non-Fused Ring Acceptors with Amplified Optoelectronic Features for Highly Efficient Organic Solar Cells: A DFT Study. Molecules, 28(8), 3625. https://doi.org/10.3390/molecules28083625