
New Indazol-Pyrimidine-Based Derivatives as Selective Anticancer Agents: Design, Synthesis, and In Silico Studies

 , , ,
, , ,
Abstract
:1. Introduction
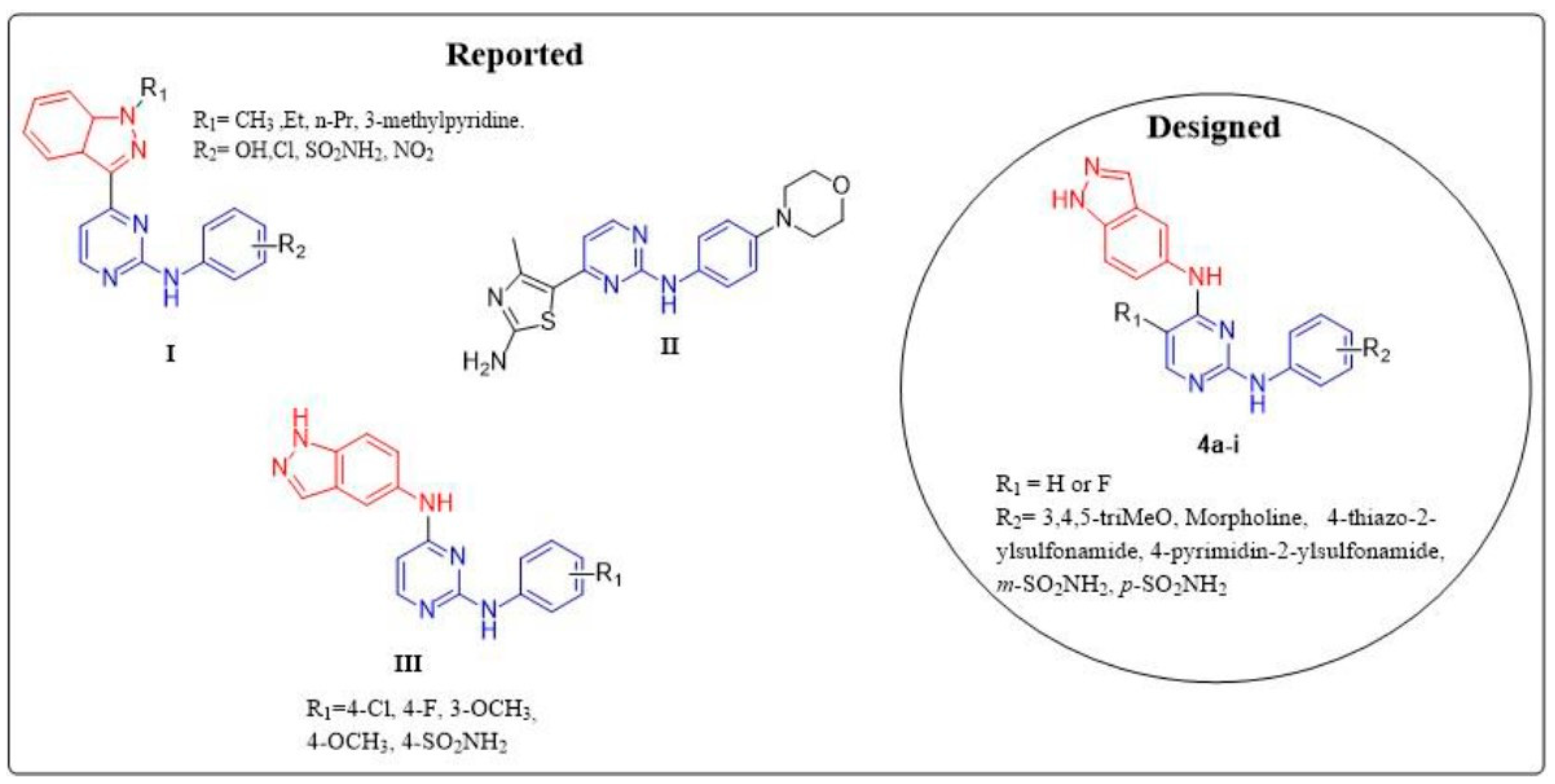
2. Results and Discussion
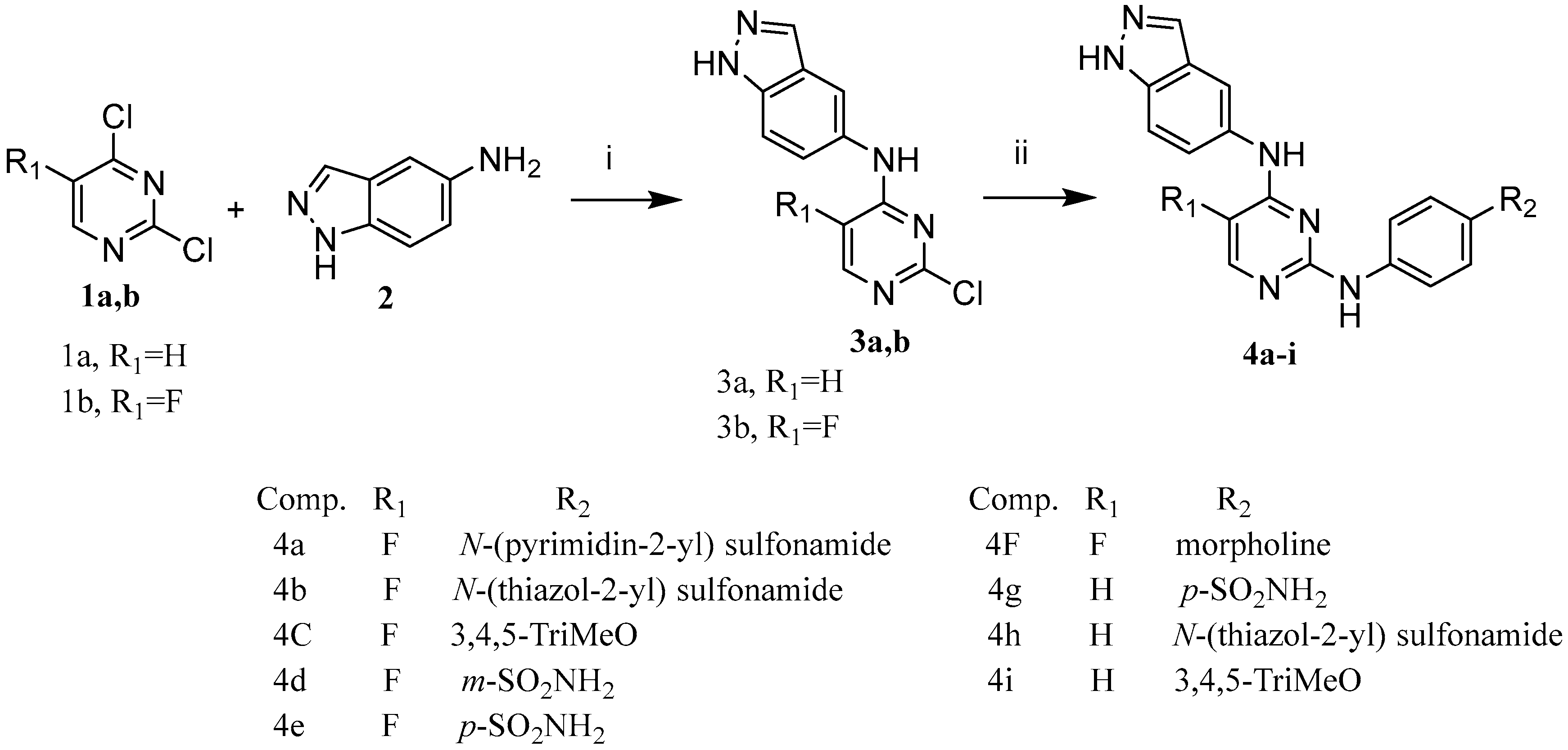
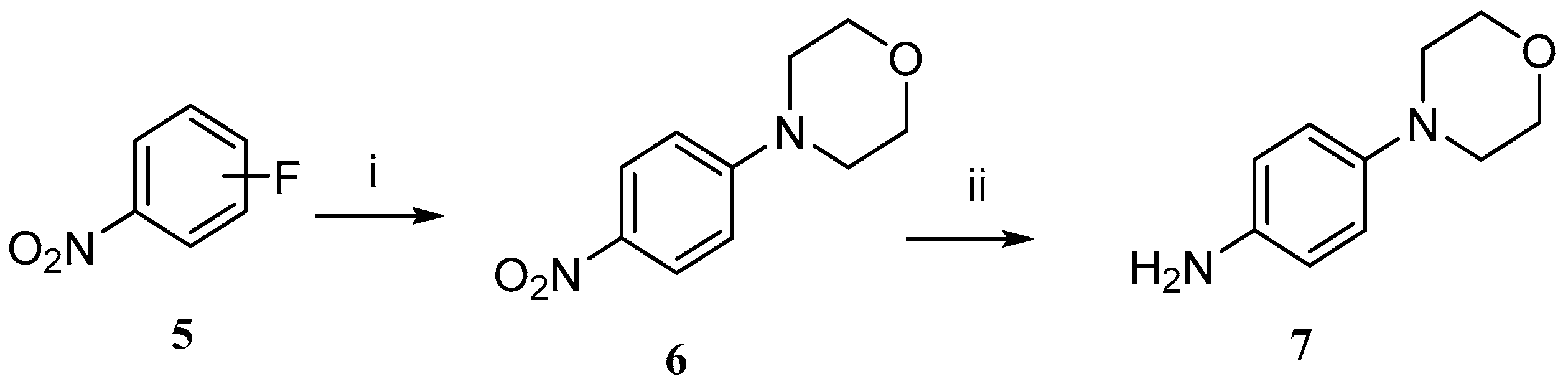
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. MTT Cytotoxicity Assay
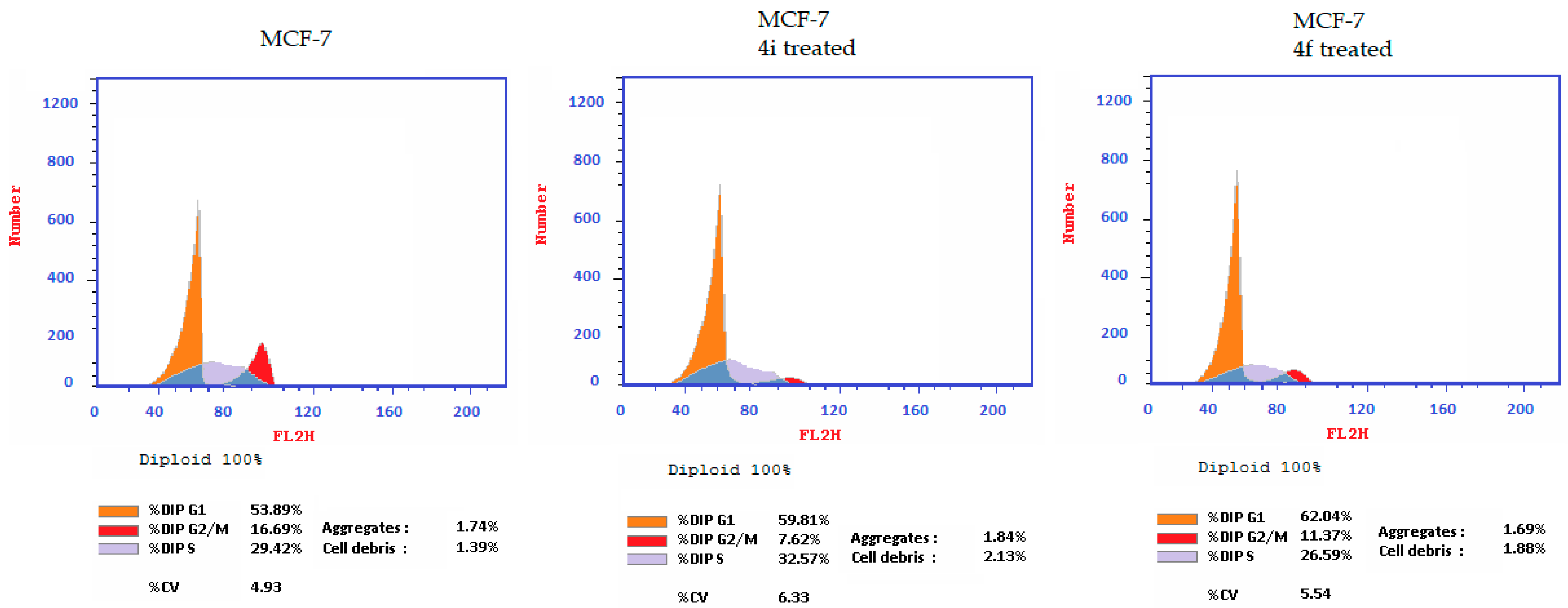
2.2.2. Cell Effects of Compounds 4f and 4i
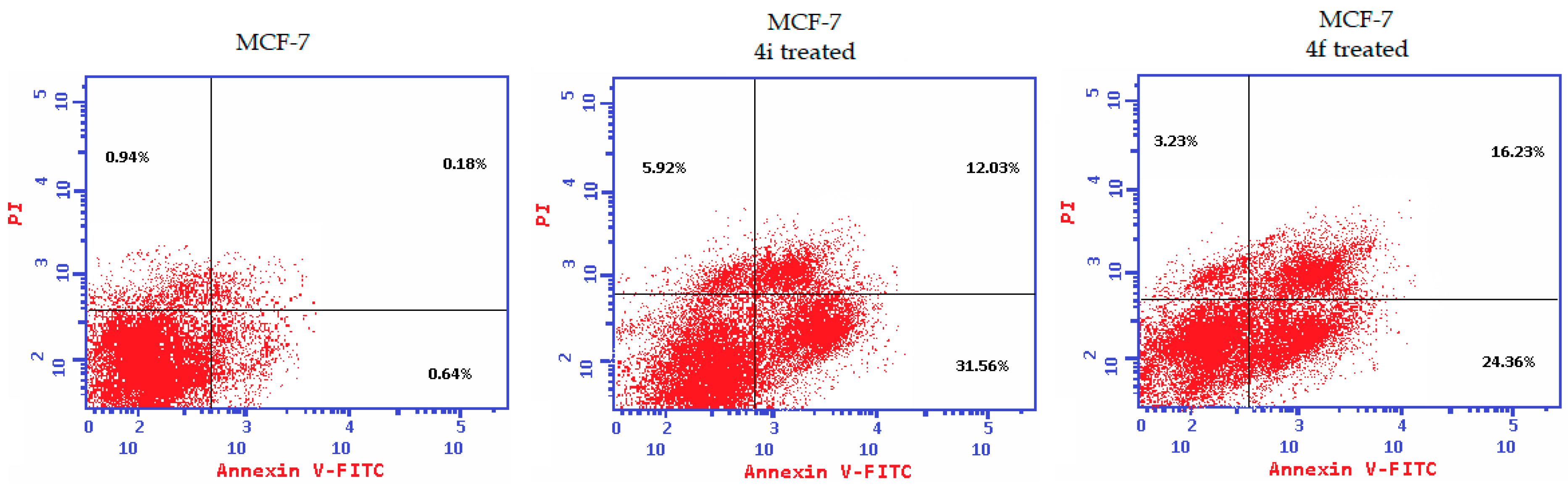
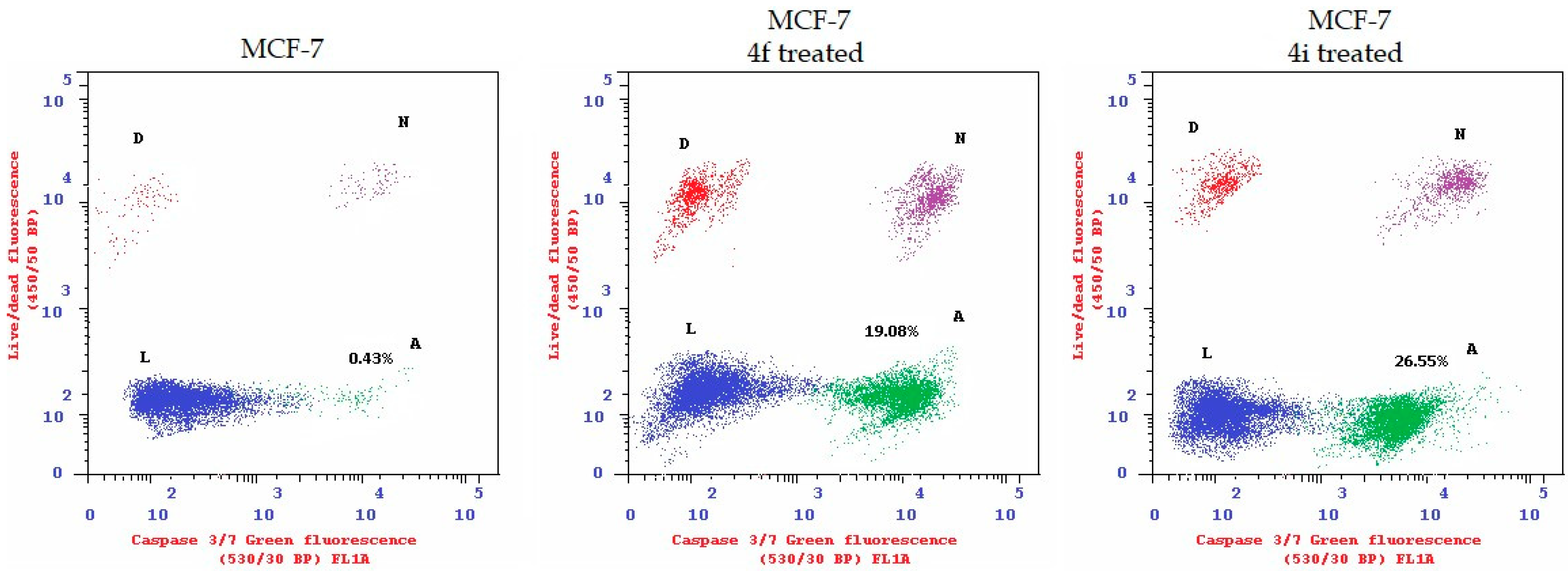
2.2.3. Apoptosis Induction and Caspase-3/7 Activation
3. Computational Studies
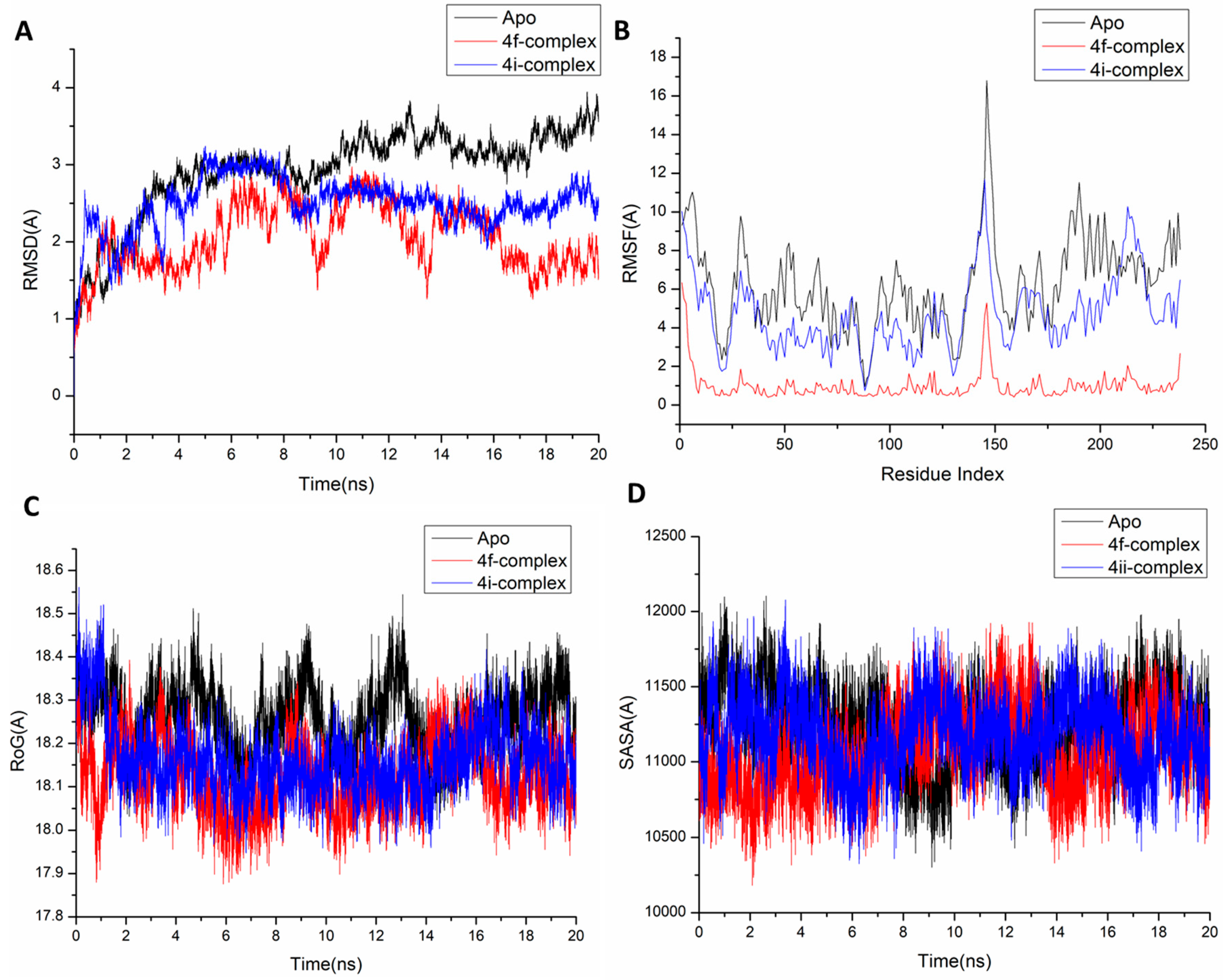
3.1. Molecular Dynamic and System Stability
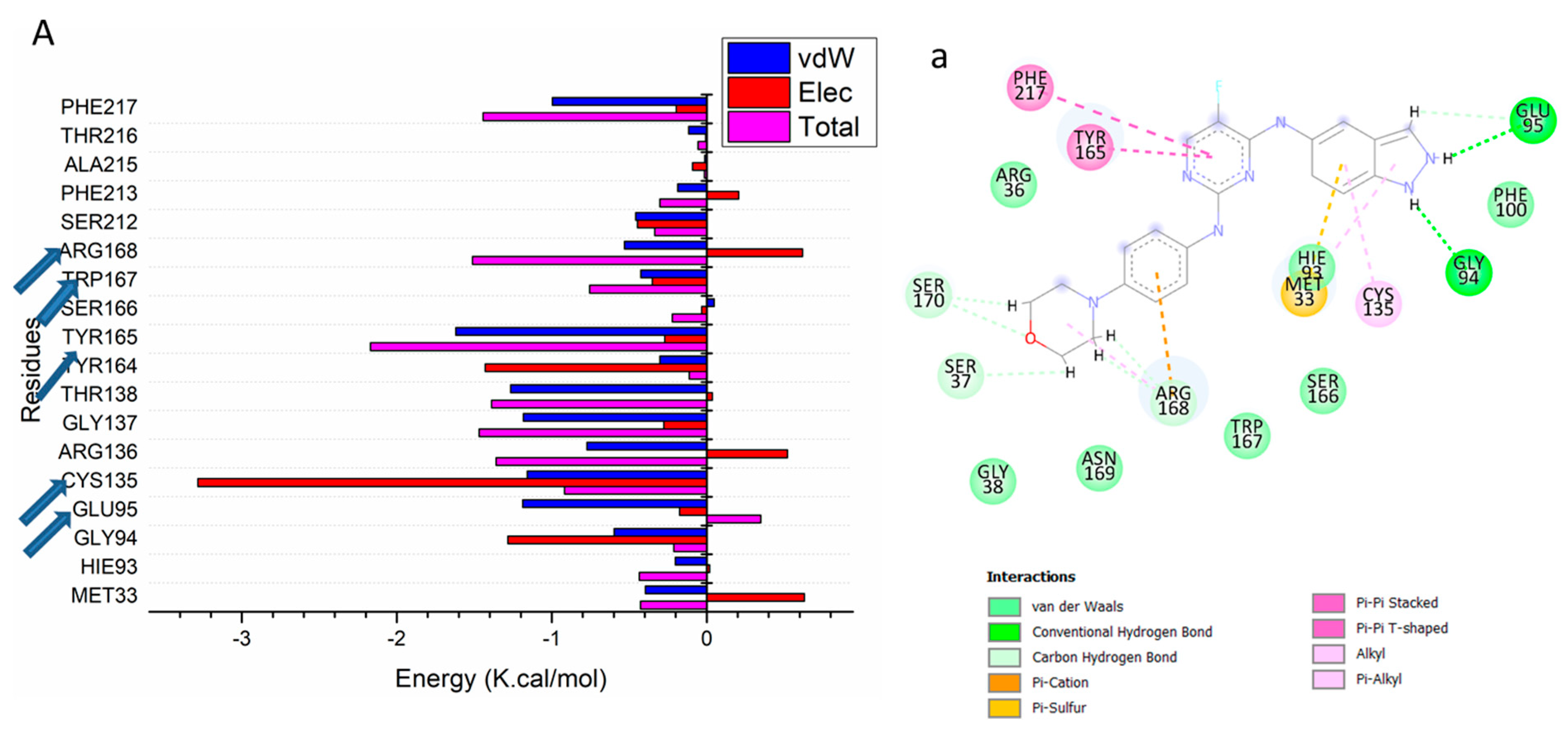
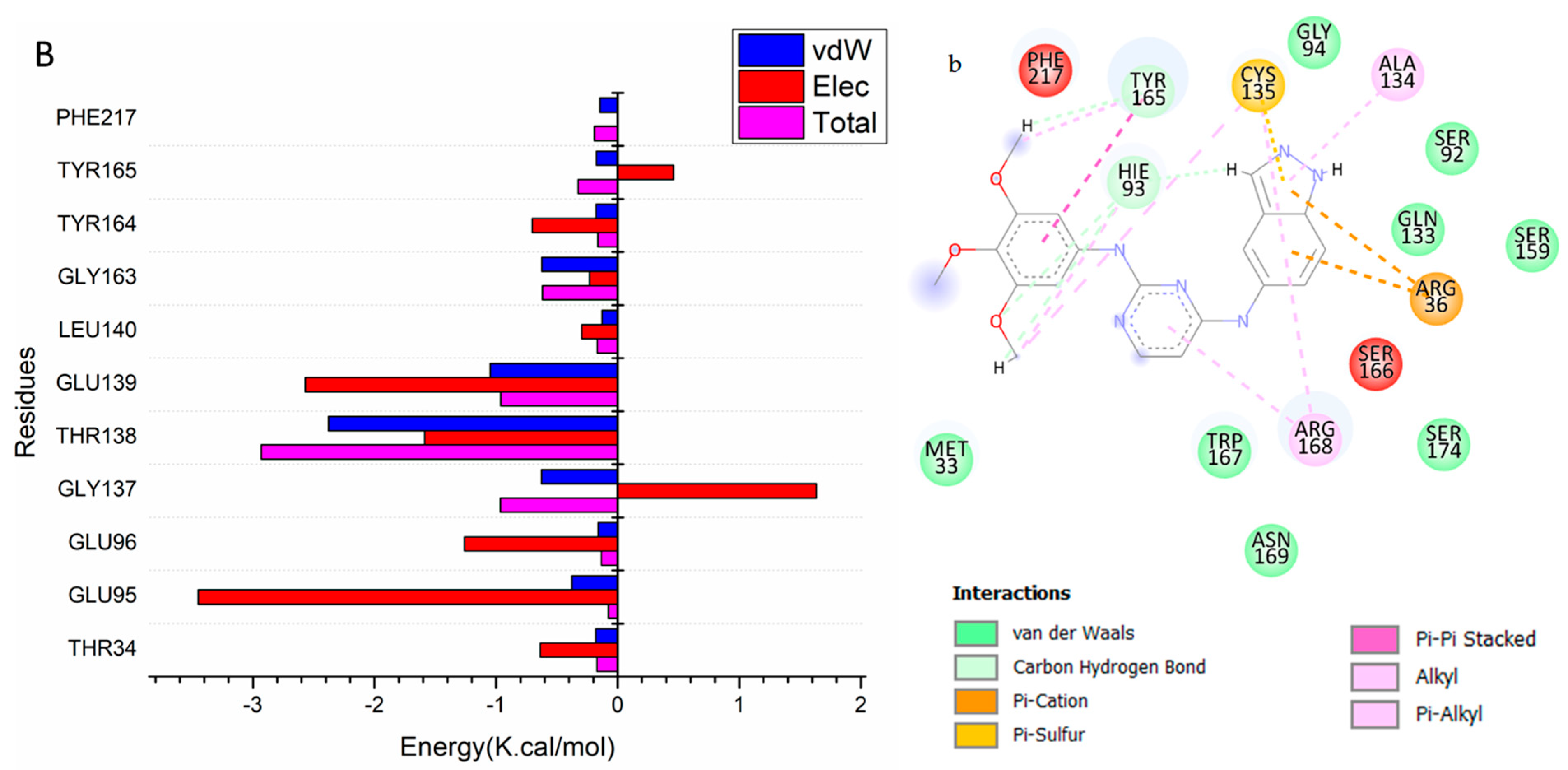
3.2. Binding Interaction Mechanism Based on Binding Free Energy Calculation
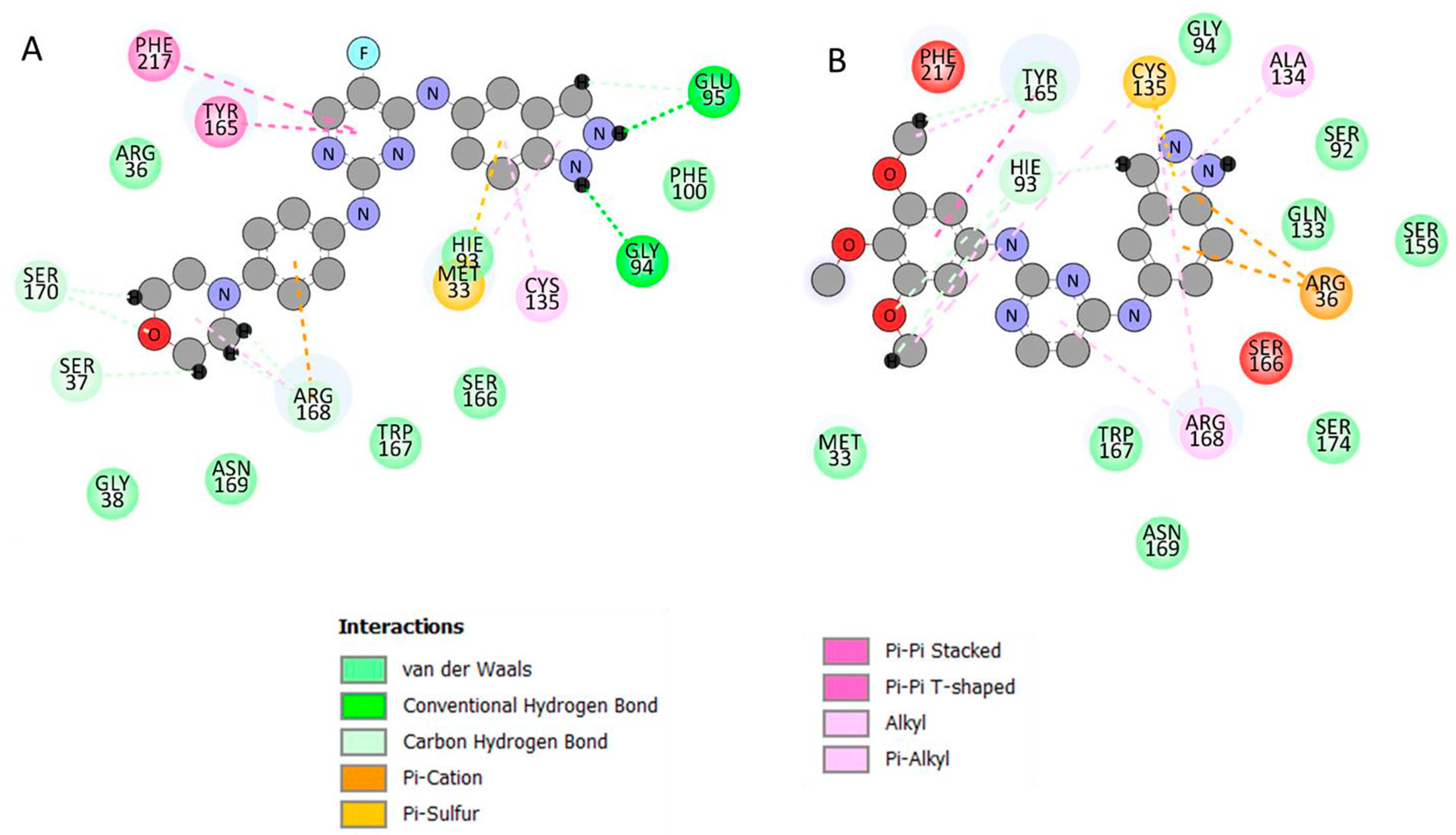
3.3. Identification of the Critical Residues Responsible for Ligand Binding
3.4. Ligand–Residue Interaction Network Profiles
3.5. In Silico ADMET Properties Prediction
4. Structure–Activity Relationship
5. Conclusions
6. Experimental Section
6.1. Chemistry
6.1.1. General Method for the Synthesis of N-(2-Chloro-5-substituted pyrimidin-4-yl)-1H -indazol-5-amine (Compounds 3a and b)
6.1.2. General Procedure for Preparation of Compounds 4a–i
6.2. Biological Assays
6.2.1. MTT Cytotoxicity Assay
6.2.2. Caspase-3/7 Assay
6.2.3. Cell Cycle Analysis
6.3. Molecular Dynamic Study
6.3.1. System Preparation and Molecular Docking
6.3.2. Molecular Docking
6.3.3. Molecular Dynamic (MD) Simulations
6.3.4. Post-MD Analysis
6.3.5. Thermodynamic Calculation
6.3.6. Computation of Drug-like Parameters and ADMET Profiling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kumar, B.; Sharma, P.; Gupta, V.P.; Khullar, M.; Singh, S.; Dogra, N.; Kumar, V. Synthesis and biological evaluation of pyrimidine bridged combretastatin derivatives as potential anticancer agent and mechanistic studies. Bioorg. Chem. 2018, 78, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sabnis, A.J.; Bivona, T.G. Principles of resistance to targeted cancer therapy: Lessons from basic and translational cancer biology. Trends Mol. Med. 2019, 25, 185–197. [Google Scholar] [CrossRef]
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Significance and biological importance of pyrimidine in the microbial world. Int. J. Med. Chem. 2014, 2014, 202784. [Google Scholar] [CrossRef]
- Dai, X.J.; Xue, L.P.; Ji, S.K.; Zhou, Y.; Gao, Y.; Zheng, Y.C.; Liu, H.M.; Liu, H.M. Triazole-fused pyrimidines in target-based anticancer drug discovery. Eur. J. Med. Chem. 2023, 249, 115101. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Wang, C.; Xin, X.; Li, S.; Li, Z.; Zhao, Y.; Gong, P. Design, synthesis and biological evaluation of novel 2, 4-diaminopyrimidine derivatives as potent antitumor agents. New J. Chem. 2019, 43, 10190–10202. [Google Scholar] [CrossRef]
- Xu, Y.; Hao, S.-Y.; Zhang, X.-J.; Li, W.-B.; Qiao, X.-P.; Wang, Z.-X.; Chen, S.-W. Discovery of novel 2, 4-disubstituted pyrimidines as Aurora kinase inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126885. [Google Scholar] [CrossRef]
- Naika, N.S.; Shastri, L.A.; Chougalaa, B.M.; Samundeeswari, S.; Holiyachi, M.; Joshi, S.D.; Sunagar, V. Synthesis of novel aryl and coumarin substituted pyrazolo[1,5-a]pyrimidine derivatives as potent anti-inflammatory and anticancer agents. Chem. Data Collect. 2020, 30, 100550. [Google Scholar] [CrossRef]
- Zhan, Z.; Ai, J.; Liu, Q.; Ji, Y.; Chen, T.; Xu, Y.; Geng, M.; Duan, W. Discovery of anilinopyrimidines as dual inhibitors of c-Met and VEGFR-2: Synthesis, SAR, and cellular activity. ACS Med. Chem. Lett. 2014, 5, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Tang, Z.; Lao, K.; Li, X.; You, Q.; Xiang, H. Structure-activity relationships of 2, 4-disubstituted pyrimidines as dual ERα/VEGFR-2 ligands with anti-breast cancer activity. Eur. J. Med. Chem. 2018, 150, 783–795. [Google Scholar] [CrossRef]
- Jo, J.; Kim, S.H.; Kim, H.; Jeong, M.; Kwak, J.-H.; Han, Y.T.; Jeong, J.-Y.; Jung, Y.-S.; Yun, H. Discovery and SAR studies of novel 2-anilinopyrimidine-based selective inhibitors against triple-negative breast cancer cell line MDA-MB-468. Bioorg. Med. Chem. Lett. 2019, 29, 62–65. [Google Scholar] [CrossRef] [PubMed]
- AboulWafa, O.M.; Daabees, H.M.; Badawi, W.A. 2-Anilinopyrimidine derivatives: Design, synthesis, in vitro anti-proliferative activity, EGFR and ARO inhibitory activity, cell cycle analysis and molecular docking study. Bioorg. Chem. 2020, 99, 103798. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Volov, A.N.; Volov, N.A.; Platonova, Y.B. Design and synthesis of novel 5-alkynyl pyrimidine nucleosides derivatives: Influence of C-6-substituent on antituberculosis activity. Bioorg. Med. Chem. Let. 2021, 48, 128261. [Google Scholar] [CrossRef]
- Haiba, M.E.; Fathalla, O.A.; Zeid, I.F.; Soliman, A.-M.M.; El-Moez, S.I.A.; El-Serwy, W.S. Synthesis and evaluation of some novel tetrahydropyrimidine derivatives as antimicrobial and cytotoxic agent. Res. Chem. Intermed. 2013, 39, 3763–3774. [Google Scholar] [CrossRef]
- Denya, I.; Malan, S.F.; Joubert, J. Indazole derivatives and their therapeutic applications: A patent review. Expert Opin. Ther. Pat. 2018, 28, 441–453. [Google Scholar] [CrossRef]
- Cerecetto, H.; Gerpe, A.; Gonzalez, M.; Aran, V.J.; Ocariz, C.O. Pharmacological properties of indazole derivatives: Recent developments. Mini-Rev. Med. Chem. 2005, 5, 869–878. [Google Scholar] [CrossRef]
- Thangadurai, A.; Minu, M.; Wakode, S.; Agrawal, S.; Narasimhan, B. Indazole: A medicinally important heterocyclic moiety. Med. Chem. Res. 2012, 21, 1509–1523. [Google Scholar] [CrossRef]
- Murugavel, S.; Deepa, S.; Ravikumar, C.; Ranganathand, R.; Alagusundaram, P. Synthesis, structural, spectral and antibacterial activity of 3,3a,4,5-tetrahydro-2H-benzo[g]indazole fused carbothioamide derivatives as antibacterial agents. J. Mol. Struct. 2020, 1222, 128961. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Kevlishvili, I.; Feng, S.; Liu, P.; Buchwald, S.L. Highly enantioselective synthesis of indazoles with a C3-quaternary chiral center using CuH catalysis. J. Am. Chem. Soc. 2020, 142, 10550–10556. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, N.M.; Serya, R.A.; Tolba, M.F.; Ahmed, M.; Barakat, K.; El Ella, D.A.A.; Abouzid, K.A. Design, synthesis, biological evaluation and dynamics simulation of indazole derivatives with antiangiogenic and antiproliferative anticancer activity. Bioorg. Chem. 2019, 82, 340–359. [Google Scholar] [CrossRef] [PubMed]
- Lukasik, P.M.; Elabar, S.; Lam, F.; Shao, H.; Liu, X.; Abbas, A.Y.; Wang, S. Synthesis and biological evaluation of imidazo[4,5-b]pyridine and 4-heteroaryl-pyrimidine derivatives as anti-cancer agents. Eur. J. Med. Chem. 2012, 57, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Elsayed, N.M.; Abou El Ella, D.A.; Serya, R.A.; Tolba, M.F.; Shalaby, R.; Abouzid, K.A. Design, synthesis and biological evaluation of indazole–pyrimidine based derivatives as anticancer agents with anti-angiogenic and antiproliferative activities. Med. Chem. Comm. 2016, 7, 881–899. [Google Scholar] [CrossRef]
- Saleh, N.M.; El-Gaby, M.S.; El-Adl, K.; Abd El-Sattar, N.E. Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors. Bioorg. Chem. 2020, 104, 104350. [Google Scholar] [CrossRef]
- Naaz, F.; Srivastava, R.; Singh, A.; Singh, N.; Verma, R.; Singh, V.K.; Singh, R.K. Molecular modeling, synthesis, antibacterial and cytotoxicity evaluation of sulfonamide derivatives of benzimidazole, indazole, benzothiazole and thiazole. Bioorg. Med. Chem. 2018, 26, 3414–3428. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Li, C.; Wu, X. An efficient and practical synthesis of antibacterial linezolid. J. Chem. Res. 2009, 12, 739–740. [Google Scholar]
- Haiba, M.E.; Al-Abdullah, E.S.; Ahmed, N.S.; Ghabbour, H.A.; Awad, H.M. Efficient and easy synthesis of New benzo[h]chromene and benzo[h]quinoline derivatives as a new class of cytotoxic agents. J. Mol. Struct. 2019, 1195, 702–711. [Google Scholar] [CrossRef]
- Hassan, A.S.; Mostafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef]
- Hamdy, N.A.; Anwar, M.M.; Abu-Zied, K.M.; Awad, H.M. Synthesis, tumor inhibitory and antioxidant activity of new polyfunctionally 2-substituted 5,6,7,8-tetrahydronaphthalene derivatives containing pyridine, thioxopyridine and pyrazolopyridine moieties. Acta Pol. Pharm.-Drug Res. 2013, 70, 987–1001. [Google Scholar]
- Mirzaei, S.; Eisvand, F.; Hadizadeh, F.; Mosaffa, F.; Ghasemi, A.; Ghodsi, R. Design, synthesis and biological evaluation of novel 5,6,7-trimethoxy-N-aryl-2-styrylquinolin-4-amines as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 98, 103711. [Google Scholar] [CrossRef]
- Hasanin, M.; Hashem, A.H.; El-Rashedy, A.A.; Kamel, S. Synthesis of novel heterocyclic compounds based on dialdehyde cellulose: Characterization, antimicrobial, antitumor activity, molecular dynamics simulation and target identification. Cellulose 2021, 28, 8355–8374. [Google Scholar] [CrossRef]
- Machaba, K.E.; Mhlongo, N.N.; Soliman, M.E.S. Induced Mutation Proves a Potential Target for TB Therapy: A Molecular Dynamics Study on LprG. Cell Biochem. Biophys. 2018, 76, 345–356. [Google Scholar] [CrossRef]
- Pan, L.; Patterson, J.C.; Deshpande, A.; Cole, G.; Frautschy, S. Molecular Dynamics Study of Zn(Aβ) and Zn(Aβ)2. PLoS ONE 2013, 8, 70681–70688. [Google Scholar] [CrossRef]
- Wijffels, G.; Dalrymple, B.; Kongsuwan, K.; Dixon, N. Conservation of Eubacterial Replicases. IUBMB Life 2005, 57, 413–419. [Google Scholar] [CrossRef]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins: Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Nassar, A.E.F.; Kamel, A.M.; Clarimont, C. Improving the decision-making process in the structural modification of drug candidates: Enhancing metabolic stability. Drug Discov. Today 2004, 9, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Thornberry, N.A. Caspases: Killer proteases. Trends Biochem. Sci. 1997, 22, 299–306. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–13126. [Google Scholar] [CrossRef]
- Lee, D.; Long, S.A.; Adams, J.L.; Chan, G.; Vaidya, K.S.; Francis, T.A.; Kikly, K.; Winkler, J.D.; Sung, C.-M.; Debouck, C.; et al. Potent and selective nonpeptide inhibitors of caspases 3 and 7 inhibit apoptosis and maintain cell functionality. J. Biol. Chem. 2000, 275, 16007–160014. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Halford, B. Reflections On ChemDraw. Chem. Eng. News Arch. 2014, 92, 26–27. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M. Using Autodock with Autodocktools: A Tutorial; The Scripps Research Institute Molecular Graphics Laboratory: San Diego, CA, USA, 2008; pp. 1–56. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar]
- Lee, T.S.; Cerutti, D.S.; Mermelstein, D.; Lin, C.; LeGrand, S.; Giese, T.J.; Roitberg, A.; Case, D.A.; Walker, R.C.; York, D.M. GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J. Chem. Inf. Model. 2018, 58, 2043–2050. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatm, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Seifert, E. OriginPro 9.1: Scientific data analysis and graphing software—Software review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Archontis, G. MM-GB(PB)SA Calculations of Protein-Ligand Binding Free Energies. In Molecular Dynamics—Studies of Synthetic and Biological Macromolecules; InTech: London, UK, 2012; pp. 1–22. [Google Scholar]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Wolf, R.M. MM/GBSA binding energy prediction on the PDBbind data set: Successes, failures, and directions for further improvement. J. Chem. Inf. Model. 2013, 53, 201–209. [Google Scholar] [CrossRef]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Saluja, S.; Zou, R.; Drach, J.C.; Townsend, L.B. Structure-Activity Relationships among 2-Substituted 5,6-Dichloro-, 4,6-Dichloro-, and 4,5-Dichloro-1-[(2-hydroxyethoxy)methyl]- and -1-[(1,3-dihydroxy-2-propoxy)methyl]benzimidazoles. J. Med. Chem. 1996, 39, 881–891. [Google Scholar] [CrossRef] [PubMed]
- El-Kashef, H.S.; El-Emary, T.I.; Gasquet, M.; Timon-David, P.; Maldonado, J.; Vanelle, P. New pyrazolo[3,4-b]pyrazines: Synthesis and biological activity. Pharmazie 2000, 55, 572–576. [Google Scholar]
- Ben Hadda, T.; Rastija, V.; AlMalki, F.; Titi, A.; Touzani, R.; Mabkhot, Y.N.; Khalid, S.; Zarrouk, A.; Siddiqui, B.S. Petra/Osiris/Molinspiration and Molecular Docking Analyses of 3-Hydroxy-Indolin-2-one Derivatives as Potential Antiviral Agents. Curr. Comput. Aided Drug 2019, 17, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of Free Online ADMET Tools for Academic or Small Biotech Environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef]
- Molecular Properties Prediction—Osiris Property Explorer [Internet]. Available online: https://www.organic-chemistry.org/prog/peo/ (accessed on 16 April 2023).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | R1 | R2 | Cytotoxicity IC50 (μM) | ||
|---|---|---|---|---|---|
| MCF-7 | A549 | Caco2 | |||
| 4a | F | N-(pyrimidin-2-yl) sulfonamido | 2.958 ± 0.14 | 3.204 ± 0.18 | 10.35 ± 0.51 |
| 4b | F | N-(thiazol-2-yl) sulfonamido | 14.28 ± 0.68 | 52.86 ± 2.92 | 31.23 ± 1.55 |
| 4c | F | 3,4,5-TriMeO | 29.12 ± 1.4 | 58.79 ± 3.25 | 67.8 ± 3.36 |
| 4d | F | m-SO2NH2 | 4.798 ± 0.23 | 13.47 ± 0.74 | 9.632 ± 0.48 |
| 4e | F | p-SO2NH2 | 25.09 ± 1.2 | 46.61 ± 2.57 | 7.172 ± 0.36 |
| 4f | F | 4-morpholino | 1.629 ± 0.08 | 12.8 ± 0.71 | 22.33 ± 1.11 |
| 4g | H | p-SO2NH2 | 4.68 ± 0.22 | 11.21 ± 0.62 | 6.909 ± 0.34 |
| 4h | H | N-(thiazol-2-yl)sulfonamido | 21.14 ± 1.01 | 23.5 ± 1.3 | 17.28 ± 0.86 |
| 4i | H | 3,4,5-TriMeO | 1.841 ± 0.09 | 2.305 ± 0.13 | 4.99 ± 0.25 |
| Staurosporine | 8.029 ± 0.38 | 7.354 ± 0.41 | 11.29 ± 0.56 | ||
| Compound | Cytotoxicity IC50 (μM) | SI | |
| MCF-10a | MCF-7 | ||
| 4f | 23.67 ± 1.17 | 1.629 ± 0.08 | 14.5 |
| 4i | 29.52 ± 1.46 | 1.841 ± 0.09 | 16.03 |
| Staurosporine | 34.86 ± 1.73 | 8.029 ± 0.38 | 4.34 |
| Energy Components (kcal/mol) | |||||
|---|---|---|---|---|---|
| Complex | ΔEvdW | ΔEelec | ΔGgas | ΔGsolv | ΔGbind |
| 4f | −30.00 ± 0.35 | −58.56 ± 0.49 | −88.57 ± 0.50 | 63.01 ± 0.42 | −25.56 ± 0.32 |
| 4i | −17.00 ± 0.21 | −76.65 ± 1.18 | −93.65 ± 1.76 | 78.02 ± 1.59 | −15.63 ± 0.25 |
| Comp. | miLog p a | Log S b (mol/L) | TPSA c (Å2) | MW d | nON e | nOHNH f | Nviolation g | Nrot h | Vol |
|---|---|---|---|---|---|---|---|---|---|
| 4a | 3.36 | −5.05 | 150.47 | 477.49 | 11 | 4 | 1 | 7 | 980.04 |
| 4b | 4.23 | −6.02 | 137.58 | 482.53 | 10 | 4 | 0 | 7 | 374.90 |
| 4c | 4.05 | −5.01 | 106.22 | 410.41 | 9 | 3 | 0 | 7 | 349.74 |
| 4d | 3.08 | −4.85 | 138.69 | 399.41 | 9 | 5 | 0 | 5 | 315.82 |
| 4e | 3.11 | −4.85 | 138.69 | 399.41 | 9 | 5 | 0 | 5 | 315.82 |
| 4f | 4.36 | −5.05 | 90.99 | 405.44 | 8 | 3 | 0 | 5 | 351.24 |
| 4g | 2.80 | −4.54 | 138.69 | 381.42 | 9 | 5 | 0 | 5 | 310.89 |
| 4h | 3.92 | −5.7 | 137.58 | 464.54 | 10 | 4 | 0 | 7 | 369.97 |
| 4i | 3.74 | −4.69 | 106.22 | 392.42 | 9 | 3 | 0 | 7 | 344.81 |
| Compound | Human Intestinal Absorption (HIA, %) | In Vitro Caco2 Cell Permeability (nm/s) | In Vitro MDCK Cell Permeability (nm/s) | In Vitro Plasma Protein Binding (%) | In Vivo Blood–Brain Barrier Penetration (C. Brain/C. Blood) | Pgp Inhibition |
|---|---|---|---|---|---|---|
| 4a | 89.849338 | 1.21442 | 0.677897 | 100.00 | 0.0558867 | None |
| 4b | 89.849338 | 1.21442 | 0.677897 | 100.00 | 0.0558867 | None |
| 4c | 91.547903 | 48.4113 | 40.1723 | 85.937587 | 0.677977 | None |
| 4d | 88.031819 | 2.73202 | 1.91443 | 99.873126 | 0.0665592 | None |
| 4e | 88.031819 | 0.727024 | 0.964024 | 92.385127 | 0.060827 | None |
| 4f | 92.405990 | 42.7836 | 39.0133 | 90.835546 | 0.869463 | None |
| 4g | 88.003231 | 0.869472 | 10.3103 | 86.366615 | 0.0536349 | None |
| 4h | 92.965896 | 0.792525 | 0.268974 | 98.188783 | 0.0192336 | None |
| 4i | 91.522703 | 24.5724 | 1.15396 | 83.422482 | 0.551769 | None |
| Comp | GPCR Ligand | Ion-Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Mutagenic | Tumorigenic | Reproductive Effective | Irritant |
|---|---|---|---|---|---|---|---|---|---|
| 4a | 0.10 | −0.10 | 0.73 | −0.42 | −0.06 | None | None | None | None |
| 4b | −0.04 | −0.22 | 0.74 | −0.54 | −0.03 | None | Medium | Medium | None |
| 4c | 0.11 | 0.06 | 0.93 | −0.34 | −0.07 | None | None | None | None |
| 4d | 0.10 | 0.05 | 0.97 | −0.48 | 0.16 | None | None | None | High |
| 4e | 0.11 | 0.07 | 0.99 | −0.45 | 0.18 | None | None | None | None |
| 4f | 0.19 | 0.10 | 0.99 | −0.26 | 0.03 | High | High | None | None |
| 4g | 0.15 | 0.13 | 0.97 | −0.42 | 0.15 | None | None | None | None |
| 4h | −0.00 | −0.17 | 0.71 | −0.52 | −0.07 | None | Medium | Medium | None |
| 4i | 0.15 | 0.12 | 0.92 | −0.32 | −0.11 | None | None | None | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Tuwaijri, H.M.; Al-Abdullah, E.S.; El-Rashedy, A.A.; Ansari, S.A.; Almomen, A.; Alshibl, H.M.; Haiba, M.E.; Alkahtani, H.M. New Indazol-Pyrimidine-Based Derivatives as Selective Anticancer Agents: Design, Synthesis, and In Silico Studies. Molecules 2023, 28, 3664. https://doi.org/10.3390/molecules28093664
Al-Tuwaijri HM, Al-Abdullah ES, El-Rashedy AA, Ansari SA, Almomen A, Alshibl HM, Haiba ME, Alkahtani HM. New Indazol-Pyrimidine-Based Derivatives as Selective Anticancer Agents: Design, Synthesis, and In Silico Studies. Molecules. 2023; 28(9):3664. https://doi.org/10.3390/molecules28093664
Chicago/Turabian StyleAl-Tuwaijri, Hanaa M., Ebtehal S. Al-Abdullah, Ahmed A. El-Rashedy, Siddique Akber Ansari, Aliyah Almomen, Hanan M. Alshibl, Mogedda E. Haiba, and Hamad M. Alkahtani. 2023. "New Indazol-Pyrimidine-Based Derivatives as Selective Anticancer Agents: Design, Synthesis, and In Silico Studies" Molecules 28, no. 9: 3664. https://doi.org/10.3390/molecules28093664
APA StyleAl-Tuwaijri, H. M., Al-Abdullah, E. S., El-Rashedy, A. A., Ansari, S. A., Almomen, A., Alshibl, H. M., Haiba, M. E., & Alkahtani, H. M. (2023). New Indazol-Pyrimidine-Based Derivatives as Selective Anticancer Agents: Design, Synthesis, and In Silico Studies. Molecules, 28(9), 3664. https://doi.org/10.3390/molecules28093664