
Dimer Rhenium Tetrafluoride with a Triple Bond Re-Re: Structure, Bond Strength



Abstract
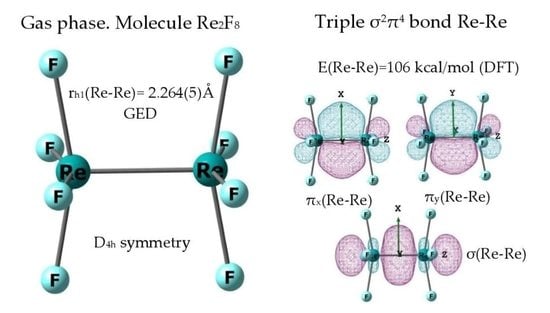
:
1. Introduction
2. Results
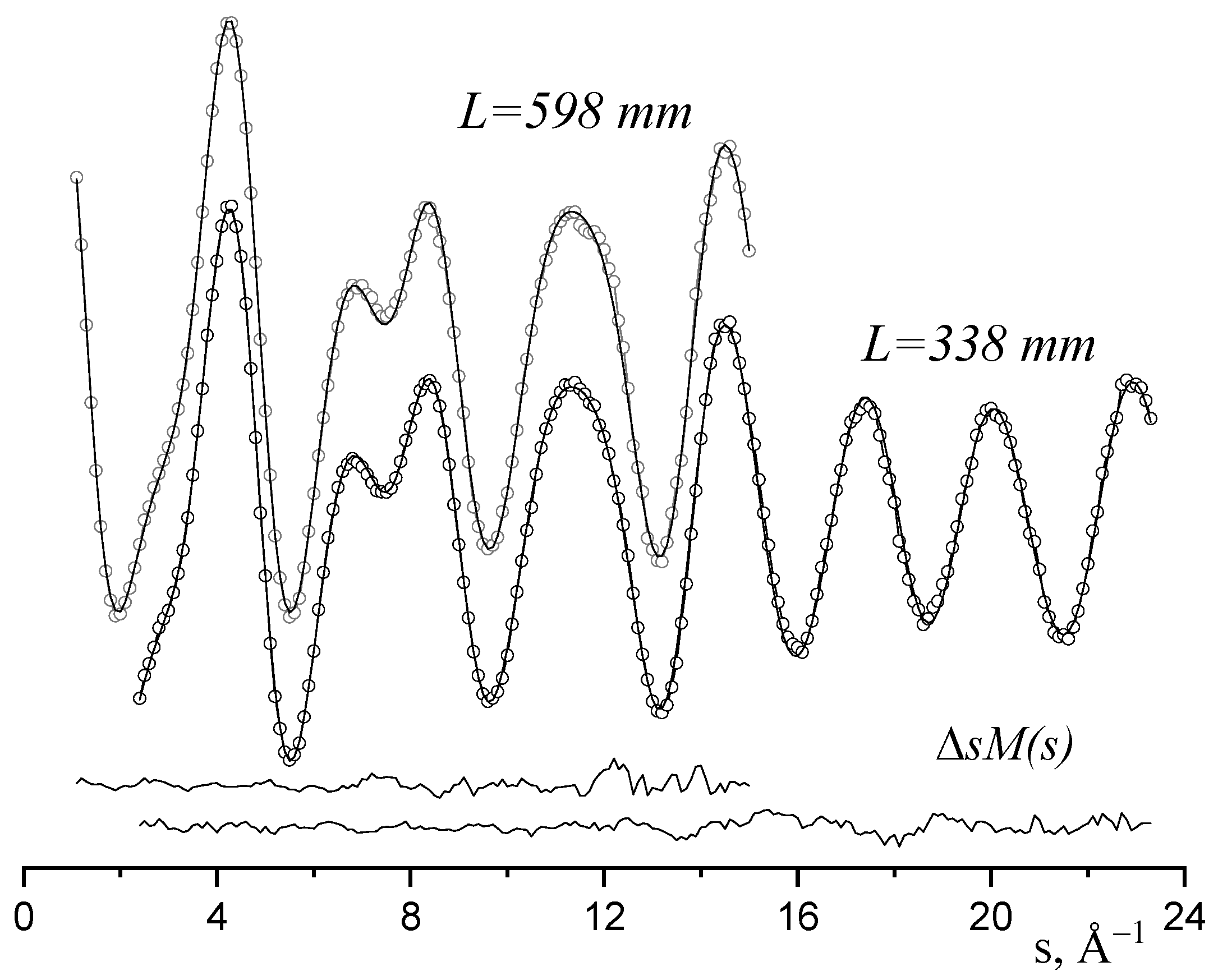
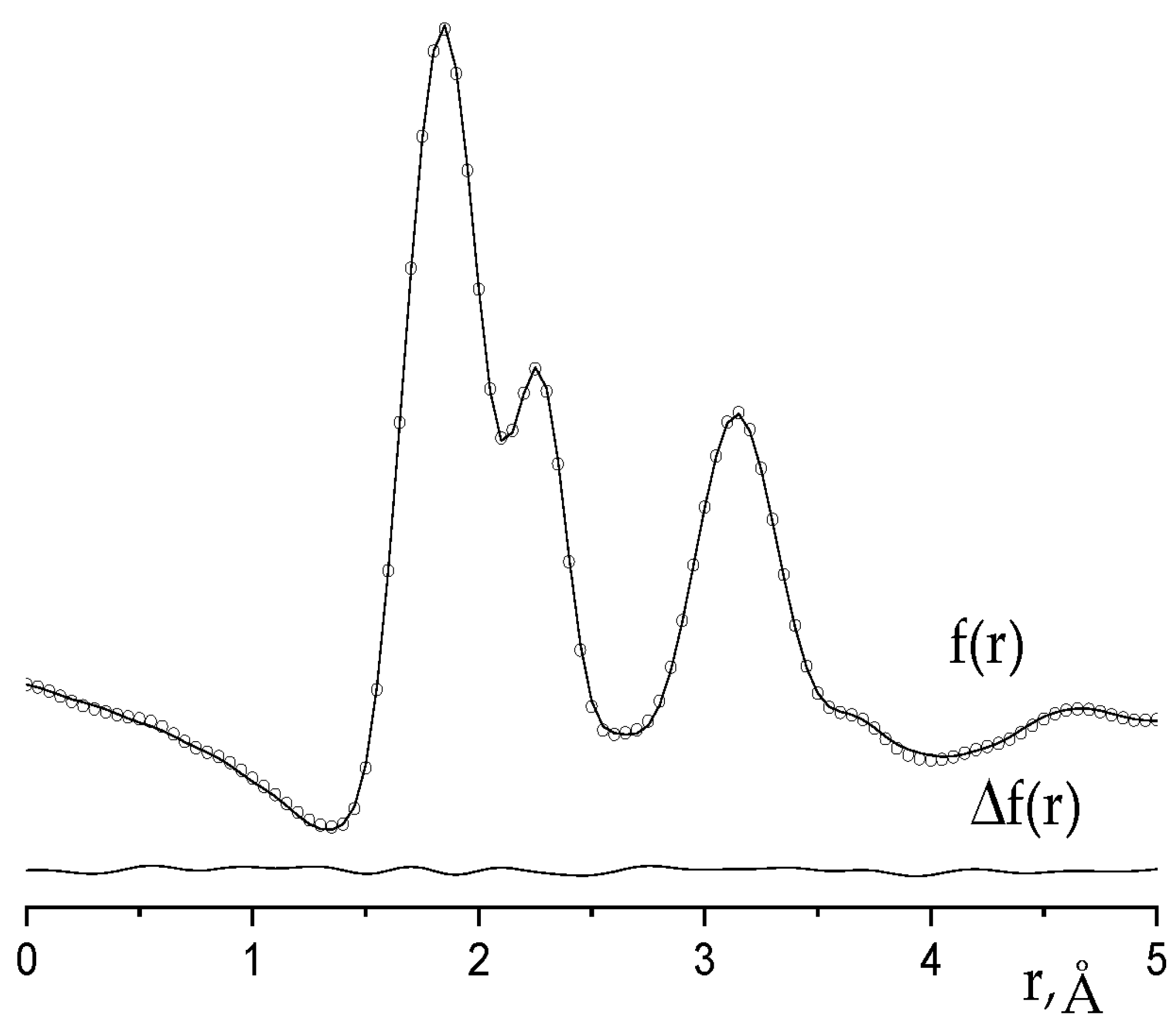
2.1. Analysis of GED/MS Data
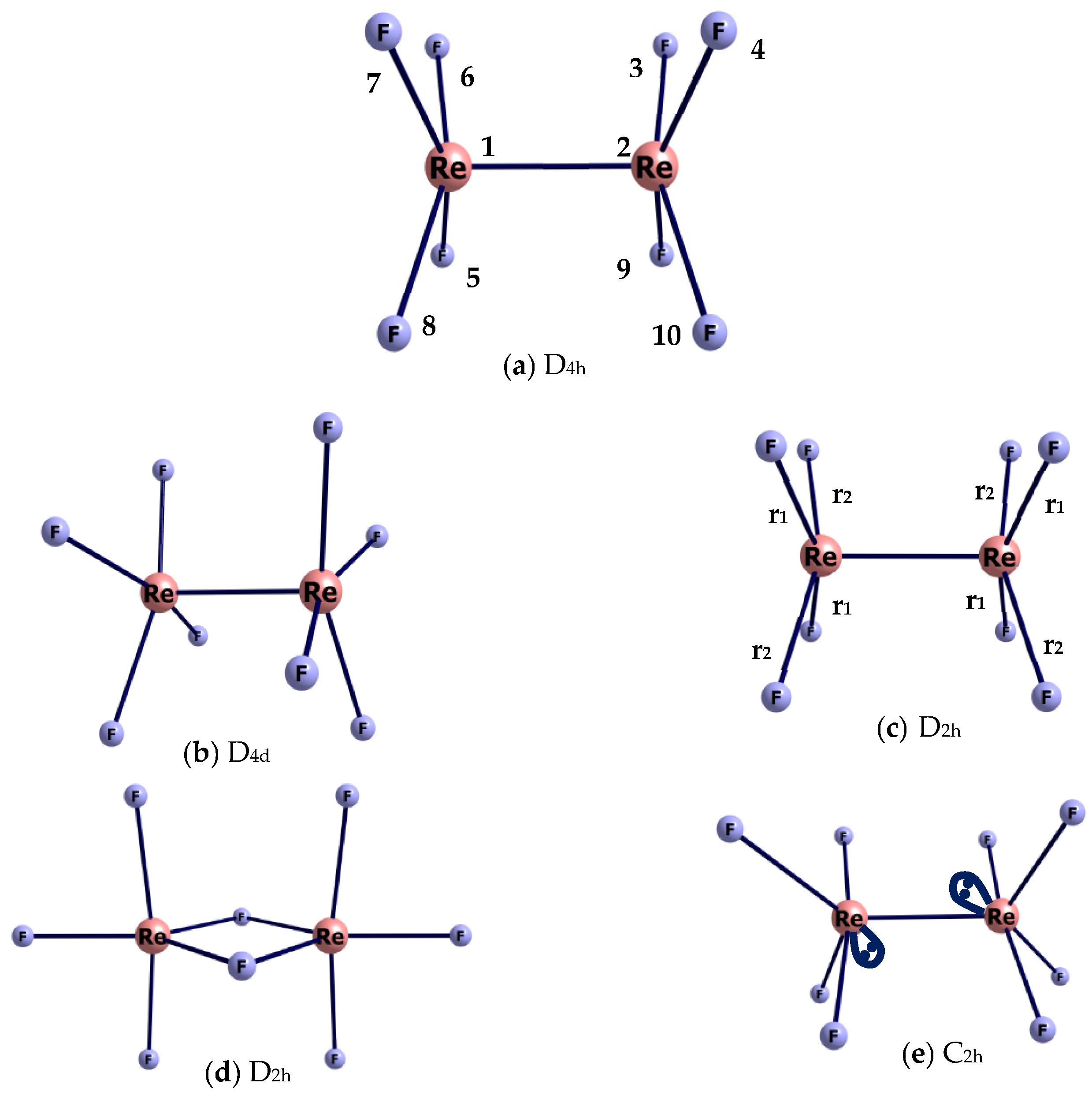
2.2. Geometric Structure of the Re2F8 Molecule
2.3. GED Data and Frequency of Torsional Vibration of the Re2F8 Molecule
2.4. Comparison of the Results of Two Methodology Approaches to LS-Analysis
3. Discussion
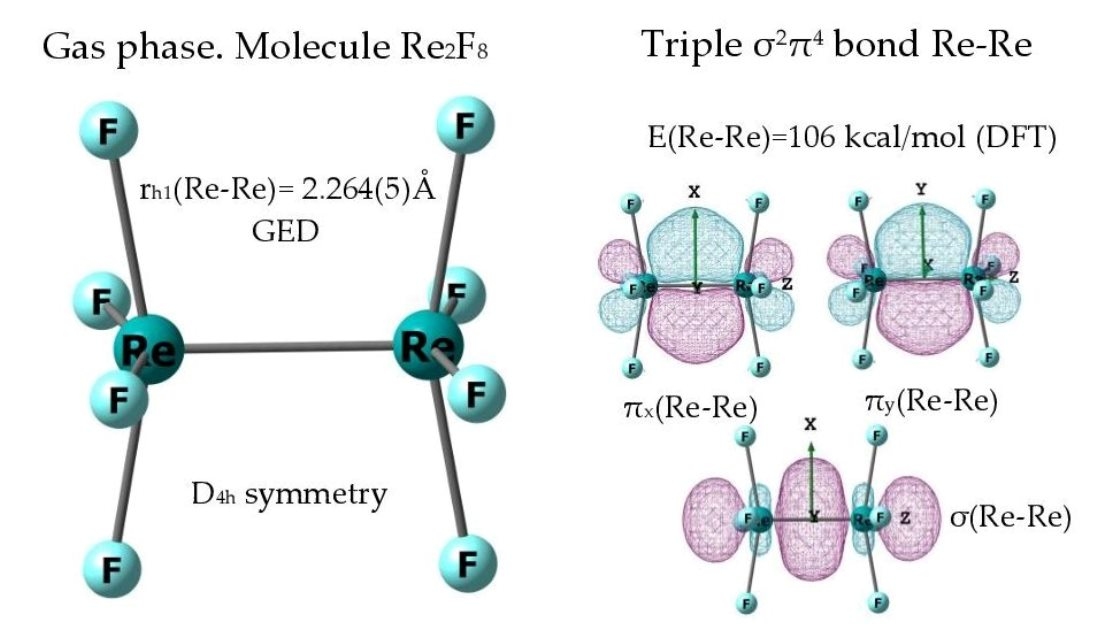
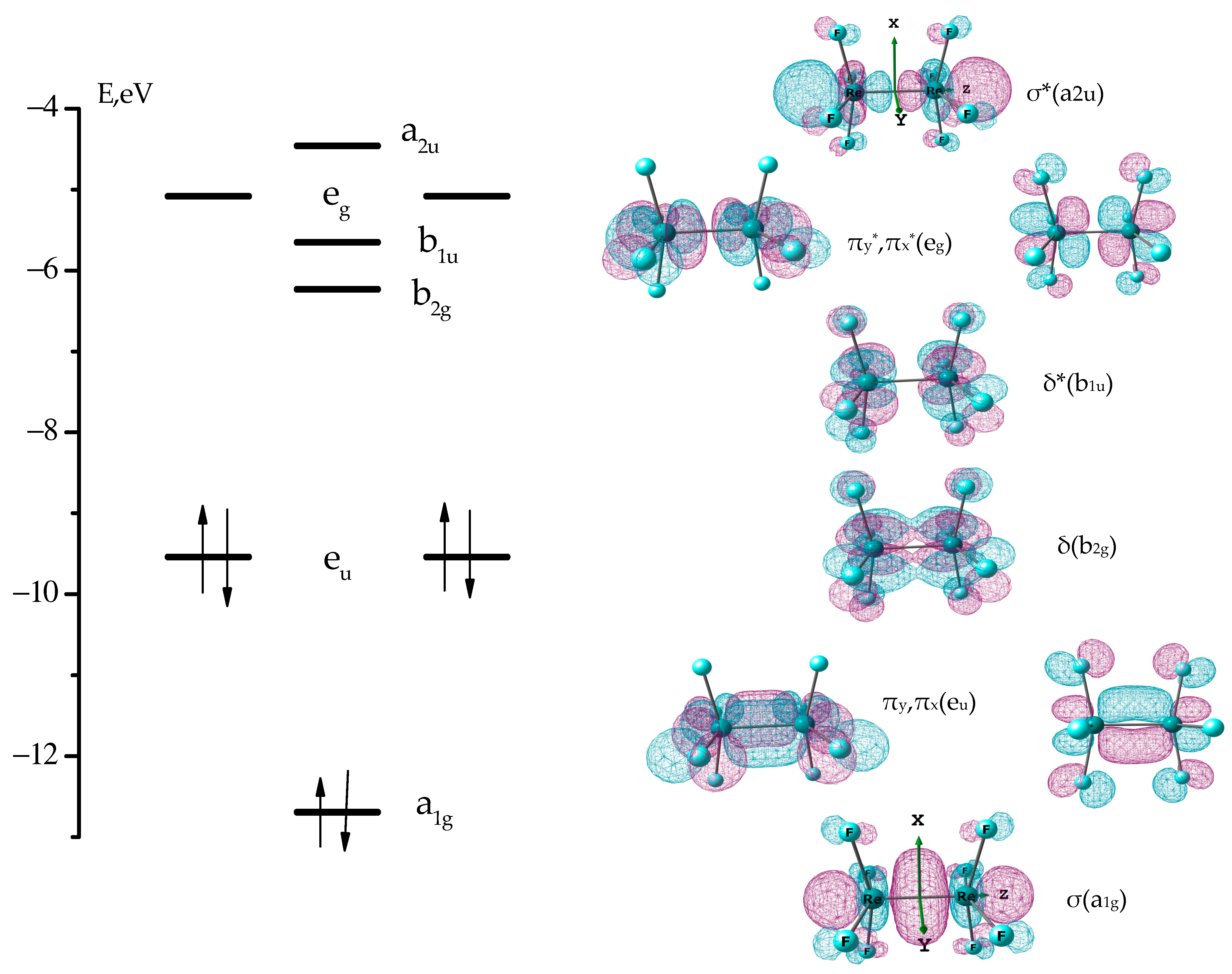
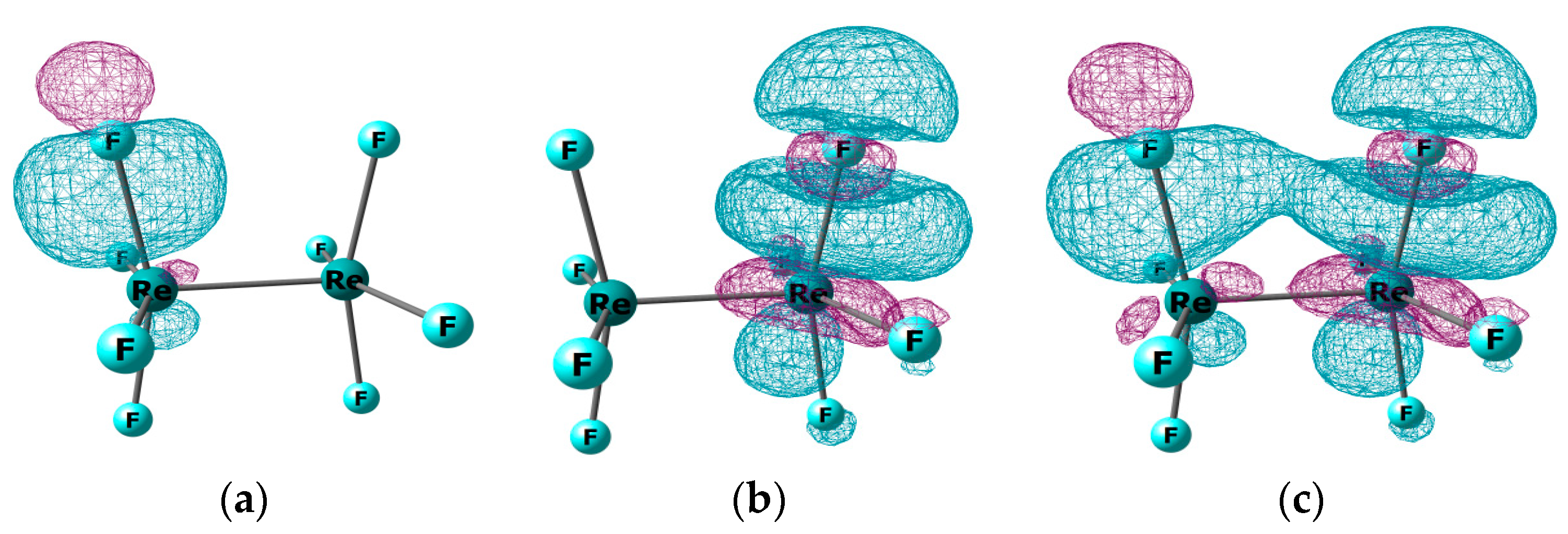
3.1. The Nature of the Re-Re Chemical Bond in the Re2F8 Molecule
3.2. Free Re2F8 Molecule in the Gas Phase and [Re2F8]2− Dianion in Crystals; the ReIV-ReIV Bond vs. the ReIII-ReIII Bond
3.3. Structural Changes in the Series Re2F8 → Re2Cl8 → Re2Br8
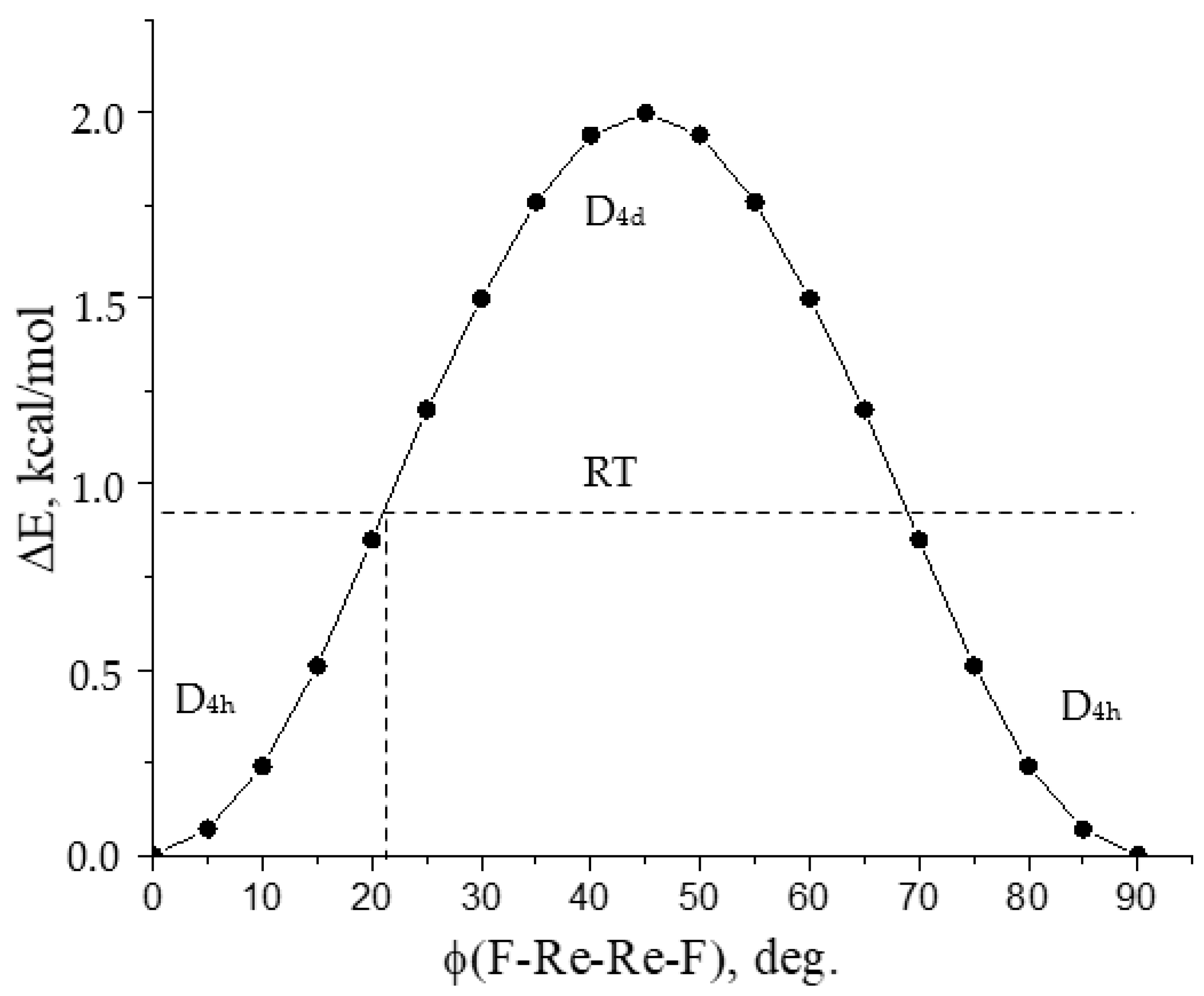
3.4. D4h or D4d Symmetry of Re2X8 Molecules?
3.5. Re2F8 Dissociation Enthalpy and Re-Re and Re-X Bond Energy in Re2X8 Molecules
4. Materials and Methods
4.1. Details of Calculations
4.2. Features of Structural Analysis of GED/MS Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sasaki, Y. Recent Progress in the Chemistry of Rhenium Cluster Complexes and Its Relevance to the Prospect of Corresponding Technetium Chemistry. J. Nucl. Radiochem. Sci. 2005, 6, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Valdés, D.H.; Alberto, R.; Jáuregui-Haza, U. Quantum chemistry calculations of technetium and rhenium compounds with application in radiopharmacy: Review. RSC Adv. 2016, 6, 107127–107140. [Google Scholar] [CrossRef]
- Demoin, D.W.; Li, Y.; Jurisson, S.S.; Deakyne, C.A. Method and basis set analysis of oxorhenium(V) complexes for theoretical calculations. Comp. Theor. Chem. 2012, 997, 34–41. [Google Scholar] [CrossRef]
- Li, Y.; Ma, L.; Gaddam, V.; Gallazzi, F.; Hennkens, H.M.; Harmata, M.; Lewis, M.R.; Deakyne, C.A.; Jurisson, S.S. Synthesis, Characterization, and In Vitro Evaluation of New 99mTc/Re(V)-Cyclized Octreotide Analogues: An Experimental and Computational Approach. Inorg. Chem. 2016, 55, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Noor, A.; Huff, G.S.; Kumar, S.V.; Lewis, J.E.M.; Paterson, B.M.; Schieber, C.; Donnelly, P.S.; Brooks, H.J.L.; Gordon, K.C.; Moratti, S.C.; et al. [Re(CO)3]+ Complexes of exo-Functionalized Tridentate “Click” Macrocycles: Synthesis, Stability, Photophysical Properties, Bioconjugation, and Antibacterial Activity. Organometallics 2014, 33, 7031–7043. [Google Scholar] [CrossRef]
- Hernández-Valdés, D.; Rodríguez-Riera, Z.; Díaz-García, A.; Benoist, E.; Jáuregui-Haza, U. Influence of the chelator structures on the stability of Re and Tc tricarbonyl complexes with iminodiacetic acid tridentate ligands: A computational study. J. Mol. Model. 2016, 22, 179. [Google Scholar] [CrossRef]
- Cotton, F.A.; Harris, C.B. The Crystal and Molecular Structure of Dipotassium Qctachlorodirhenate(III) Dihydrate, K2[Re2Cl8]·2H2O. Inorg. Chem. 1965, 4, 330–333. [Google Scholar] [CrossRef]
- Cotton, F.A.; DeBoer, B.G.; Jeremic, M. Some Reactions of the Octahalodirhenate(III) Ions. Definitive Structural Characterization of the Octabromodirhenate(III) Ion. Inorg. Chem. 1970, 9, 2143–2146. [Google Scholar] [CrossRef]
- Cotton, F.A.; Frenz, B.A.; Stults, B.R.; Webb, T.R. Investigations of Quadruple Bonds by Polarized Crystal Spectra. I. The Interpretation of the Spectrum of Tetra(n-butylammonium) Octachlorodirhenate. The Disordered Crystal Structure. J. Am. Chem. Soc. 1976, 98, 2768–2773. [Google Scholar] [CrossRef]
- Cotton, F.A.; Hall, W.T. Cesium Octachlorodirhenate(II1) Hydrate. The Correct Structure and Its Significance with Respect to the Nature of Metal-Metal Multiple Bonding. Inorg. Chem. 1977, 16, 1867–1871. [Google Scholar] [CrossRef]
- Huang, H.W.; Martin, D.S. Single-crystal polarized spectra for the .delta. .fwdarw. .delta.* band of tetrabutylammonium octachlorodirhenate(III) and tetrabutylammonium octabromodihenate(III). Crystal structure of the octabromo salt. Inorg. Chem. 1985, 24, 96–101. [Google Scholar] [CrossRef]
- Conradson, S.D.; Sattelberger, A.P.; Woodruff, W.H. X-ray Absorption Study of Octafluorodirhenate(III): EXAFS Structures and Resonance Raman Spectroscopy of Octahalodirhenates. J. Am. Chem. Soc. 1988, 110, 1309–1311. [Google Scholar] [CrossRef]
- Cotton, F.A.; Daniels, L.M.; Vidyasagar, K. The crystal structure of [(n-C4H9)4N]2Re2I8: Three-fold disorder of the effectively cubic anion. Polyhedron 1988, 7, 1667–1672. [Google Scholar] [CrossRef]
- Saito, K. Theoretical Studies on Electronic Excited States of Transition Metal Complexes: Explanation and Understanding Based on Molecular Geometries and Electronic Structures. Ph.D. Thesis, Kyoto University, Kyoto, Japan, 2012; pp. 1–152. [Google Scholar] [CrossRef]
- Henkel, G.; Peters, G.; Preetz, W.; Skowronek, J. Kristallstruktur und Schwingungsspektren von [(n-C4H9)4N]2[Re2F8]·2(C2H5)2O. Z. Naturforsch. B 1990, 45, 469–475. [Google Scholar] [CrossRef]
- Giricheva, N.I.; Girichev, G.V.; Lapshina, S.B.; Shlykov, S.A.; Politov, Y.A.; Butskii, V.D.; Pervov, V.S. Geometric structure and vibration frequencies of Re2F8. J. Struct. Chem. 1993, 34, 214–224. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.G. 2004. Available online: http://www.chem.wisc.edu/~nbo5 (accessed on 20 March 2023).
- Keith, T.A. AIMAll; TK Gristmill Software: Overland Park, KS, USA, 2016. [Google Scholar]
- Sipachev, V.A. Calculation of shrinkage corrections in harmonic approximation. J. Mol. Struct. (Theochem.) 1985, 121, 143–151. [Google Scholar] [CrossRef]
- Sipachev, V.A. Vibrational effects in diffraction and microwave experiments: A start on the problem. In Advances in Molecular Structure Research; Hargittai, I., Hargittai, M., Eds.; JAI Press: New York, NY, USA, 1999; pp. 263–311. [Google Scholar]
- Sipachev, V.A. Local centrifugal distortions caused by internal motions of molecules. J. Mol. Struct. 2001, 67–72, 567–568. [Google Scholar] [CrossRef]
- Politov, Y.A.; Alikhanjan, A.S.; Butskiy, V.D.; Pervov, V.S.; Buslaev, Y.A. Polimeric molecules of lower rhenium fluorides. Proc. USSR Acad. Sci. 1987, 296, 1385–1387. (In Russian) [Google Scholar]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Nakano, H. Quasidegenerate perturbation theory with multiconfigurational self-consistent-field reference functions. J. Chem. Phys. 1993, 99, 7983–7992. [Google Scholar] [CrossRef]
- Witek, H.A.; Choe, Y.-K.; Finley, J.P.; Hirao, K. Intruder state avoidance multireference Møller–Plesset perturbation theory. J. Comput. Chem. 2002, 23, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Granovsky, A.A. Firefly. Version 8. 2013. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 20 March 2023).
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Tatewaki, H.; Koga, T. Contracted Gaussian-type basis functions revisited. J. Chem. Phys. 1996, 104, 8493–8499. [Google Scholar] [CrossRef]
- Noro, T.; Sekiya, M.; Koga, T. Contracted polarization functions for the atoms helium through neon. Theor. Chem. Acc. 1997, 98, 25–32. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3101. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Version D.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Figgen, D.; Peterson, K.A.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials and correlation consistent basis sets for the 5d elements Hf-Pt. J. Chem. Phys. 2009, 130, 164108. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Wilson, A.K.; Woon, D.E.; Peterson, K.A.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. IX. The atoms gallium through krypton. J. Chem. Phys. 1999, 110, 7667–7676. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibsom, T.D.; Windus, T.L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Zhurko, G.A.; Zhurko, D.A. ChemCraft. Version 1.6, Build 312. Available online: https://www.chemcraftprog.com/index.html (accessed on 20 March 2023).
- Girichev, G.V.; Utkin, A.N.; Revichev, F. Modernization of electron diffraction recorder EMR-100 for gas research. Prib. Tekh. Eksp. 1984, 27, 187. (In Russian) [Google Scholar]
- Girichev, G.V.; Shlykov, S.A.; Revichev, Y.F. Equipment for studying the structure of molecules of valence-unsaturated compounds. Prib. Tekh. Eksp. 1986, 4, 167. (In Russian) [Google Scholar]
- Girichev, E.G.; Zakharov, A.V.; Girichev, G.V.; Bazanov, M.I. Automation of a physicochemical experiment: Photometry and voltammetry. Izv. Vyssh. Uchebn. Zaved. Tekstiln. Prom. 2000, 2, 142. (In Russian) [Google Scholar]
- Andersen, B.; Seip, H.M.; Strand, T.G.; Stolevik, R. Procedure and Computer Programs for the Structure Determination of Gaseous Molecules from Electron Diffraction Data. Acta Chem. Scand. 1969, 23, 3224–3234. [Google Scholar] [CrossRef]
- Vishnevskiy, Y.V.; Zhabanov, Y.A. New implementation of the first-order perturbation theory for calculation of interatomic vibrational amplitudes and corrections in gas electron diffraction. J. Phys. Conf. Ser. 2015, 633, 012076. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
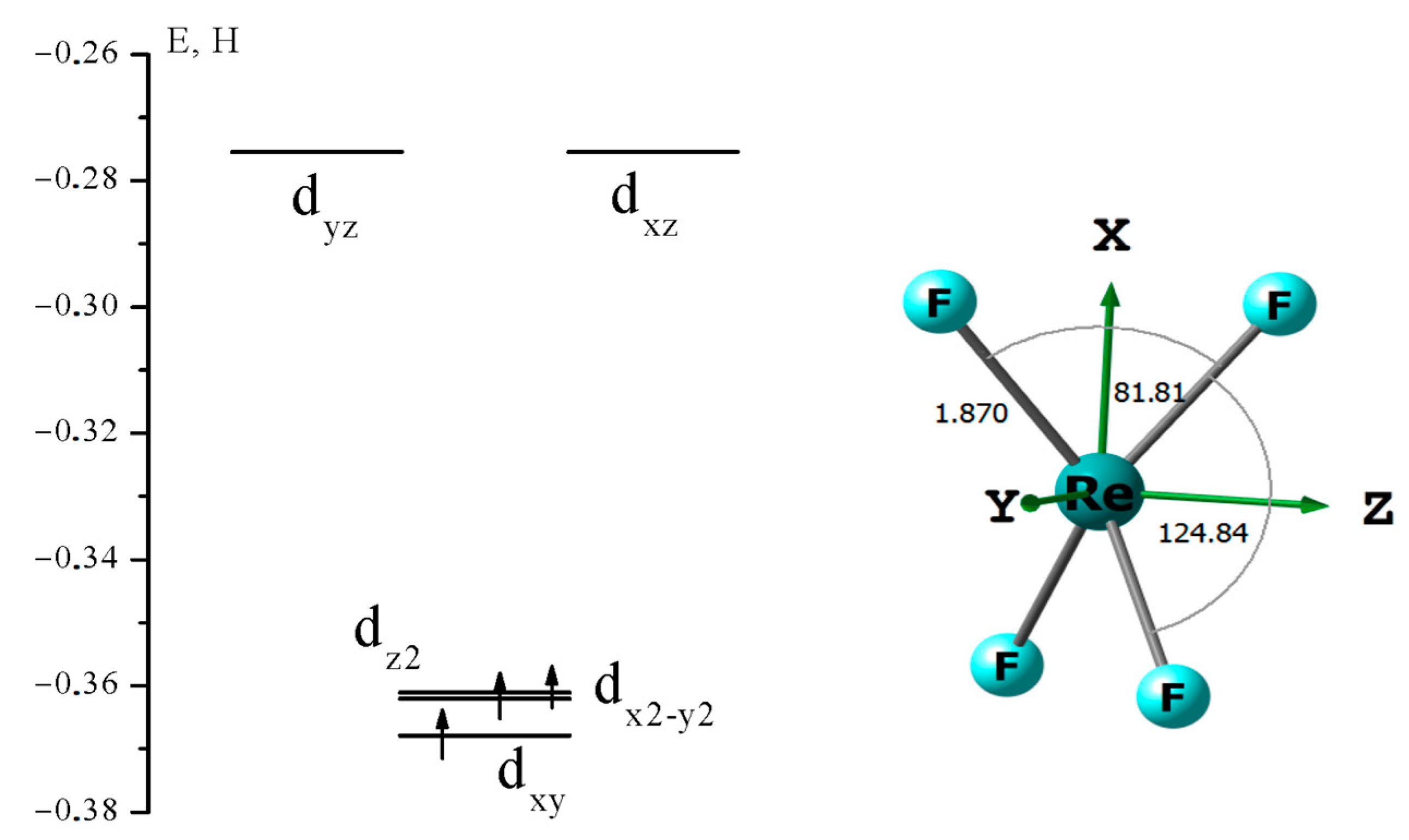{kind=link}
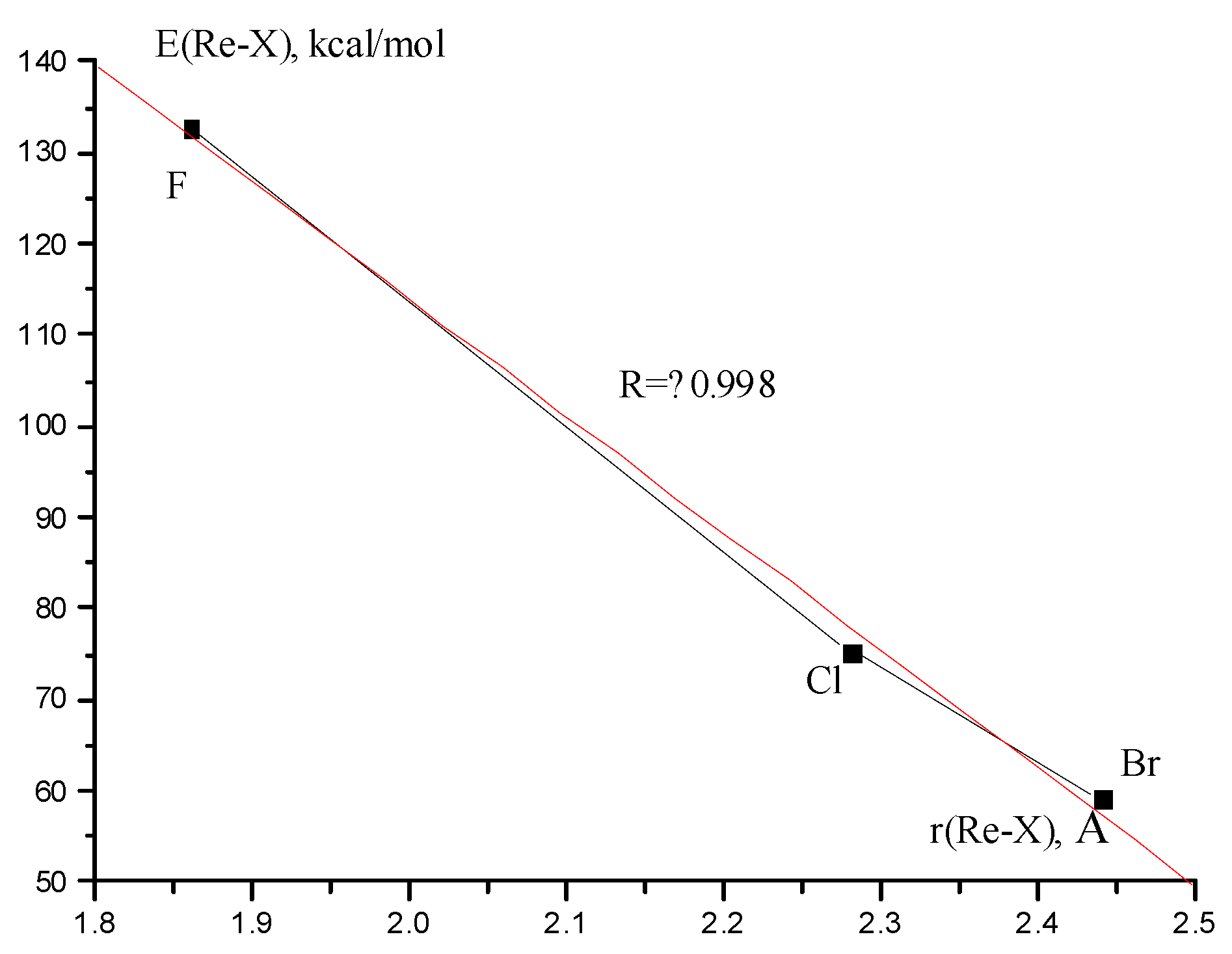{kind=link}
| Parameters | PBE | rh1 | lcalc | lexp | rα [16] | lcalc [16] | lexp [16] |
|---|---|---|---|---|---|---|---|
| Re1-Re2 | 2.259 | 2.264 (5) | 0.038 | 0.046 (2) | 2.269 (5) | 0.035 | 0.035 (2) |
| Re1-F6 | 1.861 | 1.846 (4) | 0.044 | 0.045 (4) | 1.830 (4) | 0.042 | 0.041 (5) |
| F5…F8 | 2.592 | 2.573 (6) | 0.115 | 0.123 (2) | 0.117 | 0.122 (16) | |
| F5…F9 | 2.901 | 2.887 (9) | 0.240 | 0.238 (4) | 0.199 | 0.192 (8) | |
| Re1…F9 | 3.165 | 3.153 (7) | 0.116 | 0.114 (4) | 3.123 (10) | 0.108 | 0.100 (8) |
| F5…F7 | 3.665 | 3.639 (8) | 0.071 | 0.074 (14) | 0.064 | 0.085 (30) | |
| F5…F10 | 3.890 | 3.830 (48) | 0.314 | 0.317 (14) | 0.283 ÷ 0.452 | 0.33 (8) | |
| F5…F4 | 4.674 | 4.643 (10) | 0.115 | 0.161 (34) | 0.111 | 0.18 (8) | |
| F5Re1Re2 | 99.9 | 99.7 (2) | 98.7 (7) | ||||
| F4ReReF7 | 2.4 (3.6) | ||||||
| Rf, % | 4.7 | 7.0 |
| qAIM/qNPA (Re) | 2.36/1.57 | |
| qAIM/qNPA (F) | −0.59/−0.39 | |
| bond | Re-F | Re-Re |
| ε | 0.074 | 0.000 |
| δAIM/PNBO | 0.83/0.81 | 2.28/2.24 |
| r (Re-Re), Å | r (Re-F), Å | FReRe, ° | ν (Re-Re) cm−1 | νtors cm−1 | q (Re) | q (F) | |
|---|---|---|---|---|---|---|---|
| Re2F8 GED, D4h | 2.264 (5) | 1.846 (4) | 99.7 (2) | 345 a | 30 | 1.57 a | −0.39 a |
| [Re2F8]2− X-ray, D4h | 2.188 (3) [15] 2.20 [12] | 1.95 [12] | 104.5 a | 353 a | 69 a | 1.23 a | −0.56 a |
| Molecular Characteristics | Re2F8 D4h | Re2Cl8 D4h | Re2Br8 D4h/D4d |
|---|---|---|---|
| r (Re-Re), Å | 2.259 | 2.317 | 2.336/2.300 |
| r (Re-X), Å | 1.860 | 2.280 | 2.441/2.445 |
| Re-Re-X, ° | 99.9 | 101.6 | 102.6/102.0 |
| X-Re-X, ° | 88.3 | 87.7 | 87.3/87.5 |
| r (X…X), Å | 2.901 | 3.234 | 3.402/3.786 |
| ΣrVdV (X), Å b | 2.94 | 3.5 | 3.9 |
| ν (Re-Re), cm−1 | 338 | 268 | 261/275 |
| νtor (Re-Re), cm−1 | 42.6 | 20.2 | 12.5i/9.1 |
| E (D4d)-E (D4h), kcal/mol | 2.0 | 1.1 | −1.0 |
| E (Re-Re), kcal/mol | 106.1 | 101.1 | 99.8 |
| E (Re-X), kcal/mol | 132.7 | 75.4 | 59.1 |
| re (Re-Re) Å | re (Re-F) Å | αe (ReReF) ° | ν (Re-Re) cm−1 | νtors (Re-Re) cm−1 | ν (Re-F) a cm−1 | |
|---|---|---|---|---|---|---|
| PBE0/qRECP-1 | 2.226 | 1.843 | 100.1 | 370 | 40 | 617–749 |
| B3LYP/qRECP-2 | 2.288 | 1.871 | 99.7 | 345 | 10 | 594–710 |
| PBE/RECP-3 | 2.259 | 1.860 | 99.9 | 338 | 43 | 597–704 |
| GED | 2.264 (5) | 1.846 (4) | 99.8 (0.2) | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giricheva, N.I.; Tverdova, N.V.; Sliznev, V.V.; Girichev, G.V. Dimer Rhenium Tetrafluoride with a Triple Bond Re-Re: Structure, Bond Strength. Molecules 2023, 28, 3665. https://doi.org/10.3390/molecules28093665
Giricheva NI, Tverdova NV, Sliznev VV, Girichev GV. Dimer Rhenium Tetrafluoride with a Triple Bond Re-Re: Structure, Bond Strength. Molecules. 2023; 28(9):3665. https://doi.org/10.3390/molecules28093665
Chicago/Turabian StyleGiricheva, Nina I., Natalia V. Tverdova, Valery V. Sliznev, and Georgiy V. Girichev. 2023. "Dimer Rhenium Tetrafluoride with a Triple Bond Re-Re: Structure, Bond Strength" Molecules 28, no. 9: 3665. https://doi.org/10.3390/molecules28093665
APA StyleGiricheva, N. I., Tverdova, N. V., Sliznev, V. V., & Girichev, G. V. (2023). Dimer Rhenium Tetrafluoride with a Triple Bond Re-Re: Structure, Bond Strength. Molecules, 28(9), 3665. https://doi.org/10.3390/molecules28093665