
Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
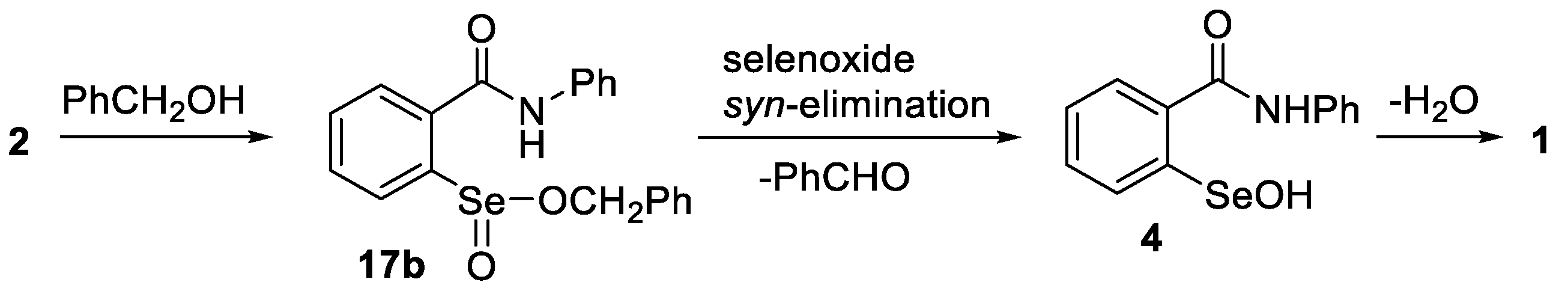{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
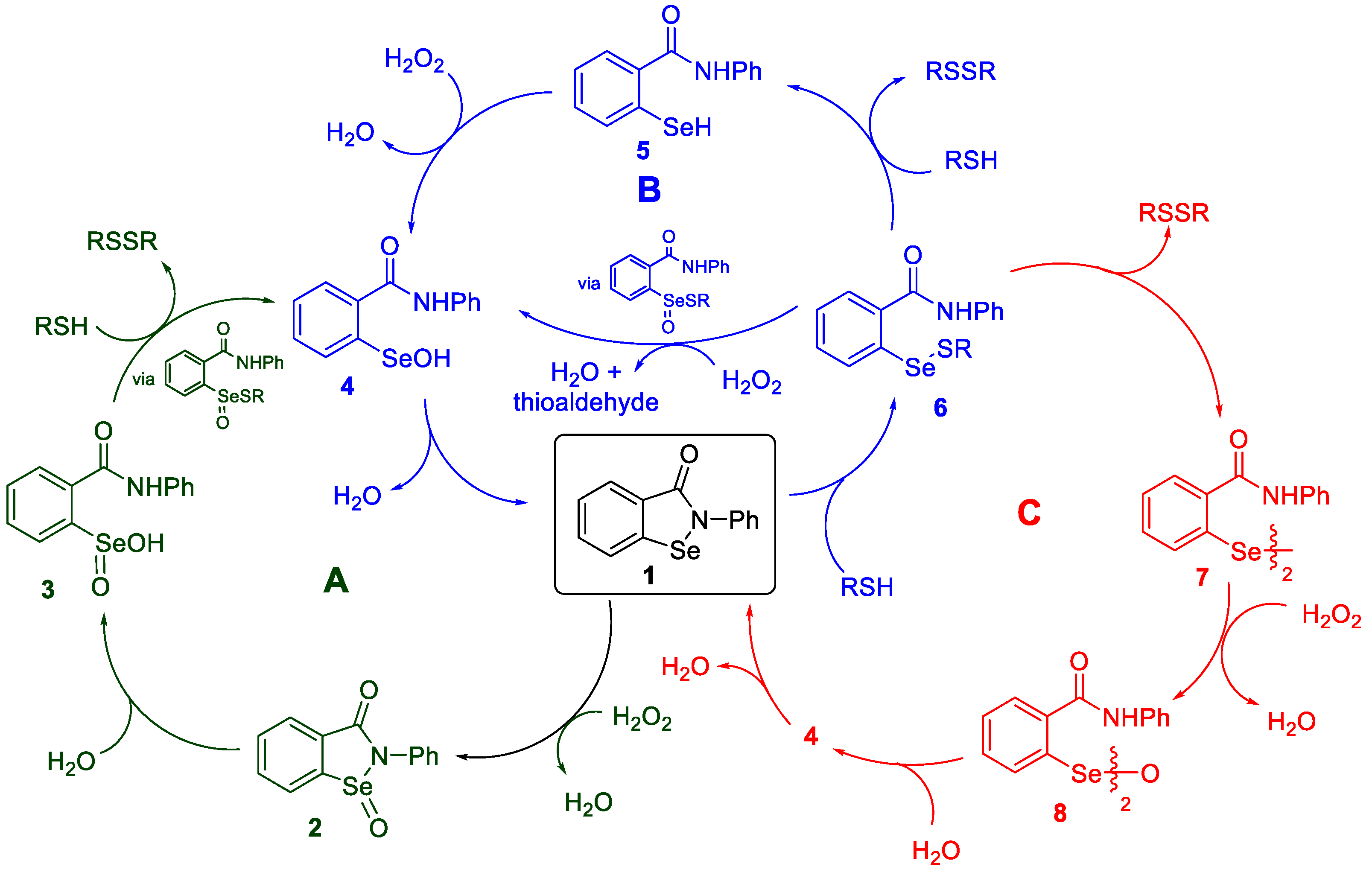
2. Results and Discussion
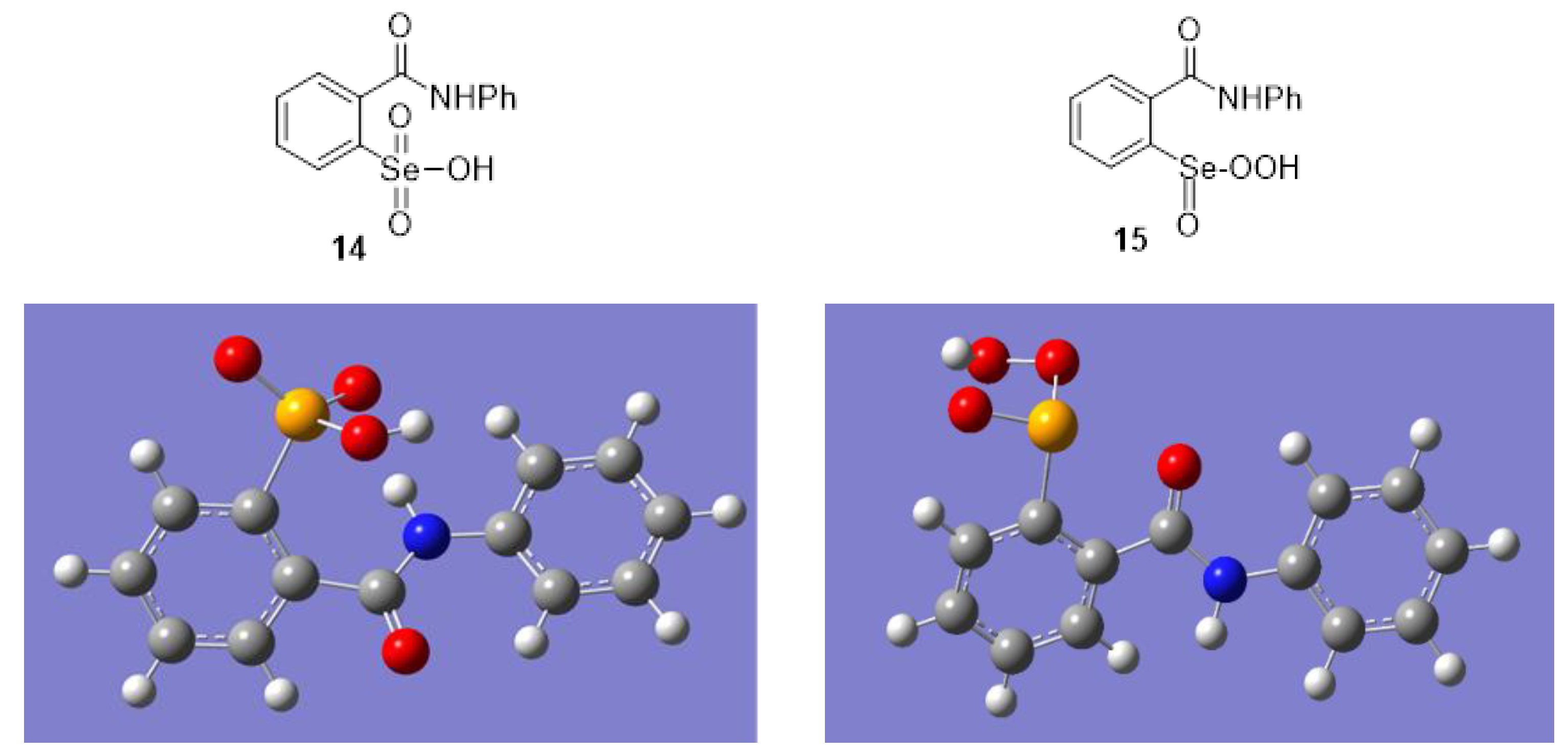
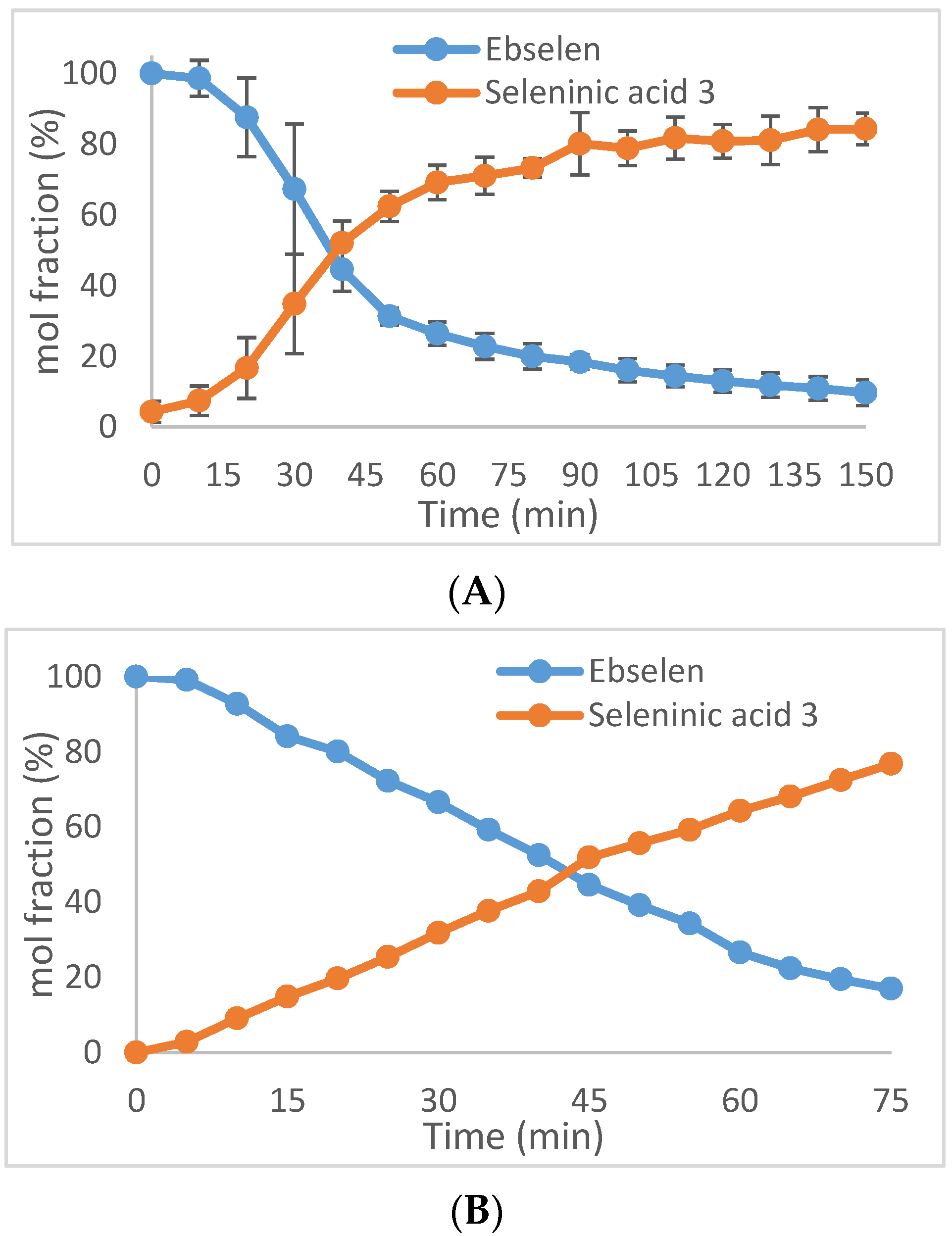
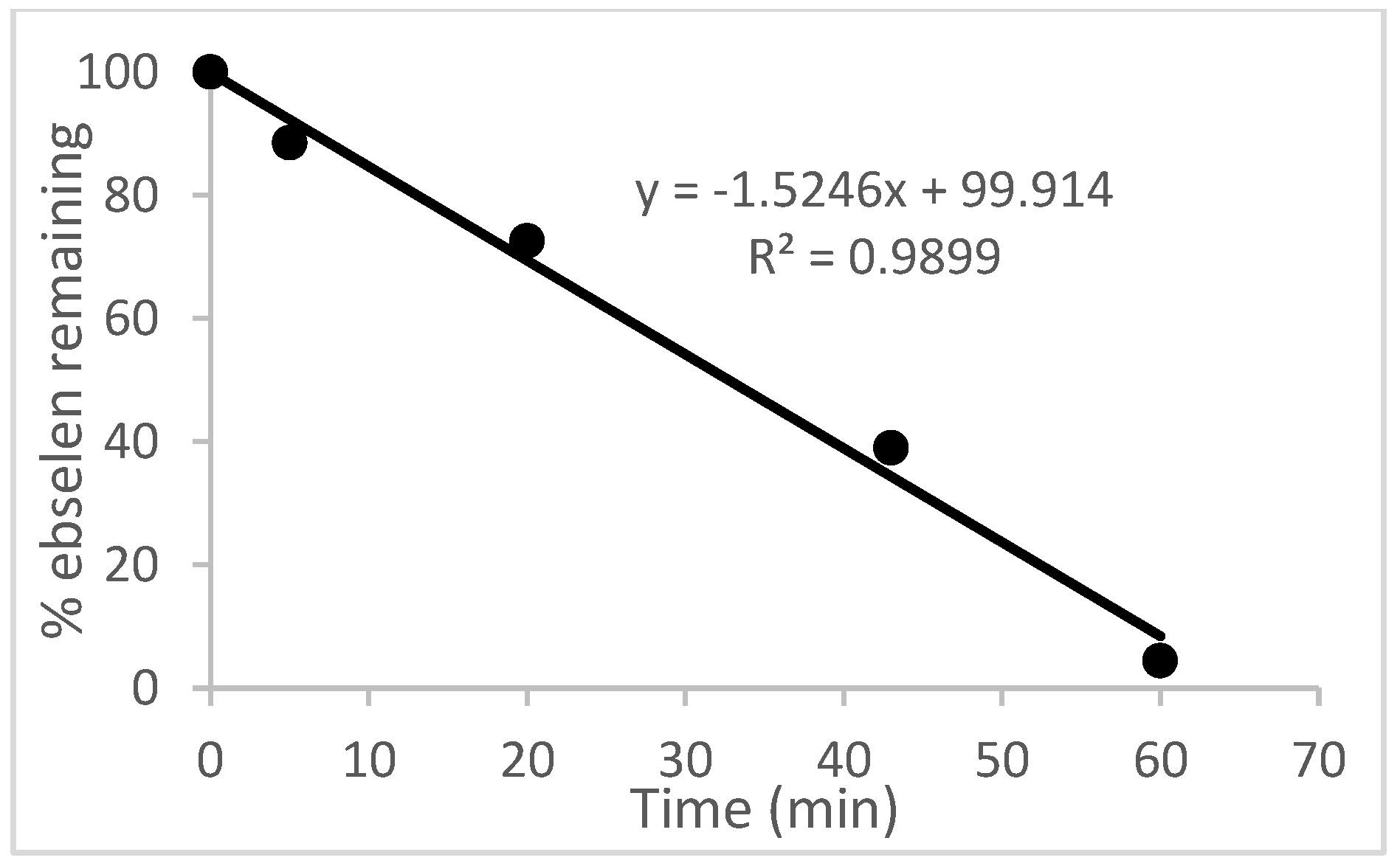
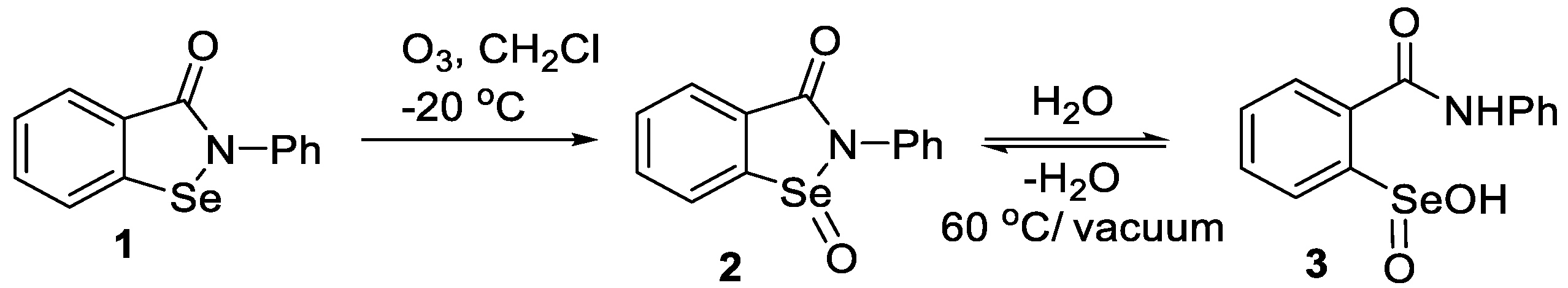
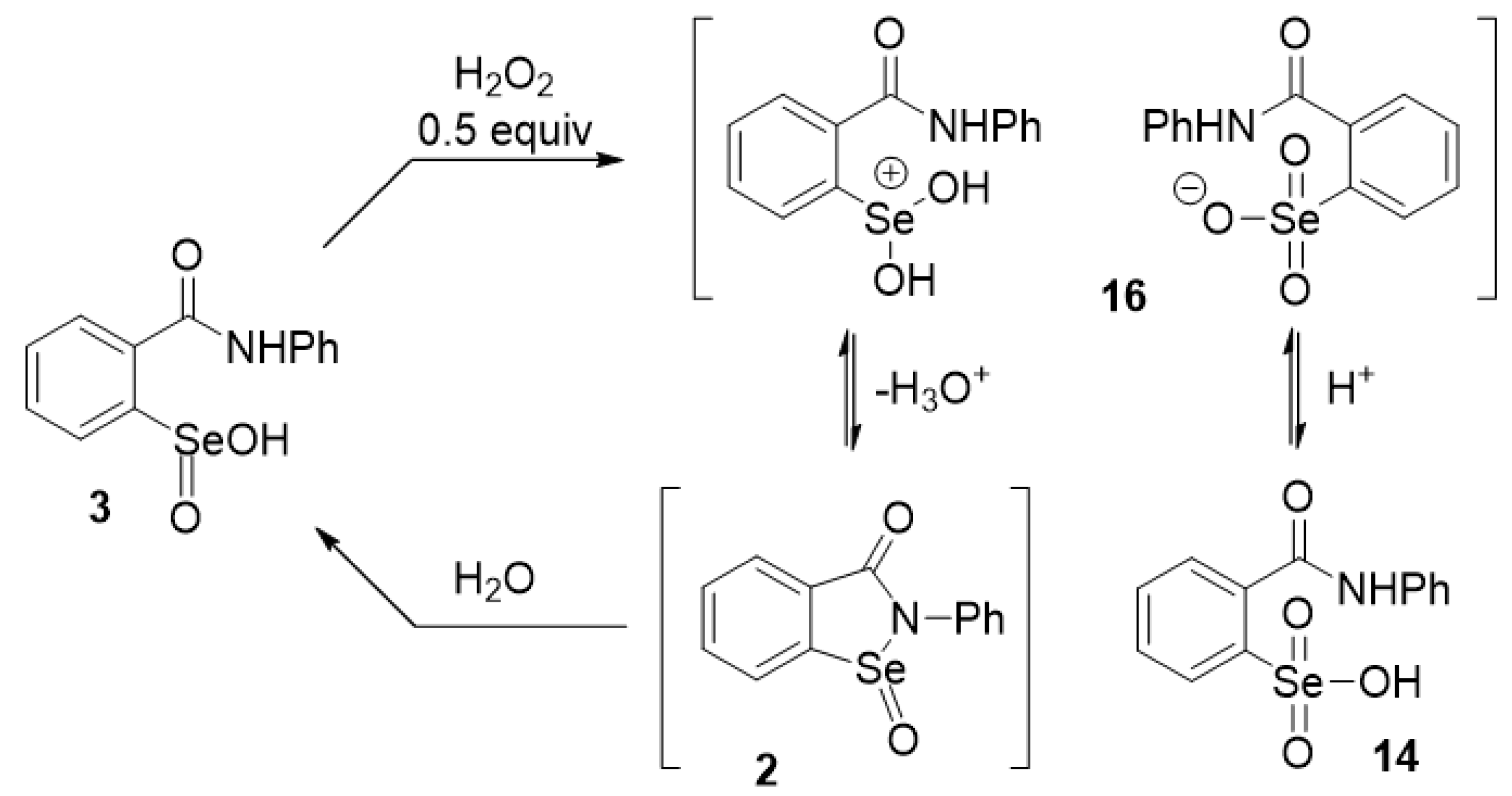
2.1. Oxidation of Ebselen
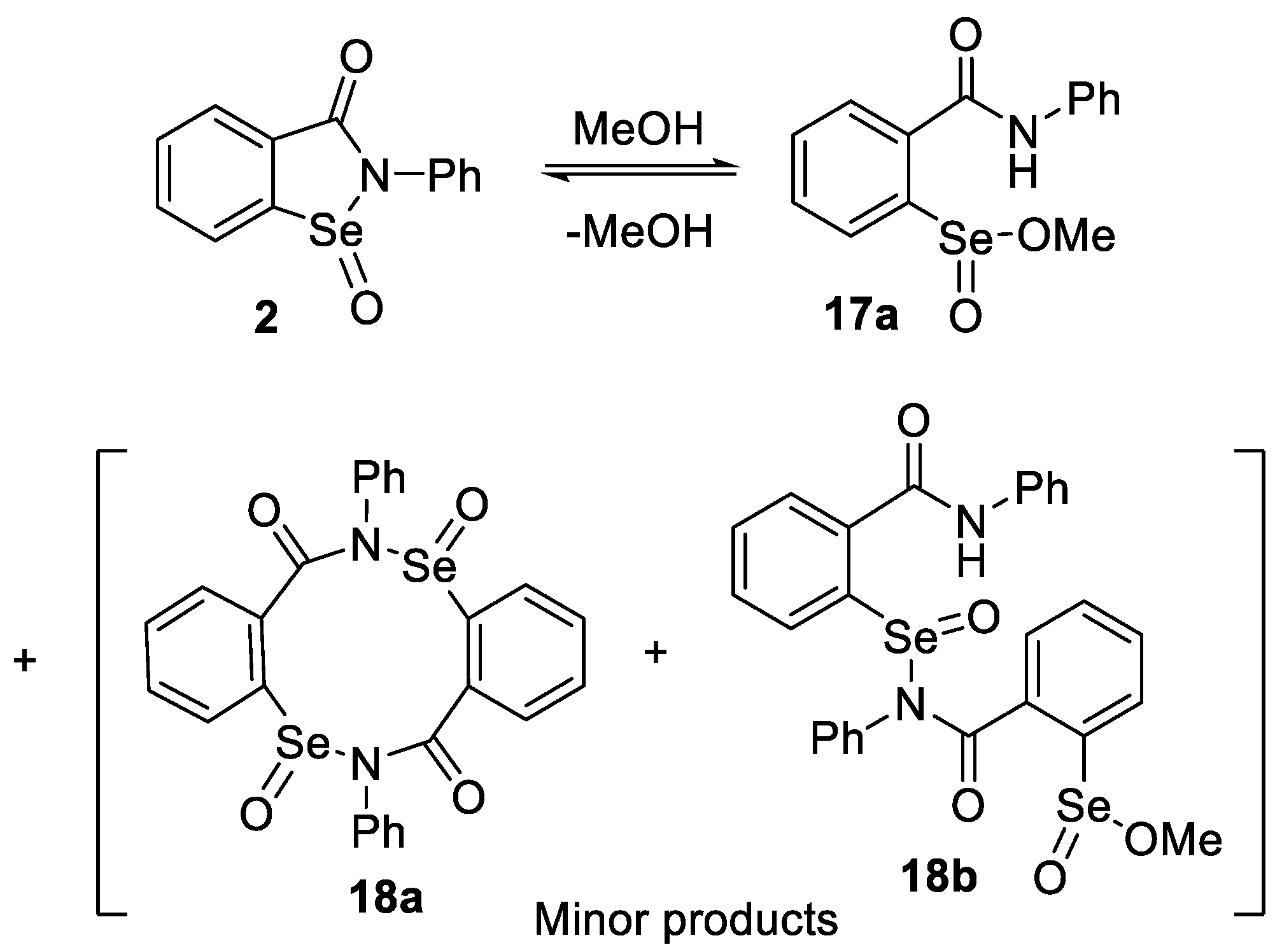
2.2. Formation of Methyl Seleninates from Seleninic Acid 3
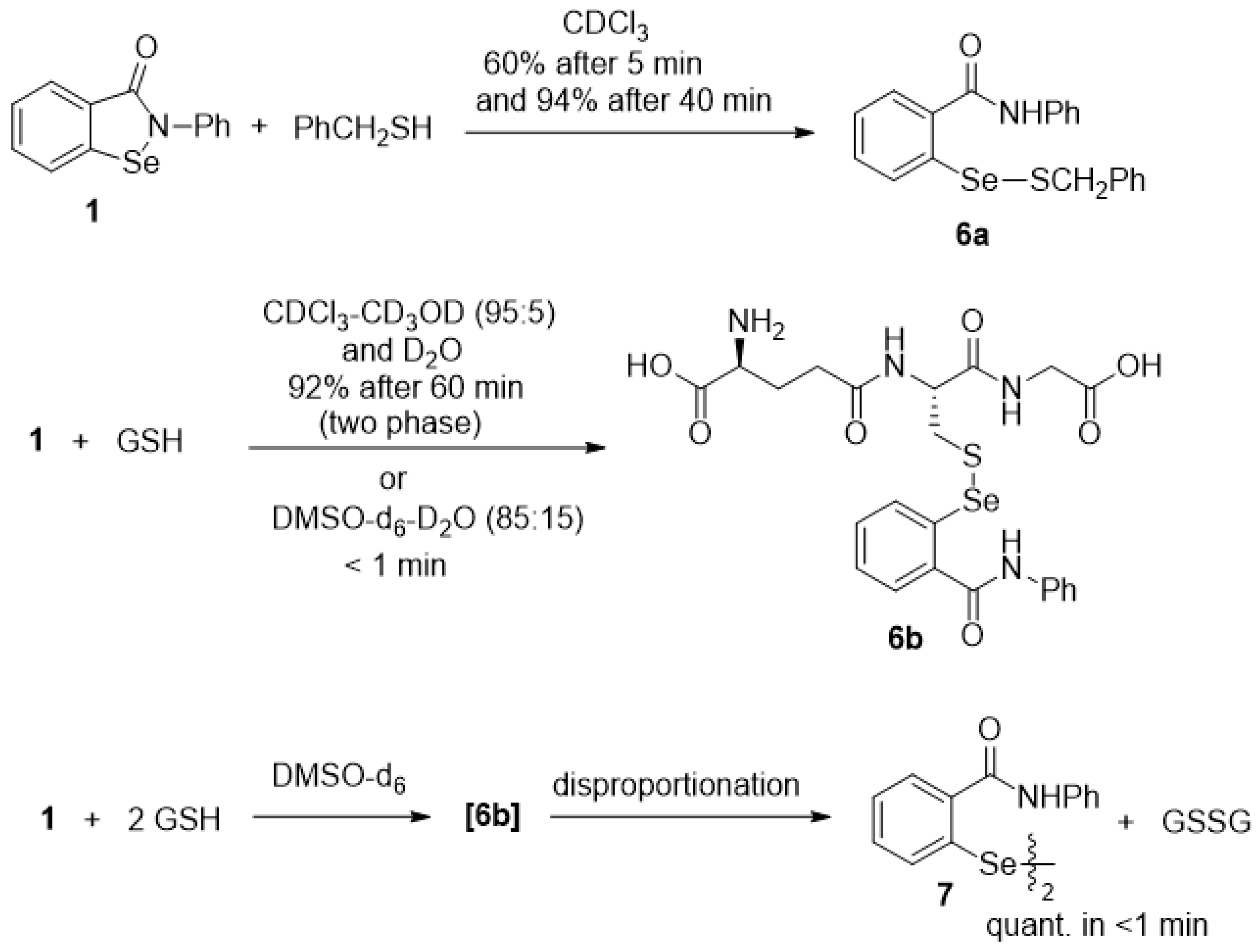
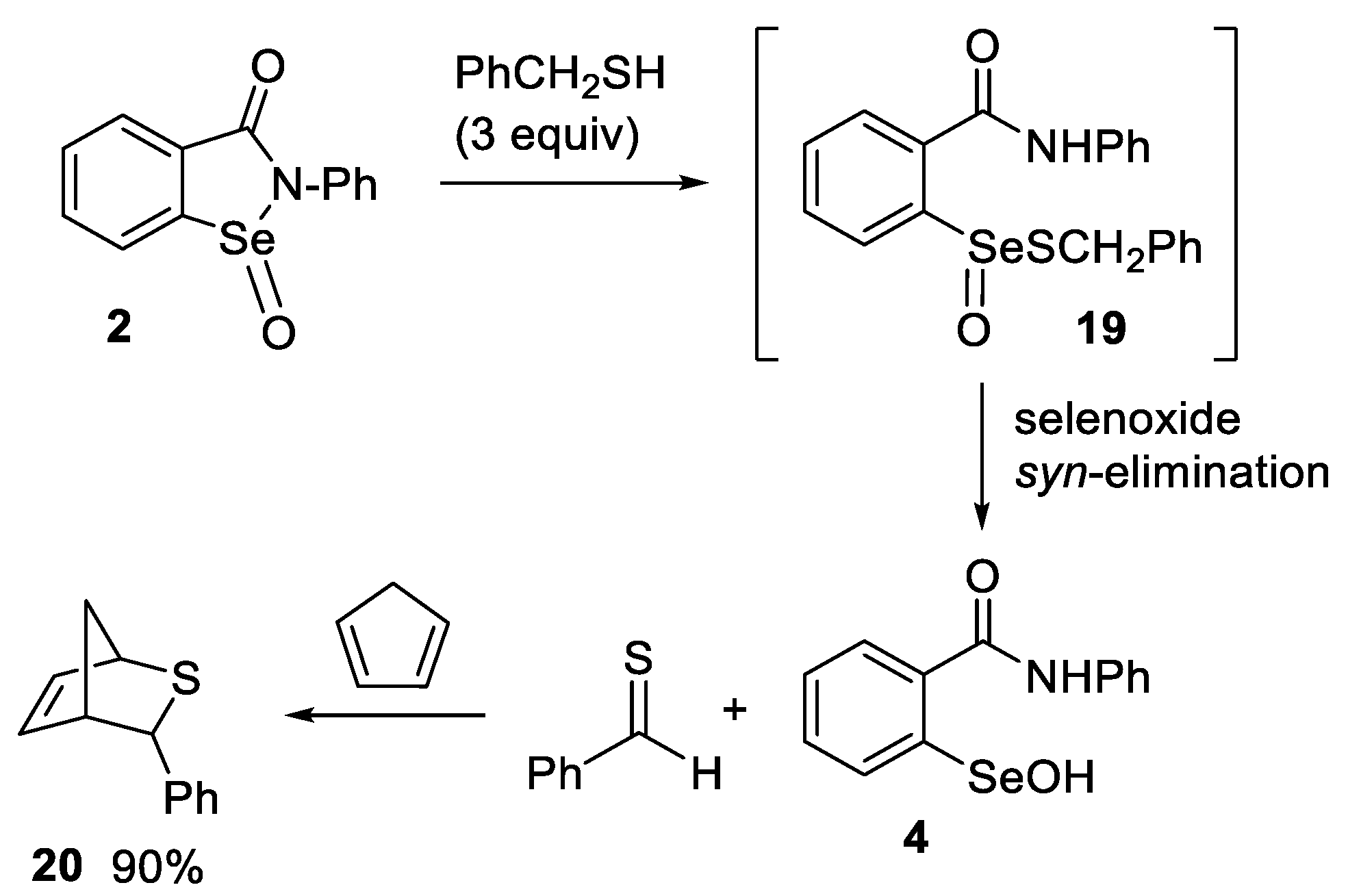
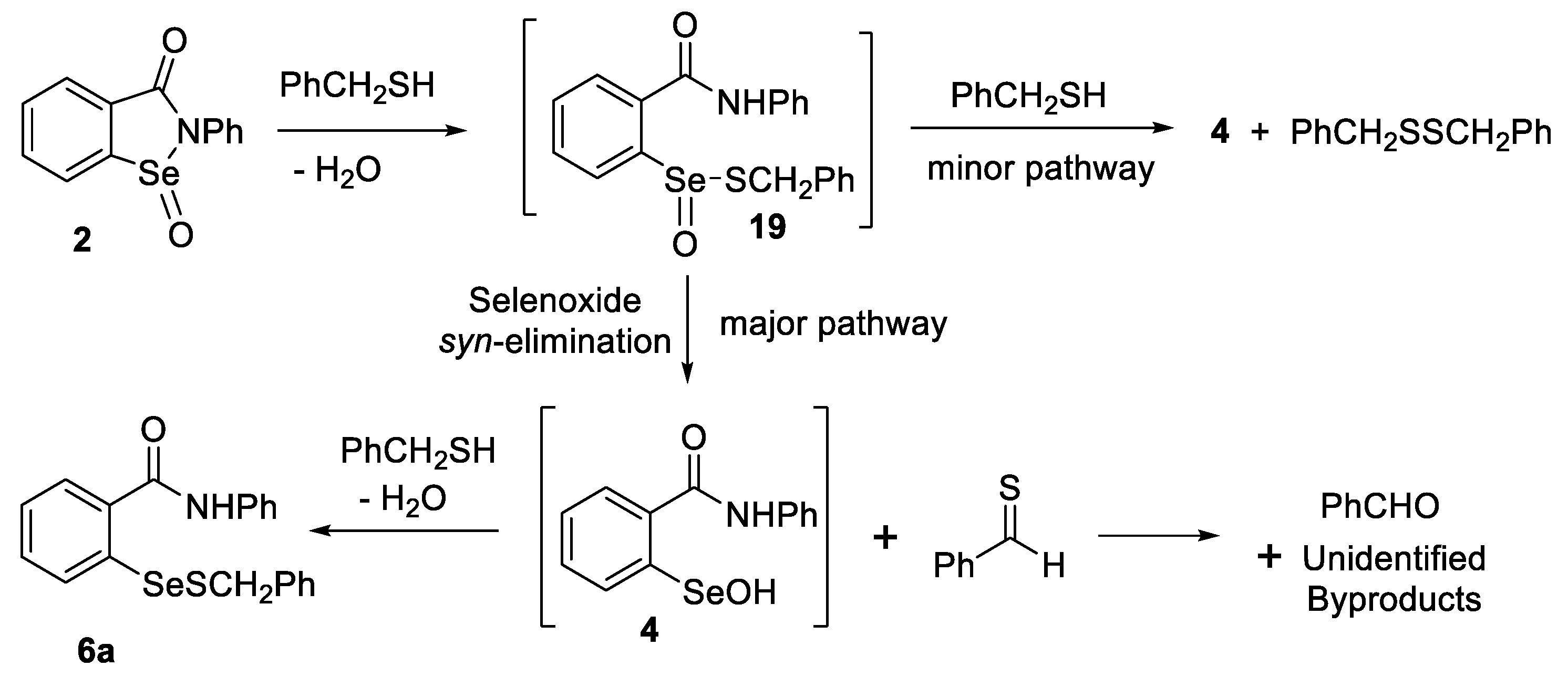
2.3. Thiolysis of Ebselen
2.4. Reduction of Seleninamide 2 with Benzyl Thiol
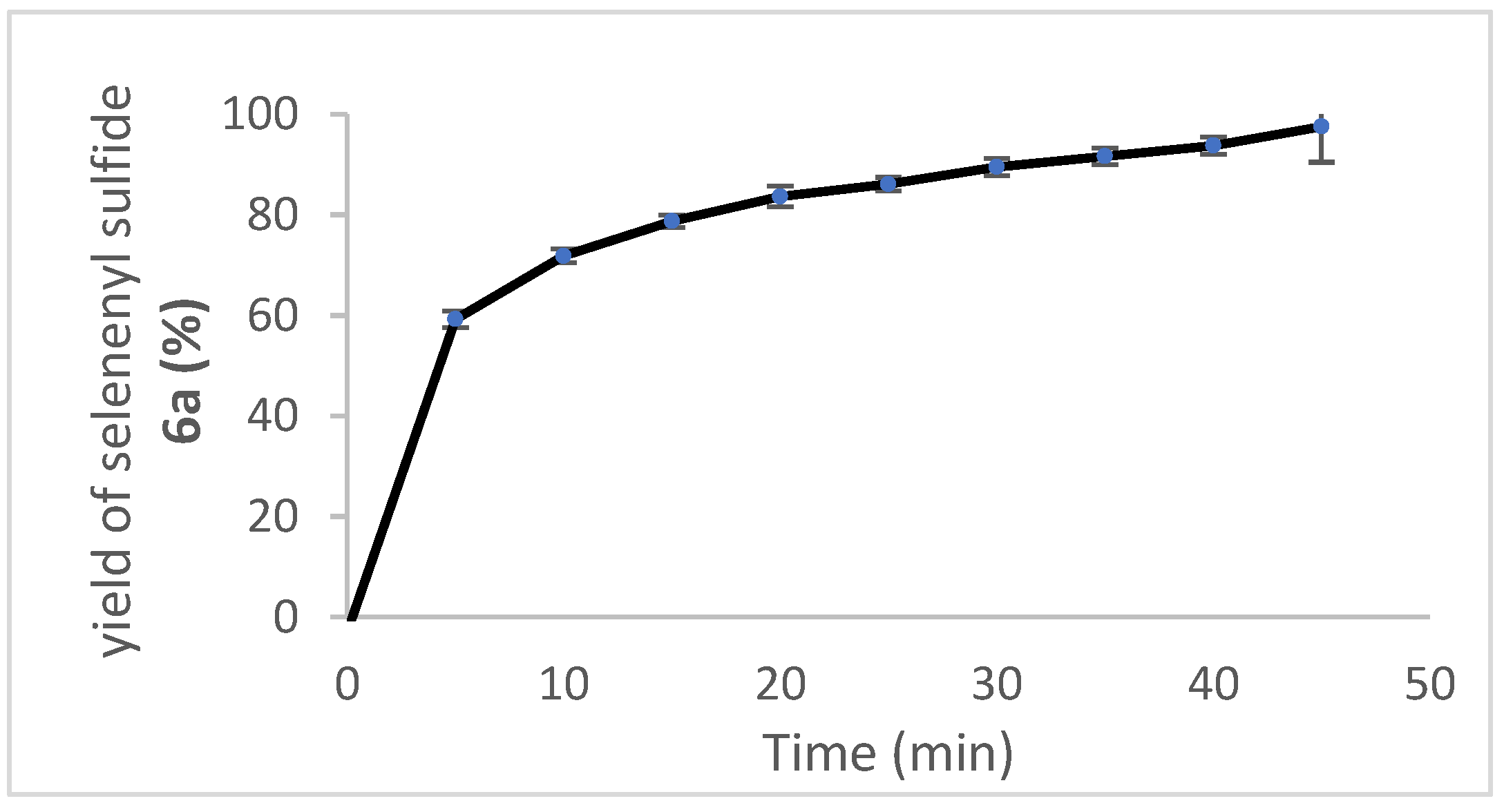
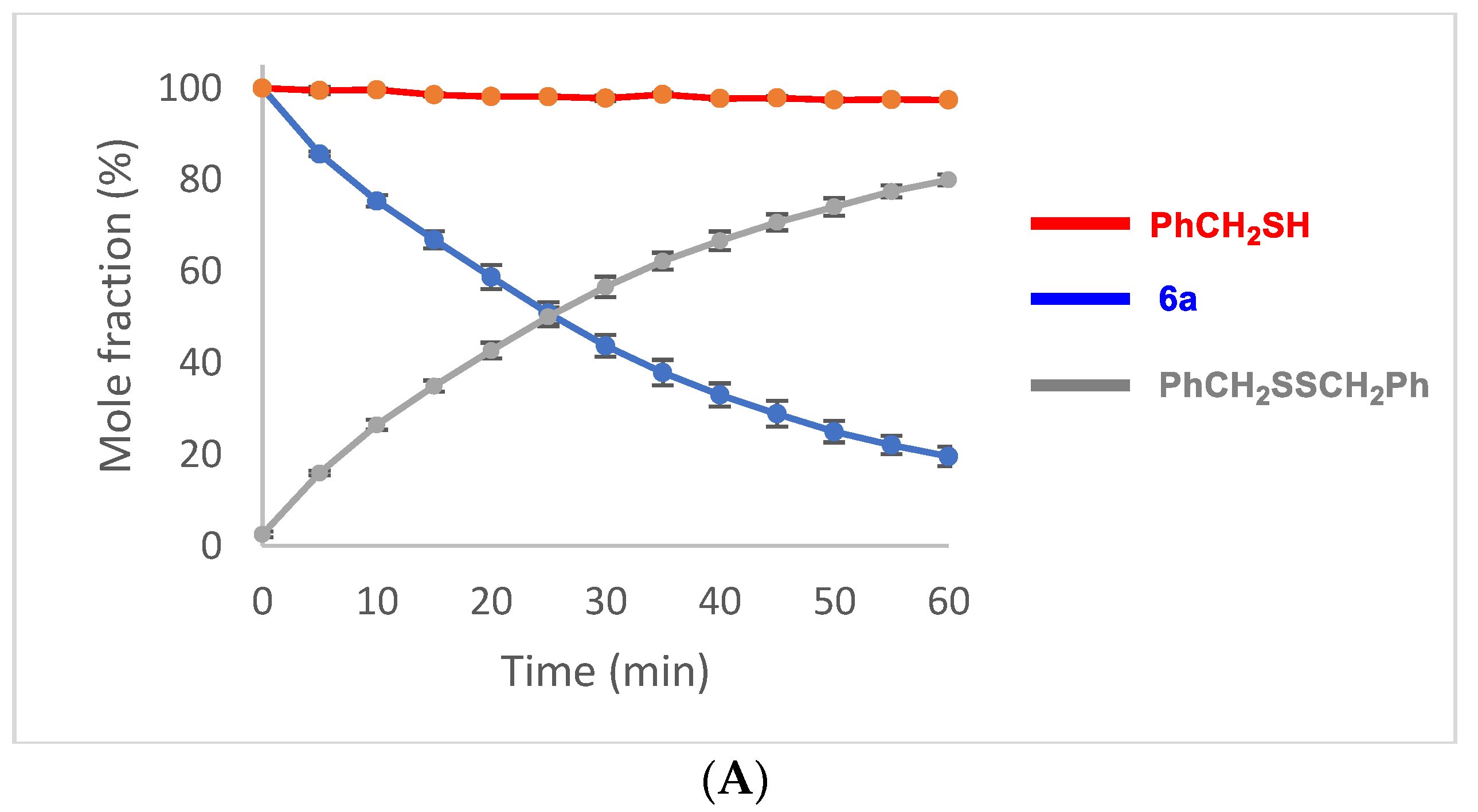
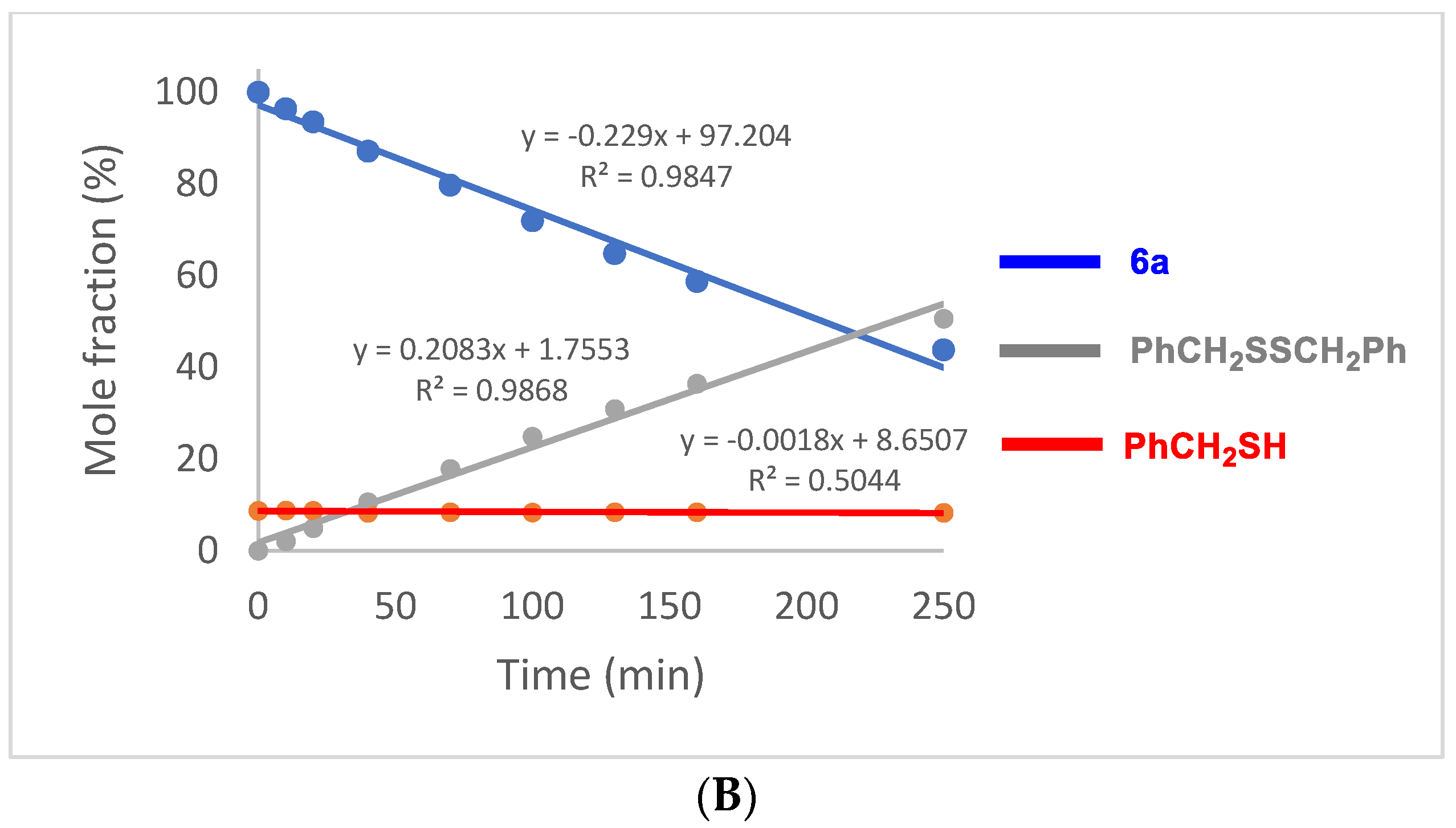
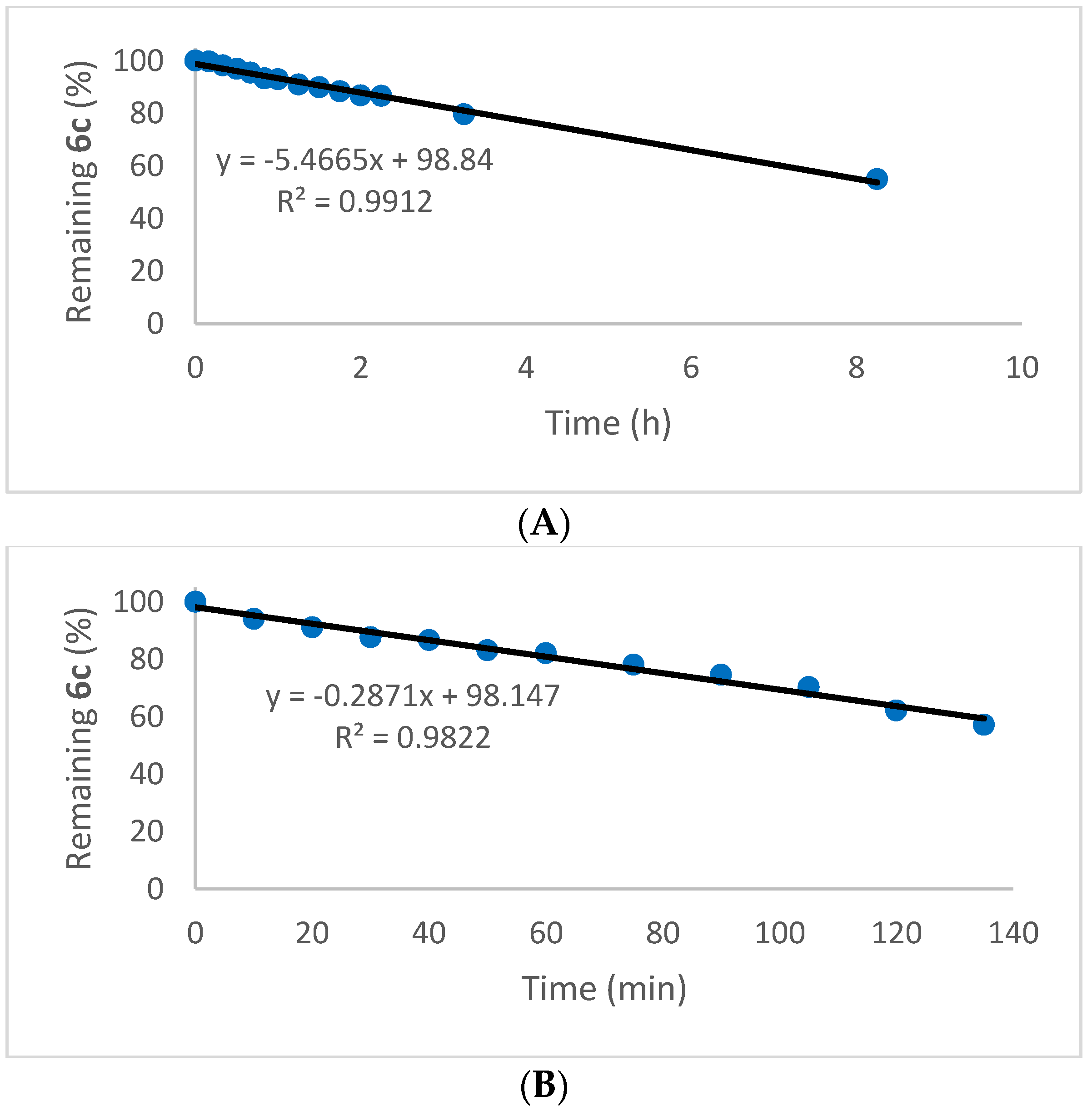
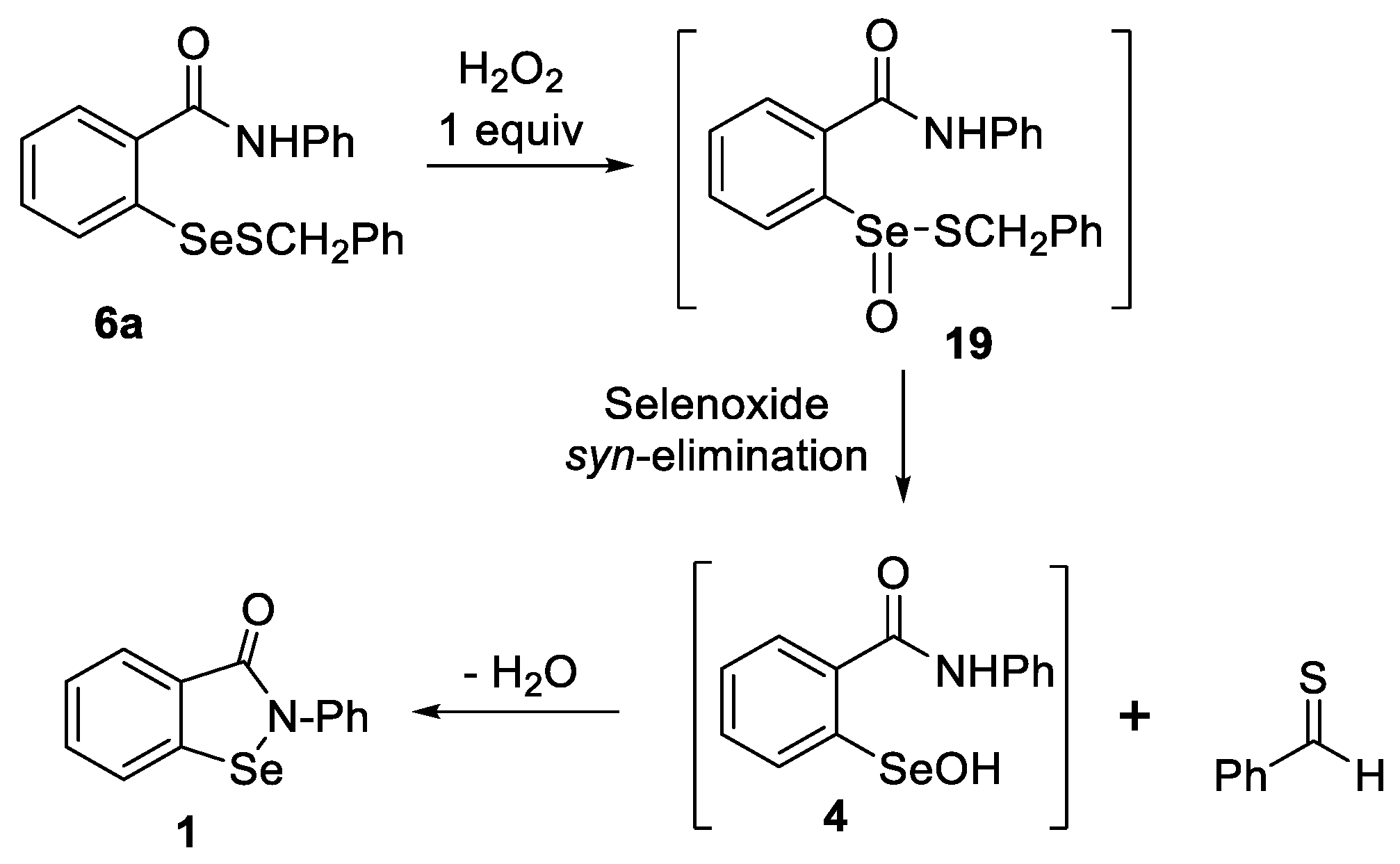
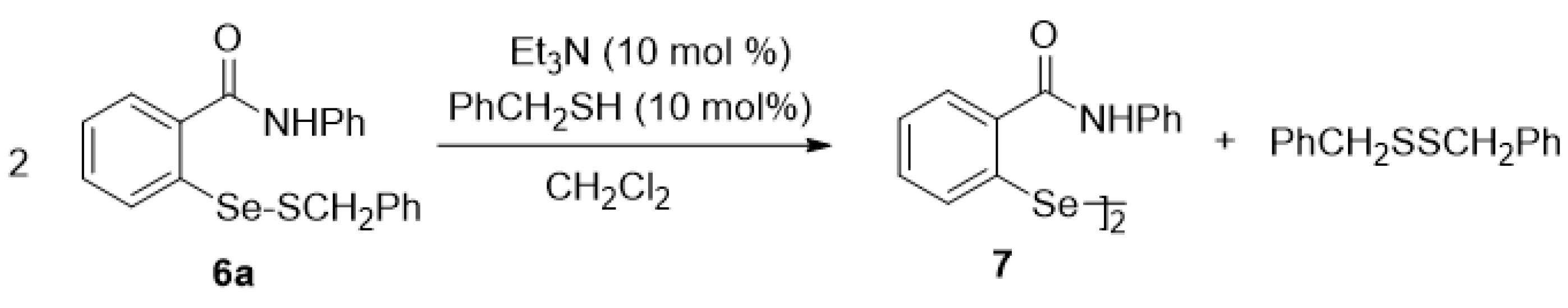
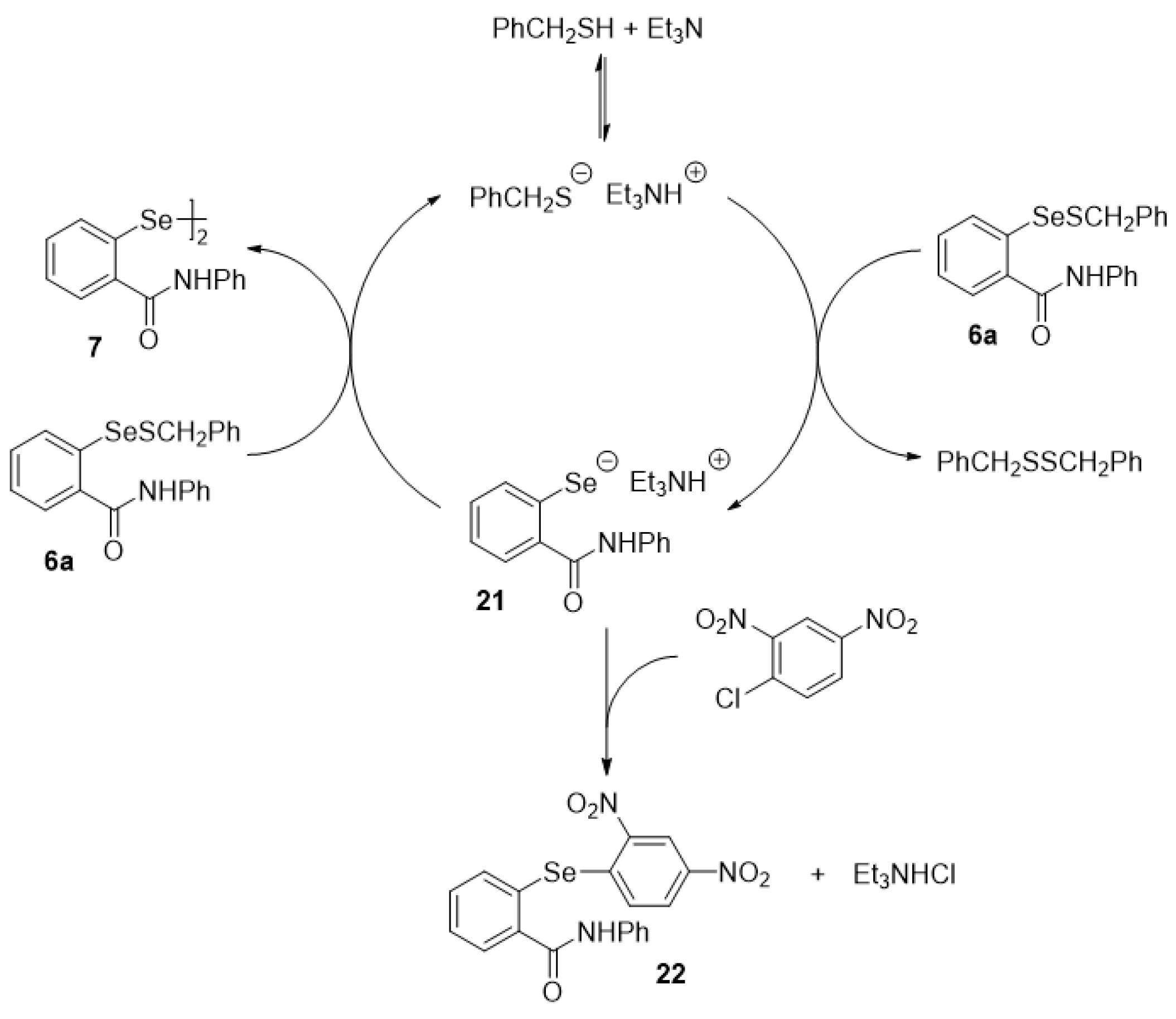
2.5. Oxidation of Selenenyl Sulfides 6a
3. Summary and Conclusions
4. Materials and Methods
4.1. General Experimental
4.2. Preparation of Ebselen (1)
4.3. Preparation of 2-Phenylbenzo[d][1,2]selenazol-3(2H)-one 1-Oxide (2)
4.4. Preparation of 2-(Phenylcarbamoyl)benzeneseleninic Acid (3)
4.5. Preparation of 2-[(Benzylthio)selanyl]-N-phenylbenzamide (6a)
4.6. Preparation of 2-[(Glutathionyl)selanyl]-N-phenylbenzamide (6b)
4.7. Preparation of 2-[(Phenylthio)selanyl]-N-phenylbenzamide (6c)
4.8. Preparation of 2,2′-Diselenobis(benzanilide) (7)
4.9. General Procedure for Kinetic Experiments
4.10. Oxidation of Ebselen with Hydrogen Peroxide (See Figure 2)
4.11. Further Oxidation of Seleninamide 2 with 0.5 Equiv of Hydrogen Peroxide
4.12. Esterification of Seleninamide 2 with Methanol: Formation of Methyl 2-(Phenylcarbamoyl)benzeneseleninate (17a) and Its Dimers 18a and 18b
4.13. Esterification of Seleninamide 2 with Benzyl Alcohol
4.14. Thiolysis of Ebselen with Benzyl Thiol (Figure 3)
4.15. Thiolysis of Ebselen with Glutathione (Figure 4)
4.16. Reaction of Seleninamide 2 with Benzyl Thiol
4.17. Oxidation of Selenenyl Sulfide 6a with Hydrogen Peroxide
4.18. Disproportionation of Selenenyl Sulfide 6a
4.19. Trapping of Selenolate 21 [65]
4.20. Photolytic Disproportionation of Selenenyl Sulfide 6a
4.21. Photolytic Disproportionation of Selenenyl Sulfide 6c
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mills, G.C. Hemoglobin catabolism. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium. Biochemical role as a component of glutathione peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Ganther, H.E. Selenoproteins. Chem. Scr. 1975, 8, 79–84. [Google Scholar]
- Ganther, H.E.; Kraus, R.J. Oxidation states of glutathione peroxidase. Methods Enzymol. 1984, 107, 593–602. [Google Scholar]
- Tappel, A.L. Selenium-glutathione peroxidase: Properties and synthesis. In Current Topics in Cellular Regulation; Academic Press: Cambridge, MA, USA, 1984; Volume 24, pp. 87–97. [Google Scholar]
- Flohé, L. The discovery of glutathione peroxidases: Milestones in understanding the biological role of selenium und sulfur. In Chalcogen Chemistry: Fundamentals and Applications; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistry: London, UK, 2023; Chapter 23; pp. 603–624. [Google Scholar]
- Flohé, L.; Günzler, W.A.; Schock, H.H. Glutathione peroxidase. Selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L. The glutathione peroxidase reaction: Molecular basis of the antioxidant function of selenium in mammals. In Current Topics in Cellular Regulation; Academic Press: Cambridge, MA, USA, 1985; Volume 27, pp. 473–478. [Google Scholar]
- Epp, O.; Ladenstein, R.; Wendel, A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983, 133, 51–69. [Google Scholar] [CrossRef]
- Stadtman, T.C. Biosynthesis and function of selenocysteine-containing enzymes. J. Biol. Chem. 1991, 266, 16257–16260. [Google Scholar] [CrossRef]
- Ren, B.; Huang, W.; Åkesson, B.; Ladenstein, R. The crystal structure of seleno-glutathione peroxidase from human plasma at 2.9 Å resolution. J. Mol. Biol. 1997, 268, 869–885. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Kipp, A.P. Physiological functions of GPx2 and its role in inflammation-triggered carcinogenesis. Ann. N. Y. Acad. Sci. 2012, 1259, 19–25. [Google Scholar] [CrossRef]
- Bhuyan, B.J.; Mugesh, G. Biological and biochemical aspects of selenium compounds. In Organoselenium Chemistry—Synthesis and Reactions; Wirth, T., Ed.; Wiley-VCH: Weinheim, Germany, 2012; Chapter 9; pp. 361–396. [Google Scholar]
- Santi, C.; Marini, F.; Lenardão, E.J. Looking beyond the traditional idea of glutathione peroxidase mimics as antioxidants. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; Royal Society of Chemistry: London, UK, 2018; Chapter 2; pp. 37–76. [Google Scholar]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Loschen, G.; Flohé, L.; Chance, B. Respiratory chain-linked hydrogen peroxide production in pigeon heart mitochondria. FEBS Lett. 1971, 18, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Synthesis of organoselenium compounds with potential biological activities. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; Royal Society of Chemistry: London, UK, 2018; Chapter 3; pp. 77–121. [Google Scholar]
- Carroll, L.D.; Davies, M.J. Reaction of selenium compounds with reactive oxygen species and the control of oxidative stress. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; Royal Society of Chemistry: London, UK, 2018; Chapter 9; pp. 254–276. [Google Scholar]
- Nasim, M.J.; Ali, W.; Domínguez-Álvarez, E.; da Silva Júnior, E.N.; Saleem, R.S.Z.; Jacob, C. Reactive Selenium Species: Redox Modulation, Antioxidant, Antimicrobial and Anticancer Activities. In Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; Royal Society of Chemistry: London, UK, 2018; Chapter 10; pp. 277–302. [Google Scholar]
- Redox Signaling and Regulation in Biology and Medicine; Jacob, C.; Winyard, P.G. (Eds.) Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Jamier, V.; Ba, L.A.; Jacob, C. Selenium- and tellurium-containing multifunctional redox agents as biochemical redox modulators with selective cytotoxicity. Chem. Eur. J. 2010, 16, 10920–10928. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef]
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef]
- Moutet, M.; d’Alessio, P.; Malette, P.; Devaux, V.; Chaudière, J. Glutathione peroxidase mimics prevent TNFα- and neutrophil-induced endothelial alterations. Free Radic. Biol. Med. 1998, 25, 270–281. [Google Scholar] [CrossRef]
- Sands, K.N.; Tuck, T.A.; Back, T.G. Cyclic Seleninate esters, spirodioxyselenuranes and related compounds—New classes of biological antioxidants that emulate glutathione peroxidase. Chem. Eur. J. 2018, 24, 9714–9728. [Google Scholar] [CrossRef]
- Wirth, T. Small organoselenium compounds: More than just glutathione peroxidase, mimics. Angew. Chem. Int. Ed. 2015, 54, 10074–10076. [Google Scholar] [CrossRef]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol. 2009, 77, 285–296. [Google Scholar] [CrossRef]
- Bhowmick, D.; Mugesh, G. Insights into the catalytic mechanism of synthetic glutathione peroxidase mimetics. Org. Biomol. Chem. 2015, 13, 10262–10272. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Mugesh, G. Functional mimics of glutathione peroxidase: Bioinspired synthetic antioxidants. Acc. Chem. Res. 2010, 43, 1408–1419. [Google Scholar] [CrossRef]
- Mugesh, G.; Singh, H.B. Synthetic organoselenium compounds as antioxidants: Glutathione peroxidase activity. Chem. Soc. Rev. 2000, 29, 347–357. [Google Scholar] [CrossRef]
- Mugesh, G.; du Mont, W.-W.; Sies, H. Chemistry of Biologically important synthetic organoselenium compounds. Chem. Rev. 2001, 101, 2125–2179. [Google Scholar] [CrossRef]
- Barbosa, N.V.; Nogueira, C.W.; Nogara, P.A.; de Bern, A.F.; Aschner, M.; Rocha, J.B.T. Organoselenium compounds as mimics of selenoproteins and thiol modifier agents. Metallomics 2017, 9, 1703–1734. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Santi, C.; Sancineto, L. New Frontiers in Organoselenium Compounds; Royal Society of Chemistry: London, UK, 2018; pp. 99–143. [Google Scholar]
- Iwaoka, M. Antioxidant organoselenium molecules. In Organoselenium Chemistry: Between Synthesis and Biochemistry; Santi, C., Ed.; Bentham Books; Bentham Science Publishers: Sharjah, United Arab Emirates, 2014; pp. 361–378. [Google Scholar]
- Sancineto, L.; Nascimento, V. Chalcogen-containing therapeutic molecules. In Chalcogen Chemistry—Fundamentals and Applications; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistry: London, UK, 2023; Chapter 22; pp. 592–602. [Google Scholar]
- Arai, K. Small organoselenium catalysts as a potential manipulator for redox homeostasis and proteostasis. In Chalcogen Chemistry—Fundamentals and Applications; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistry: London, UK, 2023; Chapter 25; pp. 648–665. [Google Scholar]
- Lesser, R.; Weiss, R. Uber selenhaltige aromatische verbindungen. Dtsch. Chem. Ges. 1924, 57, 1077–1082. [Google Scholar] [CrossRef]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound—I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (ebselen). Biochem. Pharmacol. 1984, 33, 3235–3239. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Sies, H. The early research and development of ebselen. Biochem. Pharmacol. 2013, 86, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.; Sies, H. Ebselen: Prospective therapy for cerebral ischaemia. Exp. Opin. Investig. Drugs 2000, 9, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, K.; Shah, P.; Singh, H.B.; Butcher, R.J. Synthesis, structure, and glutathione peroxidase-like activity of amino acid containing ebselen analogues and diaryl diselenides. Chem. Eur. J. 2011, 17, 12741–12755. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Glutathione peroxidase (GPx)-like antioxidant activity of the organoselenium drug ebselen: Unexpected complications with thiol exchange reactions. J. Am. Chem. Soc. 2005, 127, 11477–11485. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Antioxidant activity of the anti-inflammatory compound ebselen: A reversible cyclization pathway via selenenic and seleninic acid intermediates. Chem. Eur. J. 2008, 14, 10603–10614. [Google Scholar] [CrossRef] [PubMed]
- Nogara, P.A.; Pereira, M.E.; Oliveira, C.S.; Orian, L.; da Rocha, J.B.T. The long story of ebselen: From about one century of its synthesis to clinical trials. In Chalcogen Chemistry: Fundamentals and Applications; Lippolis, V., Santi, C., Lenardão, E.J., Braga, A.L., Eds.; Royal Society of Chemistry: London, UK, 2023; Chapter 21; pp. 567–591. [Google Scholar]
- Nascimento, V.; Silva Cordeiro, P.; Arca, M.; Marini, F.; Sancineto, L.; Braga, A.L.; Lippolis, V.; Iwaoka, M.; Santi, C. Fast and easy conversion of ortho amidoaryl diselenides into the corresponding ebselen-like derivatives driven by theoretical investigations. New J. Chem. 2020, 44, 9444–9451. [Google Scholar] [CrossRef]
- Sands, K.N.; Back, T.G. Key steps and intermediates in the catalytic mechanism for the reduction of peroxides by the antioxidant ebselen. Tetrahedron 2018, 74, 4959–4967. [Google Scholar] [CrossRef]
- Santi, C.; Scimmi, C.; Sancineto, L. Ebselen and analogues: Pharmacological properties and synthetic strategies for their preparation. Molecules 2021, 26, 4230. [Google Scholar] [CrossRef]
- Sies, H. Ebselen, a selenoorganic compound as glutathione peroxidase mimic. Free Radic. Biol. Med. 1993, 14, 313–323. [Google Scholar] [CrossRef]
- Schewe, T. Molecular actions of ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef]
- Ogawa, A.; Yoshimoto, T.; Kikuchi, H.; Sano, K.; Saito, I.; Yamaguchi, T.; Yasuhara, H. Ebselen in acute middle cerebral artery occlusion: A placebo-controlled, double-blind clinical trial. Cerebrovasc. Dis. 1999, 9, 112–118. [Google Scholar] [CrossRef]
- Kil, J.; Lobarinas, E.; Spankovich, C.; Griffiths, S.K.; Antonelli, P.J.; Lynch, E.D.; Le Prell, C.G. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2017, 390, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. Sound medicine. Nat. Med. 2012, 18, 642–645. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Halliday, A.C.; Thomas, J.M.; Kuznetsova, O.V.; Baldwin, R.; Woon, E.C.Y.; Aley, P.K.; Antoniadou, I.; Sharp, T.; Vasudevan, S.R.; et al. A safe lithium mimetic for bipolar disorder. Nat. Commun. 2013, 4, 1332. [Google Scholar] [CrossRef]
- Masaki, C.; Sharpley, A.L.; Cooper, C.M.; Godlewska, B.R.; Singh, N.; Vasudevan, S.R.; Harmer, C.J.; Churchill, G.C.; Sharp, T.; Rogers, R.D.; et al. Effects of the potential lithium-mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacology 2016, 233, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Sharpley, A.L.; Williams, C.; Holder, A.A.; Godlewska, B.R.; Singh, N.; Shanyinde, M.; MacDonald, O.; Cowen, P.J. A phase 2a randomised, double-blind, placebo-controlled, parallel-group, add-on clinical trial of ebselen (SPI-1005) as a novel treatment for mania or hypomania. Psychopharmacology 2020, 237, 3773–3782. [Google Scholar] [CrossRef] [PubMed]
- Haritha, C.V.; Sharun, K.; Jose, B. Ebselen, a new candidate therapeutic against SARS-CoV-2. Int. J. Surg. 2020, 84, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.; Fausel, M.; Safayhi, H.; Tiegs, G.; Otter, R. A novel biologically active seleno-organic compound—II. Activity of PZ 51 in relation to glutathione peroxidase. Biochem. Pharmacol. 1984, 33, 3241–3245. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Dereu, N. Mechanism of the catalytic reduction of hydroperoxides by ebselen: A selenium-77 NMR study. Bull. Soc. Chim. Belg. 1987, 96, 757–768. [Google Scholar] [CrossRef]
- Maiorino, M.; Roveri, A.; Coassin, M.; Ursini, F. Kinetic mechanism and substrate specificity of glutathione peroxidase activity of ebselen (PZ51). Biochem. Pharmacol. 1988, 37, 2267–2271. [Google Scholar] [CrossRef]
- Haenen, G.R.M.M.; De Rooij, B.M.; Vermeulen, N.P.E.; Bast, A. Mechanism of the reaction of ebselen with endogenous thiols: Dihydrolipoate is a better cofactor than glutathione in the peroxidase activity of ebselen. Mol. Pharmacol. 1990, 37, 412–422. [Google Scholar]
- Cotgreave, I.A.; Morgenstern, R.; Engman, L.; Ahokas, J. Characterization and quantitation of a selenol intermediate in the reaction of ebselen with thiols. Chem.-Biol. Interact. 1992, 84, 69–76. [Google Scholar] [CrossRef]
- Morgenstern, R.; Cotgreave, I.A.; Engman, L. Determination of the relative contributions of the diselenide and selenol forms of ebselen in the mechanism of its glutathione peroxidase-like activity. Chem. Biol. Interact. 1992, 84, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Mugesh, G. Glutathione peroxidase activity of ebselen and its analogues: Some insights into the complex chemical mechanisms underlying the antioxidant activity. Curr. Chem. Biol. 2013, 7, 47–56. [Google Scholar] [CrossRef]
- Sarma, B.K.; Mugesh, G. Thiol cofactors for selenoenzymes and their synthetic mimics. Org. Biomol. Chem. 2008, 6, 965–974. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Mugesh, G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chem. Eur. J. 2007, 13, 4594–4601. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Bhowmick, D.; Mugesh, G. Synthetic glutathione peroxidase mimics: Effect of nucleophilicity of the aryl cofactor on the antioxidant activity. Indian J. Chem. 2013, 52, 1019–1025. [Google Scholar]
- Back, T.G. Oxidations with selenium reagents. In Organoselenium Chemistry—A Practical Approach; Back, T.G., Ed.; Oxford University Press: Oxford, UK, 1999; Chapter 5; pp. 93–112. [Google Scholar]
- Drabowicz, J.; Midura, W.H.; Krasowska, D. Selenium and tellurium (1,2,3)-oxygen-containing acids and derivatives. In The Chemistry of Organoselenium and Organotellurium Compounds; Part, 2; Rappoport, Z., Ed.; Wiley: Chichester, UK, 2012; Volume 3, Chapter 17; pp. 1027–1082. [Google Scholar]
- Młochowski, J.; Brzaszcz, M.; Giurg, M.; Palus, J.; Wójtowicz, H. Selenium-promoted oxidation of organic compounds: Reactions and mechanisms. Eur. J. Org. Chem. 2003, 4329–4339. [Google Scholar] [CrossRef]
- Freudendahl, D.M.; Santoro, S.; Shahzad, S.A.; Santi, C.; Wirth, T. Green chemistry with selenium reagents: Development of efficient catalytic reactions. Angew. Chem. Int. Ed. 2009, 48, 8409–8411. [Google Scholar] [CrossRef]
- Młochowski, J.; Wójtowicz-Młochowska, H. Developments in synthetic application of selenium(IV) oxide and organoselenium compounds as oxygen donors and oxygen-transfer agents. Molecules 2015, 20, 10205–10243. [Google Scholar] [CrossRef]
- Back, T.G. Oxidations catalyzed by seleninic acids and anhydrides, their precursors and congeners. Curr. Green Chem. 2016, 3, 76–91. [Google Scholar] [CrossRef]
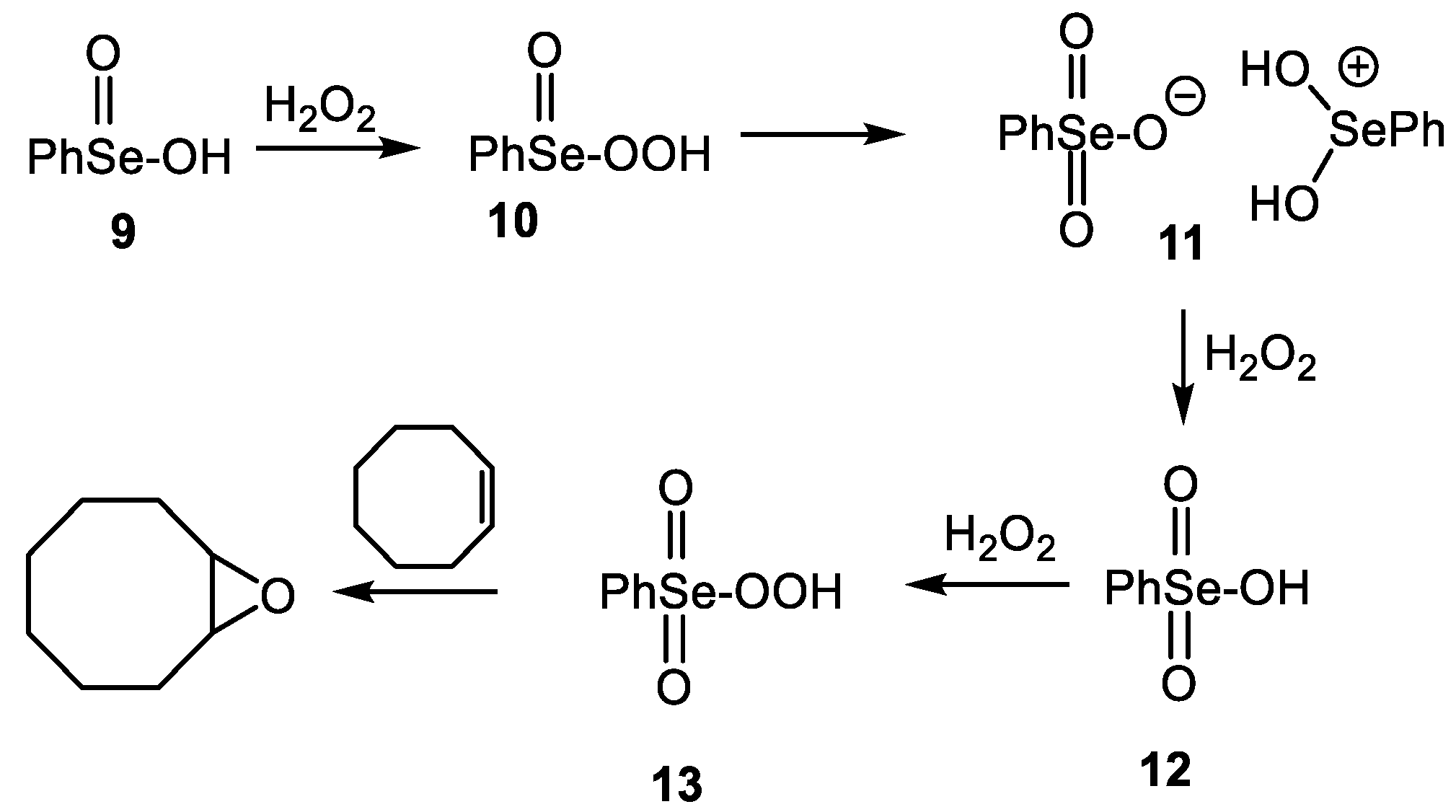
- Sands, K.N.; Mendoza Rengifo, E.; George, G.N.; Pickering, I.J.; Gelfand, B.S.; Back, T.G. The Unexpected role of SeVI species in epoxidations with benzeneseleninic acid and hydrogen peroxide. Angew. Chem. Int. Ed. 2020, 59, 4283–4287. [Google Scholar] [CrossRef]
- Grieco, P.A.; Yokoyama, Y.; Gilman, S.; Nishizawa, M. Organoselenium chemistry. Epoxidation of olefins with benzeneseleninic acid and hydrogen peroxide (“benzeneperoxyseleninic acid”). J. Org. Chem. 1977, 42, 2034–2036. [Google Scholar] [CrossRef]
- Hori, T.; Sharpless, K.B. Synthetic applications of arylselenenic and arylseleninic acids. Conversion of olefins to allylic alcohols and epoxides. J. Org. Chem. 1978, 43, 1689–1697. [Google Scholar] [CrossRef]
- Reich, H.J.; Chow, F.; Peake, S.L. Seleninic acids as catalysts for oxidations of olefins and sulfides using hydrogen peroxide. Synthesis 1978, 299–301. [Google Scholar] [CrossRef]
- Kametani, T.; Nemoto, H.; Fukumoto, K. A new method for selective epoxidation and a biogenetic-type synthesis of linalyloxides. Bioorg. Chem. 1978, 7, 215–220. [Google Scholar] [CrossRef]
- Betzemeier, B.; Lhermitte, F.; Knochel, P. A selenium-catalyzed epoxidation in perfluorinated solvents with hydrogen peroxide. Synlett 1999, 1999, 489–491. [Google Scholar] [CrossRef]
- ten Brink, G.-J.; Fernandes, B.C.M.; van Vliet, M.C.A.; Arends, I.W.C.E.; Sheldon, R.A. Selenium catalyzed oxidations with aqueous hydrogen peroxide. Part I. Epoxidation reactions in homogeneous solution. J. Chem. Soc. Perkin Trans. 1 2001, 224–228. [Google Scholar] [CrossRef]
- Paetzold, R.; Lienig, D. Benzeneselenonic acid. Z. Chem. 1964, 4, 186. [Google Scholar] [CrossRef]
- Klayman, D.L. Selenonic acids. In Organic Selenium Compounds: Their Chemistry and Biology; Klayman, D.L., Gunther, W.H.H., Eds.; J. Wiley and Sons: New York, NY, USA, 1973; pp. 141–144. [Google Scholar]
- Kamigata, N.; Takata, M.; Matsuyama, H.; Kobayashi, M. Oxidation of thiols and sulfides by 2-aryl-1,2-benzisoselenazol-3(2H)-one 1-oxide. Sulfur Lett. 1986, 5, 1–7. [Google Scholar]
- Satheeshkumar, K.; Mugesh, G. Synthesis and antioxidant activity of peptide-based ebselen analogues. Chem.-Eur. J. 2011, 17, 4849–4857. [Google Scholar] [CrossRef] [PubMed]
- Haas, A.; Weiler, H.U. (Trifluoromethyl)selenium(VI) compounds, synthesis and properties of tetrafluoro(trifluoromethyl)selenium halides and trifluoromethaneselenonates. Chem. Ber. 1985, 118, 943–951. [Google Scholar] [CrossRef]
- Haas, A.; Schinkel, K. Synthesis and chemical properties of pentafluorobenzeneselenonic acid and derivatives of trifluoromethaneselenonic acid. Chem. Ber. 1990, 123, 685–689. [Google Scholar] [CrossRef]
- Boese, R.; Haas, A.; Herkt, S.; Pryka, M. Perfluoroorganochalcogenic acids in higher oxidation states. Chem. Ber. 1995, 128, 423–428. [Google Scholar] [CrossRef]
- Abdo, M.; Knapp, S. Biomimetic seleninates and selenonates. J. Am. Chem. Soc. 2008, 130, 9234–9235. [Google Scholar] [CrossRef]
- Satheeshkumar, K.; Raju, S.; Singh, H.B.; Butcher, R.J. Reactivity of selenocystine and tellurocystine: Structure and antioxidant activity of the derivatives. Chem. Eur. J. 2018, 24, 17513–17522. [Google Scholar] [CrossRef] [PubMed]
- Sands, K.N.; Gelfand, B.S.; Back, T.G. One-pot synthesis of aryl selenonic acids and some unexpected byproducts. J. Org. Chem. 2021, 86, 9938–9944. [Google Scholar] [CrossRef] [PubMed]
- Syper, L.; Młochowski, J. Benzeneperoxyseleninic acids-synthesis and properties. Tetrahedron 1987, 43, 207–213. [Google Scholar]
- Antony, S.; Bayse, C.A. Modeling the mechanism of the glutathione peroxidase mimic ebselen. Inorg. Chem. 2011, 50, 12075–12084. [Google Scholar] [CrossRef]
- McCullough, J.D.; Gould, E.S. The dissociation constants of some monosubstituted benzeneselenic acids. J. Am. Chem. Soc. 1949, 71, 674–676. [Google Scholar] [CrossRef]
- Ayrey, G.; Barnard, D.; Woodbridge, D.T. Oxidation of organoselenium compounds by ozone. J. Chem. Soc. 1962, 2089–2099. [Google Scholar] [CrossRef]
- Piotto, M.; Bourdonneau, M.; Elbayed, K.; Wieruszeski, J.-M.; Lippens, G. New DEFT sequences for the acquisition of one-dimensional carbon NMR spectra of small unlabelled molecules. Magn. Reson. Chem. 2006, 44, 943–947. [Google Scholar] [CrossRef]
- Kice, J.L.; Lee, T.W.S. Oxidation-reduction reactions of organoselenium compounds. 1. Mechanism of the reaction between seleninic acids and thiols. J. Am. Chem. Soc. 1978, 100, 5094–5102. [Google Scholar] [CrossRef]
- Kice, J.L.; Purkiss, D.W. The induced decomposition of S-tert-butyl benzenethioseleninate. J. Org. Chem. 1987, 52, 3448–3451. [Google Scholar] [CrossRef]
- Glass, R.S.; Farooqui, F.; Sabahi, M.; Ehler, K.W. Formation of thiocarbonyl compounds in the reaction of ebselen oxide with thiols. J. Org. Chem. 1989, 54, 1092–1097. [Google Scholar] [CrossRef]
- Reich, H.J.; Jasperse, C.P. Organoselenium chemistry. Redox chemistry of selenocysteine model systems. J. Am. Chem. Soc. 1987, 109, 5549–5551. [Google Scholar] [CrossRef]
- Pearson, J.K.; Boyd, R.J. Density functional theory study of the reaction mechanism and energetics of the reduction of hydrogen peroxide by ebselen, ebselen diselenide, and ebselen selenol. J. Phys. Chem. A 2007, 111, 3152–3160. [Google Scholar] [CrossRef]
- Pearson, J.K.; Boyd, R.J. Modeling the reduction of hydrogen peroxide by glutathione peroxidase mimics. J. Phys. Chem. A 2006, 110, 8979–8985. [Google Scholar] [CrossRef]
- Singh, R.; Whitesides, G.M. Selenols catalyze the interchange reactions of dithiols and disulfides in water. J. Org. Chem. 1991, 56, 6931–6933. [Google Scholar] [CrossRef]
- Rabenstein, D.L.; Scott, T.M.; Guo, W. Nuclear magnetic resonance study of the kinetics of the penicillamine/bis(penicillamine) selenide symmetrical exchange reaction. J. Org. Chem. 1991, 56, 4176–4181. [Google Scholar] [CrossRef]
- Rasmussen, B.; Sorensen, A.; Gotfredsen, H.; Pittelkow, M. Dynamic combinatorial chemistry with diselenides and disulfides in water. Chem. Commun. 2014, 50, 3716–3718. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, D.; Nauser, T.; Koppenol, W.H. Selenium and sulfur in exchange reactions: A comparative study. J. Org. Chem. 2010, 75, 6696–6699. [Google Scholar] [CrossRef] [PubMed]
- Back, T.G.; Krishna, M.V. Free-radical additions of diselenides to dimethyl acetylenedicarboxylate, methyl propiolate, and dimethyl maleate. J. Org. Chem. 1988, 53, 2533–2536. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, H.; Yokoyama, K.; Masawaki, T.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl diselenide to acetylenes. J. Org. Chem. 1991, 56, 5721–5723. [Google Scholar] [CrossRef]
- Reichert, J.S.; McNeight, S.A.; Rudel, H.W. Determination of hydrogen peroxide and some related peroxygen compounds. Ind. Eng. Chem. Anal. Ed. 1939, 11, 194–197. [Google Scholar] [CrossRef]
- Burchat, A.F.; Chong, J.M.; Nielsen, N. Titration of alkyllithiums with a simple reagent to a blue endpoint. J. Organomet. Chem. 1997, 542, 281–283. [Google Scholar] [CrossRef]
- Duddeck, H. Selenium-77 nuclear magnetic resonance spectroscopy. Prog. Nucl. Magn. Reson. Spectrosc. 1995, 27, 1–323. [Google Scholar] [CrossRef]
- Engman, L.; Hallberg, A. Expedient synthesis of ebselen and related compounds. J. Org. Chem. 1989, 54, 2964–2966. [Google Scholar] [CrossRef]
- Bhabak, K.P.; Vernekar, A.A.; Jakka, S.R.; Roy, G.; Mugesh, G. Mechanistic investigations on the efficient catalytic decomposition of peroxynitrite by ebselen analogues. Org. Biomol. Chem. 2011, 9, 5193–5200. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sands, K.N.; Burman, A.L.; Ansah-Asamoah, E.; Back, T.G. Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen. Molecules 2023, 28, 3732. https://doi.org/10.3390/molecules28093732
Sands KN, Burman AL, Ansah-Asamoah E, Back TG. Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen. Molecules. 2023; 28(9):3732. https://doi.org/10.3390/molecules28093732
Chicago/Turabian StyleSands, Kai N., Austin L. Burman, Esther Ansah-Asamoah, and Thomas G. Back. 2023. "Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen" Molecules 28, no. 9: 3732. https://doi.org/10.3390/molecules28093732
APA StyleSands, K. N., Burman, A. L., Ansah-Asamoah, E., & Back, T. G. (2023). Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen. Molecules, 28(9), 3732. https://doi.org/10.3390/molecules28093732