



Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study

 ,
,  , , ,
, , ,  , ,
, ,  and
and
Abstract
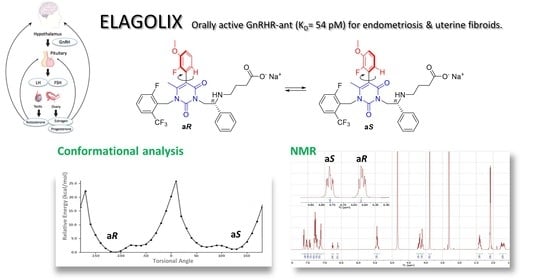
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. NMR Spectroscopy
2.3. HPLC Analyses
2.4. Structure Description
2.5. Conformational Analysis
3. Materials and Methods
3.1. General
3.2. Synthesis of 1 from 3
3.2.1. 1-(2-fluoro-6-(trifluoromethyl)benzyl) urea (4)
3.2.2. 1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (5)
3.2.3. 5-bromo-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (6)
3.2.4. 5-(2-fluoro-3-methoxyphenyl)-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine-2,4(1H,3H)-dione (7)
3.2.5. (R)-2-[(tert-butoxy-carbonyl)amino]-2-phenylethyl methane-sulfonate (10)
3.2.6. (R)-3-(amino(phenyl)methyl)-5-(2-fluoro-3-methoxyphenyl)-1-(2-fluoro-6-(trifluoromethyl)benzyl)-6-methylpyrimidine 2,4(1H,3H)-dione (8)
3.2.7. Ethyl (R)-4-(((5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-(trifluoromethyl)benzyl)-4-methyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl)(phenyl)methyl)amino)butanoate (9)
3.2.8. Elagolix Sodium Salt (1)
3.3. NMR Spectroscopy
3.4. HPLC Analyses
- −
- Chiral HPLC analysis: a Merck-Hitachi (Hitachi Ltd., Tokyo, Japan), equipped with a UV detector model L-4250, pump system model L-6200 and a chromato-integrator model D-2500. The column employed in the analyses was a Phenomenex Lux-Cellulose 1 (Phenomenex, Torrance, CA, USA). The dimension of the column is 250 mm × 4.6 mm, 3 µm. The elution was in isocratic mode with the indicated eluant and flow. All the samples were measured at λ = 254 nm and 25 °C.
- −
- RP-HPLC analysis: Agilent 1100 system (Agilent Technologies, Waldbronn, Germany) equipped with a Zorbax SB-C18 column (150 mm × 3.0 mm, 3.5 µm) for 1 and with a Supelco Discovery C18 (250 mm × 4.6 mm, 5.0 µm) for 7.
3.5. X-ray Analysis
3.6. Conformational Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lamb, Y.N. Elagolix: First Global Approval. Drugs 2018, 78, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Sara, A.R.; Al Hendy, A. Elagolix in the treatment of heavy menstrual bleeding associated with uterine fibroids in premenopausal women. Expert Rev. Clin. Pharmacol. 2021, 14, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Betz, S.F.; Zhu, Y.-F.; Chen, C.; Struthers, R.S. Non-peptide gonadotropin-releasing hormone receptor antagonists. J. Med. Chem. 2008, 51, 3331–3348. [Google Scholar] [CrossRef] [PubMed]
- Tukun, F.-L.; Olberg, D.E.; Riss, P.J.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent Development of Non-Peptide GnRH Antagonists. Molecules 2017, 22, 2188. [Google Scholar] [CrossRef]
- Tucci, F.C.; Hu, T.; Mesleh, M.F.; Bokser, A.; Allsopp, E.; Gross, T.D.; Guo, Z.; Zhu, Y.-F.; Struthers, R.S.; Ling, N.; et al. Atropisomeric property of 1-(2,6-difluorobenzyl)-3-(2R)-amino-2-phenethyl-5-(2-fluoro-3-methoxyphenyl)-6-methyluracil. Chirality 2005, 17, 559–564. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Edwards, P.J.; Fader, L.D.; Jakalian, A.; Hucke, O. Revealing atropisomer axial chirality in drug discovery. ChemMedChem 2011, 6, 505–513. [Google Scholar] [CrossRef]
- Basilaia, M.; Chen, M.H.; Secka, J.; Gustafson, J.L. Atropisomerism in the Pharmaceutically Relevant Realm. Acc. Chem. Res. 2022, 55, 2904–2919. [Google Scholar] [CrossRef]
- Toenjes, S.T.; Gustafson, J.L. Atropisomerism in medicinal chemistry: Challenges and opportunities. Future Med. Chem. 2018, 10, 409–422. [Google Scholar] [CrossRef]
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The challenge of atropisomerism in drug discovery. Angew. Chem. Int. Ed Engl. 2009, 48, 6398–6401. [Google Scholar] [CrossRef]
- Wang, J.; Zeng, W.; Li, S.; Shen, L.; Gu, Z.; Zhang, Y.; Li, J.; Chen, S.; Jia, X. Discovery and Assessment of Atropisomers of (±)-Lesinurad. ACS Med. Chem. Lett. 2017, 8, 299–303. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Huai, Q.; Cai, J.; Zoraghi, R.; Francis, S.H.; Corbin, J.D.; Robinson, H.; Xin, Z.; Lin, G.; et al. Multiple conformations of phosphodiesterase-5: Implications for enzyme function and drug development. J. Biol. Chem. 2006, 281, 21469–21479. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Huang, C.Q.; Gross, T.D.; Pontillo, J.; Rowbottom, M.W.; Saunders, J.; Struthers, S.; Tucci, F.C.; Xie, Q.; et al. Uracils as potent antagonists of the human gonadotropin-releasing hormone receptor without atropisomers. Bioorg. Med. Chem. Lett. 2005, 15, 2519–2522. [Google Scholar] [CrossRef]
- Wang, Z.; Meng, L.; Liu, X.; Zhang, L.; Yu, Z.; Wu, G. Recent progress toward developing axial chirality bioactive compounds. Eur. J. Med. Chem. 2022, 243, 114700. [Google Scholar] [CrossRef]
- Zhao, L.; Guo, Z.; Chen, Y.; Hu, T.; Wu, D.; Zhu, Y.-F.; Rowbottom, M.; Gross, T.D.; Tucci, F.C.; Struthers, R.S.; et al. 5-Aryluracils as potent GnRH antagonists-Characterization of atropisomers. Bioorg. Med. Chem. Lett. 2008, 18, 3344–3349. [Google Scholar] [CrossRef]
- Yan, W.; Cheng, L.; Wang, W.; Wu, C.; Yang, X.; Du, X.; Ma, L.; Qi, S.; Wei, Y.; Lu, Z.; et al. Structure of the human gonadotropin-releasing hormone receptor GnRH1R reveals an unusual ligand binding mode. Nat. Commun. 2020, 11, 5287. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Wu, D.; Chen, C.; Wade, W.; Dwight, W.J.; Huang, C.Q.; Tucci, F.C. Preparation of Pyrimidine-2,4(1H,3H)-dione Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. WO Patent WO2005007165, 27 January 2005. [Google Scholar]
- Chen, C.; Wu, D.; Guo, Z.; Xie, Q.; Reinhart, G.J.; Madan, A.; Wen, J.; Chen, T.; Huang, C.Q.; Chen, M.; et al. Discovery of sodium R-(+)-4-{2-5-(2-fluoro-3-methoxyphenyl)-3-(2-fluoro-6-trifluoromethylbenzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl-1-phenylethylamino}butyrate (elagolix), a potent and orally available nonpeptide antagonist of the human gonadotropin-releasing hormone receptor. J. Med. Chem. 2008, 51, 7478–7485. [Google Scholar] [CrossRef]
- Peddireddy, S.R.; Allam, S.K.; Kottur, M.K.; Oruganti, S.; Kandagatla, B. Process for the Preparation of Elagolix Sodium and Its Polymorph. WO Patent WO2017221144, 18 December 2017. [Google Scholar]
- Vlasakova, R.; Cerna, I.; Obadalova, I.; Krejcik, L.; Dammer, O.; Svobodova, J.; Sembera, F. Solid Forms of Elagolix. Wo Patent WO2018224063, 13 December 2018. [Google Scholar]
- Sulake, R.S.; Shinde, S.R.; Siyan, R.S.; Bhise, N.B.; Singh, G.P. Process for the Preparation of Elagolix and Pharmaceutically Acceptable Salts Thereof. WO Patent WO2018198086, 1 November 2018. [Google Scholar]
- Lenna, R.; Fasana, A.; Ortiz, J. Process for the Preparation of the Sodium Salt of Elagolix and Its Intermediates. Wo Patent WO2021044230, 11 March 2022. [Google Scholar]
- Siripragada, M.R.; Pendyam, K.; Kallepally, S.; Avula, S.R.; Gottapu, V.N.; Pappula, V.R. An Improved Process for the Preparation of Elagolix Sodium. WO Patent WO2021064561, 8 April 2021. [Google Scholar]
- Ballete, R.; Jimenez Alonso, O.; Garcia Garcia, E.; Dobarro Rodriguez, A. 3-((R)-2-(Amino-2-phenylethyl)-1-(2-fluoro-6-trifluoromethylbenzyl)-5-iodo-6-methyl-1H-pyrimidine-2,4-dione or a Salt Thereof, Process for its Preparation, and Its in the Synthesis of Elagolix. WO Patent WO2021083554, 6 May 2021. [Google Scholar]
- Zhong, X.; Lv, Q.; Yong, Q.; Hu, W.; Li, D.; Ji, S.; Zhan, L.; Chen, W.; Li, M.; Lin, J.; et al. Forced degradation studies of elagolix sodium with the implementation of high resolution LC-UV-PDA-MSn (n = 1,2,3…) and NMR structural elucidation. J. Pharm. Biomed. Anal. 2023, 224, 115198. [Google Scholar] [CrossRef]
- Ferraboschi, P.; Chiara Sala, M.; Stradi, R.; Ragonesi, L.; Gagliardi, C.; Lanzarotti, P.; Ragg, E.M.; Mori, M.; Meneghetti, F. Full spectroscopic characterization of two crystal pseudopolymorphic forms of the antiandrogen cortexolone 17α-propionate for topic application. Steroids 2017, 128, 95–104. [Google Scholar] [CrossRef]
- Meneghetti, F.; Ferraboschi, P.; Grisenti, P.; Reza Elahi, S.; Mori, M.; Ciceri, S. Crystallographic and NMR Investigation of Ergometrine and Methylergometrine, Two Alkaloids from Claviceps purpurea. Molecules 2020, 25, 331. [Google Scholar] [CrossRef]
- Ciceri, S.; Colombo, D.; Ferraboschi, P.; Grisenti, P.; Iannone, M.; Mori, M.; Meneghetti, F. Vecuronium bromide and its advanced intermediates: A crystallographic and spectroscopic study. Steroids 2021, 176, 108928. [Google Scholar] [CrossRef]
- Gallagher, D.; Treiber, L.; Hughes, R.; Campopiano, O.; Wang, P.; Zhao, Y.; Chou, S.; Ouellette, M.; Hettinger, D. Processes for the Preparation of Uracil Derivatives. WO Patent WO2009062087, 14 May 2009. [Google Scholar]
- Larrañaga, M.D.; Lewis, R.J.; Lewis, R.A.; Hawley, G.G. Hawley’s Condensed Chemical Dictionary, 16th ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2016; ISBN 1119312469. [Google Scholar]
- Lewis, R.J. Sax’s Dangerous Properties of Industrial Materials, 12th ed.; Wiley: Hoboken, NJ, USA, 2012; ISBN 9780471701347. [Google Scholar]
- Badertscher, M.; Bühlmann, P.; Pretsch, E. Tables of Spectral Data for Structure Determination of Organic Compounds; Springer: Berlin/Heidelberg, Germany, 1983; ISBN 978-3-662-22455-7. [Google Scholar]
- Marek, R.; Lycka, A. 15N NMR Spectroscopy in Structural Analysis. COC 2002, 6, 35–66. [Google Scholar] [CrossRef]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on chiral stationary phases: A powerful tool for the investigation of stereomutation processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Trapp, O.; Schoetz, G.; Schurig, V. Determination of enantiomerization barriers by dynamic and stopped-flow chromatographic methods. Chirality 2001, 13, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C. Stereolabile chiral compounds: Analysis by dynamic chromatography and stopped-flow methods. Chem. Soc. Rev. 2005, 34, 595–608. [Google Scholar] [CrossRef] [PubMed]
- König, W.A.; Gehrcke, B.; Runge, T.; Wolf, C. Gas chromatographic separation of atropisomeric alkylated and polychlorinated biphenyls using modified cyclodextrins. J. High Resol. Chromatogr. 1993, 16, 376–378. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, Y.-F.; Gross, T.D.; Tucci, F.C.; Gao, Y.; Moorjani, M.; Connors, P.J.; Rowbottom, M.W.; Chen, Y.; Struthers, R.S.; et al. Synthesis and structure-activity relationships of 1-arylmethyl-5-aryl-6-methyluracils as potent gonadotropin-releasing hormone receptor antagonists. J. Med. Chem. 2004, 47, 1259–1271. [Google Scholar] [CrossRef]
- Mennucci, B.; Tomasi, J.; Cammi, R.; Cheeseman, J.R.; Frisch, M.J.; Devlin, F.J.; Gabriel, S.; Stephens, P.J. Polarizable Continuum Model (PCM) Calculations of Solvent Effects on Optical Rotations of Chiral Molecules. J. Phys. Chem. A 2002, 106, 6102–6113. [Google Scholar] [CrossRef]
- Lan, C.B.; Auclair, K. Ammonium Chloride-Promoted Rapid Synthesis of Monosubstituted Ureas under Microwave Irradiation. Eur. J. Org. Chem. 2021, 2021, 5135–5146. [Google Scholar] [CrossRef]
- Capasso, C.; Winum, J.-Y. Novel method of treating macular degeneration: A patent evaluation (WO2018/107005). Expert Opin. Ther. Pat. 2019, 29, 749–752. [Google Scholar] [CrossRef]
- O’Brien, P.M.; Sliskovic, D.R.; Blankley, C.J.; Roth, B.D.; Wilson, M.W.; Hamelehle, K.L.; Krause, B.R.; Stanfield, R.L. Inhibitors of acyl-CoA:cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 8. Incorporation of amide or amine functionalities into a series of disubstituted ureas and carbamates. Effects on ACAT inhibition in vitro and efficacy in vivo. J. Med. Chem. 1994, 37, 1810–1822. [Google Scholar] [CrossRef]
- Liu, Y.; Hao, Q.; Lin, K.; Zhou, W.; Pan, J.; Chen, L.; Zhou, T. Preparation of Elagolix Intermediate. China Patent CN110498770, 30 April 2021. [Google Scholar]
- SMART & SAINT Software Reference Manual, Version 6.45; Bruker Analytical X-Ray Systems, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. SADABS, Version 2008/1; Bruker AXS Inc.: Karlsruhe, Germany, 2008.
- Sheldrick, G.M. SHELXL-2018; Universität Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Nardelli, M. Parst: A system of fortran routines for calculating molecular structure parameters from results of crystal structure analyses. Comput. Chem. 1983, 7, 95–98. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1H | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | / | / | / | / | / | / |
| 2 | / | / | / | / | / | / |
| 3 | 8.97 (brs) | Exchanged with CD3OD | 8.52 (brs) | / | / | / |
| 4 | / | / | / | / | / | / |
| 5 | 5.59 (s) | / | / | / | / | / |
| 6 | / | / | / | / | / | / |
| 7 | 5.36 (s) | 5.38 (s) | 5.47 (s) | 5.50 (m) | 5.47 (m) | 5.43 (m) |
| 8 | / | / | / | / | / | / |
| 9 | / | / | / | / | / | / |
| 10 | 7.25 (dd, J = 12.3 and 8.5 Hz) | 7.20 (dd, J= 11.7 and 8.3 Hz) | 7.28 (dd, J = 11.8 and 8.3 Hz) | 7.26 (m, overlapped with 28) | 7.29 (m, overlapped with 28) | 7.44 (m) |
| 11 | 7.42 (m) | 7.37 (m) | 7.42 (m) | 7.41 (m, overlapped with 26 and 26’) | 7.41 (m) | 7.53 (td, J = 8.1, 5.0 Hz) |
| 12 | 7.54 (d, J = 7.9 Hz) | 7.48 (d, J = 7.8 Hz) | 7.54 (d, J = 7.9 Hz) | 7.55 (d, J = 7.9 Hz) | 7.54 (d, J = 7.9 Hz) | 7.62 (d, J = 7.9 Hz) |
| 13 | / | / | / | / | / | / |
| 14 | / | / | / | / | / | / |
| 15 | 2.15 (s) | 2.35 (s) | 2.05 (s) | 2.07 (s) | 2.07 and 2.06 * | 2.085 (s) and 2.076 (s) * |
| 16 | / | / | / | / | / | / |
| 17 | / | / | / | / | / | / |
| 18 | / | / | / | / | / | / |
| 19 | / | / | 6.97 (m) | 6.97 (td, J = 8.2 and 1.3 Hz) | 6.97 (tt, J = 8.1 and 1.6 Hz) | 7.10 (n.d., overlapped with 20) |
| 20 | / | / | 7.10 (dd, J = 9.0 and 7.9 Hz) | 7.11 (tdd, J = 8.0, 2.8, and 1.3 Hz) | 7.11 (tdd, J = 8.0, 4.6, and 1.3 Hz) | 7.11 and 7.14 (n.d., overlapped with 19) |
| 21 | / | / | 6.81 (m) | 6.85 (td, J = 6.2 and 1.5 Hz) and 6.78 (td, J = 6.2 and 1.4 Hz) * | 6.83 (ddd, J = 7.7, 6.0, and 1.6 Hz) and 6.76 (ddd, J = 7.7, 6.0, and 1.6 Hz) * | 6.76 (m) and 6.61 (m) * |
| 22 | / | / | 3.88 (s) | 3.89 (s) | 3.889 (s) and 3.886 (s) * | 3.883 (s) and 3.880 (s) * |
| 23 | / | / | / | 4.29 (m o ddd, J = 16.2, 13.1, 9.8 Hz, Ha) and 4.11 (m, Hb) | 4.28 (m, Ha) and 4.04(m, Hb) | 4.26–4.07 (m) |
| 24 | / | / | / | 4.42 (dd, J = 9.8 and 4.5 Hz) | 4.12 (m) | 4.11 (n.d.) |
| 25 | / | / | / | / | / | / |
| 26 and 26′ | / | / | / | 7.41 (m, overlapped with 11) | 7.37 (m) | 7.29 (n.d., overlapped with 28) |
| 27 and 27′ | / | / | / | 7.32 (m) | 7.30 (m, overlapped with 10) | 7.27 (n.d., overlapped with 26 and 26’) |
| 28 | / | / | / | 7.27 (m, overlapped with 10) | 7.24 (m) | 7.22 (m) |
| 29 | / | / | / | 2.13 | 1.63 (brs) | / |
| 30 | / | / | / | / | 2.46 (m, Ha) and 2.38 (m, Hb) | 2.40 (m) |
| 31 | / | / | / | / | 1.68 (m) | 1.75 (m, Ha) and 1.68 (m, Hb) |
| 32 | / | / | / | / | 2.29 (m) | 2.09 (m, overlapped with 15) |
| 33 | / | / | / | / | / | / |
| CH2CH3 | / | / | / | / | 4.084 (q, J = 7.1Hz,) and 4.082 (q, J = 7.1Hz) * | / |
| CH2CH3 | / | / | / | / | 1.21 (t, J = 7.1 Hz) | / |
| 13C | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | / | / | / | / | / | / |
| 2 | 151.7 | 150.8 | 151.0 | 152.3 | 152.3 and 152.2 * | 153.3 and 153.2 * |
| 3 | / | / | / | / | / | / |
| 4 | 162.4 | 159.6 | 161.2 | 161.8 and 161.6 * | 161.7 and 161.6 * | 163.5 and 163.3 * |
| 5 | 102.5 | 99.2 | 108.8 | 108.3 and 108.2 * | 108.3 and 108.2 * | 109.3 and 109.2 * |
| 6 | 153.6 | 151.6 | 151.9 | 149.9 and 149.8 * | 149.7 and 149.6 * | 152.6 and 152.5 * |
| 7 | 41.1 (m) | 42.5 (m) | 41.8 (m) | 42.7 (brs) | 42.7 (brs) | 44.1 (brs) |
| 8 | 121.8 (d, J = 11.6 Hz) | 121.2 (d, J = 11.3 Hz) | 121.8 (d, J = 11.1 Hz) | 122.0 (d, J= 11.4 Hz) | 122.1 (d, J = 11.3 Hz) | 123.6 (overlapped with 12 and 16) |
| 9 | 161.4 (d, J = 250.0 Hz) | 161.1 (d, J = 249.6 Hz) | 161.4 (d, J = 250.0 Hz) | 161.3 (d, J = 249.5 Hz) | 161.3 (d, J = 250.0 Hz) | 162.8 (d, J =248.0 Hz) |
| 10 | 120.9 (d, J = 24.0 Hz) | 120.9 (d, J = 24.0 Hz) | 121.0 (d, J = 24.4 Hz) | 120.9 (d, J = 24.1 Hz) | 120.9 (d, J = 24.4 Hz) | 122.15 (d, J = 24.3 Hz) and 122.19 (d, J = 24.3 Hz) * |
| 11 | 129.5 (d, J = 10.0 Hz) | 129.6 (d, J = 9.7 Hz) | 129.5 (d, J = 9.8 Hz) | 129.3 (d, J = 9.8 Hz) | 129.3 (d, J = 9.6 Hz) | 130.9 and 130.8 * |
| 12 | 122.6 (m) | 122.5 (m) | 122.6 (m) | 122.6 (m) | 122.5 (m) | 123.7 (overlapped with 8 and 16) |
| 13 | 129.6 (dd, J = 30.0 and 3.6 Hz; partially hidden by 11) | 129.3 (dd, J = 30.9 and 3.7 Hz; | 129.6 (dd, J = 30.9 and 4.0 Hz) | 129.5 (dd, J = 31.0 and 3.1, partially overlapped with 11) | 129.3 (dd, J = 30.2 and 3.0 Hz) | 130.6 (dd, J = 31.1 and 3.3 Hz) |
| 14 | 123.4 (dd, J = 273.6 and 4.0 Hz) | 123.3 (dd, J = 274.3 and 4.2 Hz) | 123.5 (dd, J = 273.8 and 3.9 Hz) | 123.5 (dd, J = 273.6 and 3.8 Hz) | 123.5 (dd, J = 271.1 and 3.7 Hz) | 125.1 (dd, J = 273.3 and 3.8 Hz) |
| 15 | 20.1 | 20.1 | 17.9 | 17.8 | 17.8 | 18.1 |
| 16 | / | / | 121.5 (d, J = 13.6 Hz) | 122.3 (d, J = 13.7 Hz) and 122.2 (d, J = 13.7 Hz) * | 122.37 (d, J = 13.8 Hz) and 122.35 (d, J = 13.8 Hz) | 123.8 (overlapped with 8 and 12) |
| 17 | / | / | 149.9 (d, J = 246.4 Hz) | 150.2 (d, J = 246.3 Hz) and 150.1 (d, J = 246.3 Hz) * | 150.23 (d, J = 245.5) and 150.17 (d, J = 245.5) | 151.8 (d, J = 245.4 Hz) |
| 18 | / | / | 148.0 (d, J = 11.0 Hz) | 148.0 (d, J = 11.0 Hz) | 148.0 (d, J = 11.0 Hz) | 149.5 (d, J = 11.2 Hz) |
| 19 | / | / | 113.5 | 113.3 | 113.2 | 114.8 |
| 20 | / | / | 123.8 | 123.8 (overlapped with 21) | 123.80 (s) and 123.76 (s) * | 125.1 (s) and 125.0 (s) * |
| 21 | / | / | 123.9 (d, J = 4.6 Hz) | 123.9 and 123.8 (overlapped with 20) * | 123.92 (s) and 123.85 (s) * | 125.0 and 124.9 * |
| 22 | / | / | 56.3 | 56.2 | 56.2 | 56.8 |
| 23 | / | / | / | 49.10 and 49.14 * | 47.7 and 47.6 * | 48.1 and 47.9 * |
| 24 | / | / | / | 54.1 and 54.2 * | 60.9 and 60.8 * | 62.2 and 62.0 * |
| 25 | / | / | / | 143.7 and 143.6 * | 141.92 (s) and 141.89 (s) * | 142.1 and 141.9 * |
| 26 and 26’ | / | / | / | 126.4 and 126.3 * | 127.2 and 127.1 (overlapped with 28) * | 128.7 |
| 27 and 27’ | / | / | / | 128.5 and 128.4 * | 128.4 and 128.3 * | 129.4 |
| 28 | / | / | / | 127.3 and 127.2 * | 127.2 (overlapped with 26 and 26′) | 128.5 |
| 29 | / | / | / | / | / | / |
| 30 | / | / | / | / | 46.51 (s) and 46.47 (s) * | 48.7 |
| 31 | / | / | / | 25.4 | 27.86 (s) and 27.83 (s) * | |
| 32 | / | / | / | / | 32.0 | 37.03 (s) and 37.00 (s) * |
| 33 | / | / | / | / | 173.7 60.1 | 182.3 |
| CH2CH3 | / | / | / | / | / | |
| CH2CH3 | / | / | / | / | 14.2 | / |
| 15N | 5 | 6 | 7 | 8 | 9 | 1 |
|---|---|---|---|---|---|---|
| 1 | 136.7 | 137.9 | 134.3 | 134.3 | 135.5 | 137.1 |
| 3 | 154.3 b | 153.4 b | 151.9 b | 160.1 | 161.3 | 160.5 |
| 29 | / | / | / | 31.2 | 43.6 | 46.3 |
| 13C | HMBC (13C→1H) |
|---|---|
| C-2 | H-7 and H-23 |
| C-4 | H-23 and H-15 (weak) |
| C-5 | H-21 and H-15 |
| C-6 | H-15 |
| C-8 | H-7, H-10, and H-12 |
| C-9 | H-7, H-10, and H-11 |
| C-13 | H-7 and H-11 |
| C-14 | H-12 |
| C-16 | H-20 |
| C-17 | H-19 and H-21 |
| C-18 | H-20 and H-22 |
| C-25 | H-23, H-24, H-27, and H-27’ |
| C-33 | H-31 and H-32 |
| Conformers | τ1 (°) | τ2 (°) | τ3 (°) | τ4 (°) | Gas Phase ΔE (kcal/mol) | Gas Phase (%) |
|---|---|---|---|---|---|---|
| 1A | −81.6 | −48.5 | −132.0 | 49.1 | 4.35 | 0.0 |
| 1B | −70.0 | 147.8 | −130.0 | 49.7 | 6.84 | 0.0 |
| 1C | 96.0 | −54.0 | −125.0 | 70.0 | 0.67 | 20.5 |
| 1D | 111.8 | 134.3 | −133.0 | 52.5 | 4.60 | 0.0 |
| 1E | −80.5 | −47.0 | −61.3 | −52.4 | 4.67 | 0.0 |
| 1F | −69.4 | 150.3 | −55.0 | −55.5 | 7.09 | 0.0 |
| 1G | 113.0 | 136.0 | −60.5 | −55.6 | 5.01 | 0.0 |
| 1H | 93.2 | −62.1 | −64.1 | −55.6 | 1.90 | 2.6 |
| 1I | 94.5 | −53.6 | 61.3 | −142.6 | 2.26 | 1.4 |
| 1J | 118.5 | 147.4 | 59.4 | −140.8 | 4.86 | 0.0 |
| 1K | −74.0 | −40.3 | 67.8 | −141.2 | 4.78 | 0.0 |
| 1L | −69.1 | 148.9 | 60.5 | −142.9 | 3.69 | 0.1 |
| 1M | 92.6 | −61.9 | −110.0 | −134.5 | 0.00 | 63.9 |
| 1N | 113.6 | 146.5 | −77.5 | 147.4 | 3.98 | 0.1 |
| 1O | −85.9 | −51.6 | −115.4 | −136.4 | 3.58 | 0.2 |
| 1P | −70.9 | 148.7 | −75.9 | 146.3 | 5.33 | 0.0 |
| 1Q | −81.0 | −46.4 | −75.4 | 142.8 | 3.64 | 0.1 |
| 1R | −71.1 | 148.9 | −75.2 | 147.0 | 5.40 | 0.0 |
| 1S | 92.7 | −55.0 | −76.1 | 139.9 | 1.09 | 10.1 |
| 1T | 113.9 | 146.9 | −77.6 | 147.5 | 3.93 | 0.1 |
| 1U | −86.3 | −49.9 | 56.0 | 53.0 | 4.53 | 0.0 |
| 1V | −70.1 | 145.9 | 51.1 | 55.2 | 3.67 | 0.1 |
| 1W | 93.5 | −53.5 | 49.9 | 53.8 | 2.77 | 0.6 |
| 1X | 114.2 | 142.7 | 51.3 | 53.6 | 5.32 | 0.0 |
| Code | τ1 | τ2 | τ3 | τ4 | τ5 | Water ΔE (kcal/mol) | Water (%) |
|---|---|---|---|---|---|---|---|
| 7 (crystal structure) | −105.5 | −144.7 | 110.4 | ||||
| 1M−A | 87.7° | −63.3° | −104.7° | −133.9° | −111.8° | 0.00 | 83.6 |
| 1M−C | 91.3° | −60.2° | −106.7° | −132.6° | 75.2° | 2.07 | 2.6 |
| 1M−D | 92.9° | −60.3° | −110.0° | −135.0° | 126.3° | 1.06 | 13.8 |
| Conformer | t6 | Distance CH22 – H19 (Å) | Gas Phase ΔE (kcal/mol) | Gas Phase (%) | Methanol ΔE (kcal/mol) | Methanol (%) |
|---|---|---|---|---|---|---|
| 1M-A | 0 | 2.56 | 0.00 | 44.5 | 0 | 86.0 |
| 1M-A (I) | −66 | 3.60 | 0.29 | 27.5 | 1.62 | 5.6 |
| 1M-A (II) | +66 | 3.60 | 0.27 | 28.0 | 1.38 | 8.4 |
| 1M-D | 0 | 2.56 | 0.22 | 30.5 | 0.00 | 84.5 |
| 1M-D (I) | −66 | 3.60 | 0.32 | 25.6 | 1.36 | 8.5 |
| 1M-D (II) | +66 | 3.60 | 0.00 | 43.9 | 1.48 | 7.0 |
| Crystal Data | |
|---|---|
| Chemical formula | C20H15F5N2O3 |
| Mr | 426.34 |
| Crystal system, space group | Orthorhombic, P bca |
| a, b, c (Å) | 11.165 (2), 11.073 (2), 30.367(6) |
| V (Å3) | 3754.2(13)) |
| Z | 8 |
| F (000) | 1744 |
| Density (g/cm3) | 1.509 |
| Temperature (K) | 298 |
| Radiation type | Mo-Kα (λ = 0.71073 Å) |
| µ (mm−1) | 1.135 |
| Crystal size (mm) | 0.06 × 0.03 × 0.02 |
| Data collection | |
| Diffractometer | Bruker-Axs Smart-Apex CCD |
| Tmin, Tmax | 0.893, 1.000 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 29,138, 4175, 1715 |
| Rint | 0.0298 |
| Structure refinement | |
| R, wR2, S | 0.0437 (I > 2σ(I)) and 0.1119 (all), 0.0973 (I > 2σ(I)) and 0.1126 (all), 0.760 (all) |
| No. of parameters | 271 |
| No. of restraints | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciceri, S.; Colombo, D.; Fassi, E.M.A.; Ferraboschi, P.; Grazioso, G.; Grisenti, P.; Iannone, M.; Castellano, C.; Meneghetti, F. Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules 2023, 28, 3861. https://doi.org/10.3390/molecules28093861
Ciceri S, Colombo D, Fassi EMA, Ferraboschi P, Grazioso G, Grisenti P, Iannone M, Castellano C, Meneghetti F. Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules. 2023; 28(9):3861. https://doi.org/10.3390/molecules28093861
Chicago/Turabian StyleCiceri, Samuele, Diego Colombo, Enrico M. A. Fassi, Patrizia Ferraboschi, Giovanni Grazioso, Paride Grisenti, Marco Iannone, Carlo Castellano, and Fiorella Meneghetti. 2023. "Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study" Molecules 28, no. 9: 3861. https://doi.org/10.3390/molecules28093861
APA StyleCiceri, S., Colombo, D., Fassi, E. M. A., Ferraboschi, P., Grazioso, G., Grisenti, P., Iannone, M., Castellano, C., & Meneghetti, F. (2023). Elagolix Sodium Salt and Its Synthetic Intermediates: A Spectroscopic, Crystallographic, and Conformational Study. Molecules, 28(9), 3861. https://doi.org/10.3390/molecules28093861