1. Introduction
Leishmaniasis is a series of vector-borne diseases, transmitted through the bite of a sandfly infected by
Leishmania parasites. This disease is endemic to several regions in the world, including some developed countries [
1]. In 2020, there were over 208,000 new cases of cutaneous leishmaniasis reported worldwide and nearly 13,000 new visceral leishmaniasis cases [
2]. Current therapies include sometimes long, painful, and toxic treatments such as pentavalent antimonial, liposomal amphotericin B, miltefosine, or paromomycin [
3]. Furthermore, each therapy is effective for a limited number of
Leishmania species and, thus, specific forms of the disease.
Liposomal amphotericin B is used to treat visceral leishmaniasis [
3]. Its leishmanicidal activity is mediated through the formation of pores in the
L. mexicana and
L. donovani promastigote plasma membrane as it binds membrane sterols, namely ergosterol, and cholesterol to a lesser extent [
4,
5]. Formulations such as Kalsome TM10 were shown to depolarize the mitochondrial membrane, cause ATP levels depletion, and increase reactive oxygen species (ROS) production [
6].
Miltefosine is the only oral treatment used against cutaneous leishmaniasis as well as visceral leishmaniasis in endemic regions such as South America and India [
7]. The drug is an alkylphosphocholine that has been shown to have several targets in
Leishmania cells, although its mechanism of action is not fully elucidated [
8,
9,
10,
11].
In addition to therapy efficacy in specific forms of the disease, only, recent emergence of resistant strains and/or relapses after treatment have been reported for both treatments. For instance, mutations to the enzyme sterol 14α-demethylase, leading to a decrease in plasma-membrane ergosterol levels [
12], and higher levels of multidrug resistance 1 (MDR1), causing greater amphotericin B efflux [
13], have been shown to decrease the efficacy of amphotericin B treatments. Many in-laboratory miltefosine-resistant strains obtained through several mutations, lead to drug efflux or a reduction in uptake, and an increase in oxidative stress [
14]. However resistant strains have not yet been discovered in clinical isolates against miltefosine, though several relapses after treatment have nonetheless been documented [
15,
16].
In this context, eugenol could represent a potentially useful molecule. An eugenol oil-in-water emulsion showed beneficial immunomodulatory activity in mice infected with
L. donovani [
17]. Indeed, after 10 days of treatment with the eugenol emulsion, the hepatic and splenic parasites load was significantly decreased compared to untreated infected mice, and the host defense was stimulated as evidenced by an increase in nitric oxide, IFN-γ, and IL-2 levels, a decrease in IL-4 and IL-10 levels, as well as an increase in CD4+ and CD8+ T cells. Nonetheless, the mechanism of action of eugenol on
Leishmania promastigotes has not yet been elucidated. Previous research conducted in our group showed that eugenol has an antileishmanial activity of 2.72 µg/mL (16.59 µM) against
Leishmania mexicana mexicana (
Lmm) promastigotes, with a selectivity index above 10 against the WI38 human fibroblasts and the J774 macrophage mouse-derived cell line [
18].
The aim of the present study was to better characterize the mechanism of action of eugenol on Leishmania, other than its immunomodulatory activity. Amphotericin B and miltefosine were used as reference drugs in the different assays, not only to assess the magnitude of the activity but also to better determine underlying mechanisms of action. To this aim, morphological changes to Lmm promastigotes after treatment with increasing concentrations of eugenol during 24 h and 72 h were followed by scanning electron and fluorescence microscopy. Membrane permeability was evaluated on liposomes (LUVs mimicking Lmm membranes) by following calcein release. Membrane fluidity was studied on both LUVs and Leishmania promastigotes by measuring changes in 1,6-diphenyl-1,3,5-hexatriene (DPH) anisotropy. Apoptosis in Lmm populations was followed by FACS analysis of the calcein-acetoxymethyl ester (AM)/propidium iodide (PI) dye staining. Metabolic activity through NADH/NAD+ and cytochrome c oxidase was assessed in Leishmania by using the Alamar blue assay. Finally, in an effort to elucidate a potential target of eugenol in the metabolism of Lmm, the abundance of acidocalcisomes and lipid droplets, respectively, involved in pH and Ca2+ homeostasis and energy storage, were quantified in Lmm promastigotes using fluorescence microscopy for anti-Trypanosoma brucei vacuolar H+ pyrophosphatase (TbVP1) and Nile Red labeling.
Altogether, the results show that eugenol possesses a mechanism of action that differs from that of the two known drugs amphotericin B and miltefosine, by targeting lipid storage at concentrations below 10× IC50, leading to a stunt in cell growth while maintaining membrane integrity. At concentrations above 25× IC50, eugenol causes membrane disruption though without selectively targeting membrane sterols. These data give insight into the mode of action of eugenol to mediate its antiparasitic activity.
2. Results
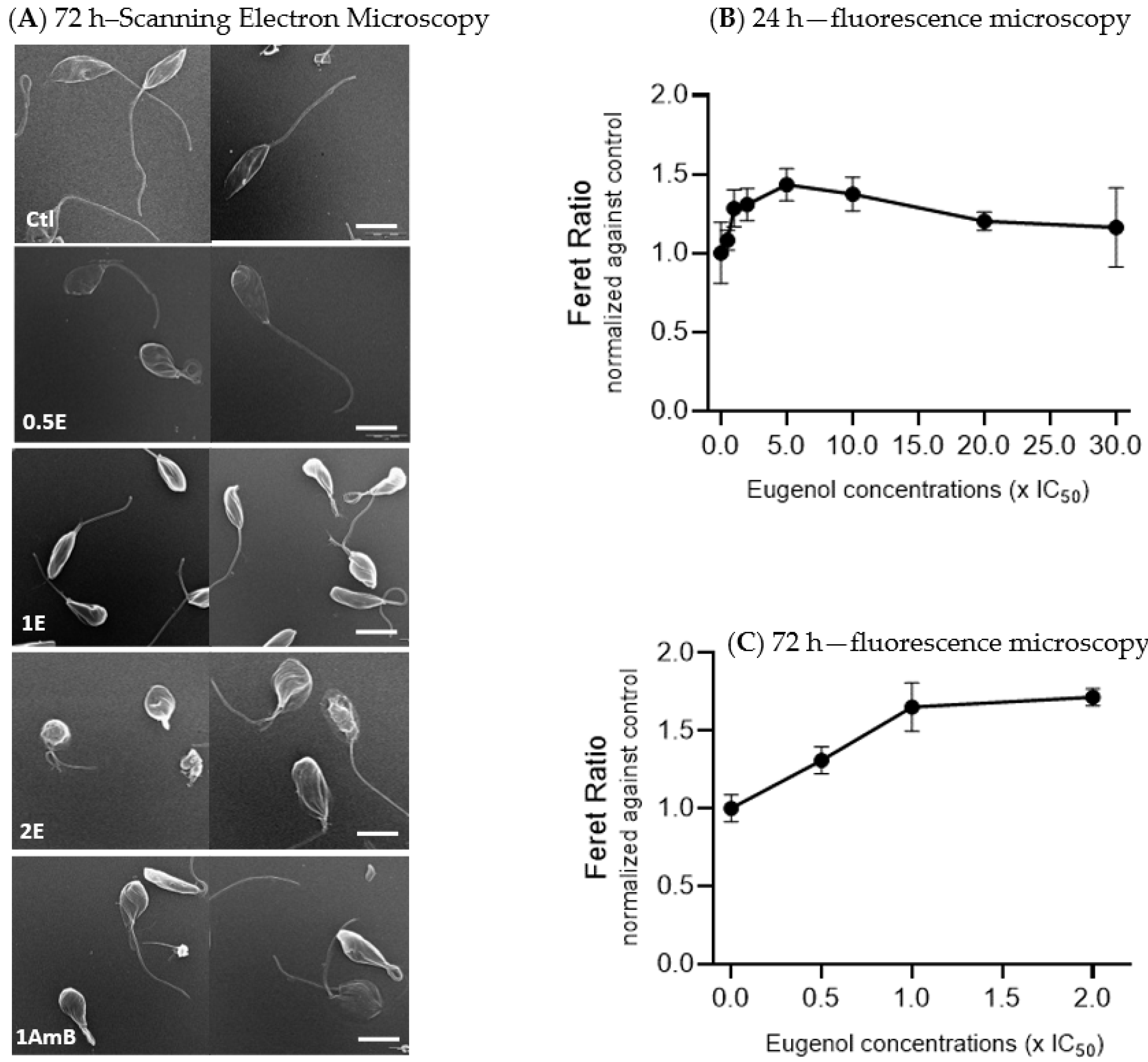
2.1. Eugenol Induces Round Shape of Lmm Promastigotes
To evaluate the general effect of eugenol on
Lmm, promastigotes were treated for 72 h with 0.01% DMSO (Ctl) or three concentrations of eugenol (0.5× IC
50, IC
50, and 2× IC
50) or amphotericin B (at IC
50). Images were recorded by using scanning electron microscopy (SEM) (
Figure 1A).
SEM analysis of untreated Leishmania promastigotes showed elongated bodies with long flagella. Promastigotes treated with eugenol at 0.5× IC50 appeared more swollen and at IC50 and 2× IC50 showed alterations in both shape and size, as observed with gradually rounding promastigote bodies and loss or shortening of their flagella. At 2× IC50, folds appeared on the body of the Lmm and their flagella became stumpy. Amphotericin B, at IC50, also showed more round-shaped bodies compared to the control.
Feret ratio values, which describe the shape of the promastigotes (the higher the value, the rounder the object), were calculated. Data revealed an increase of 1.4 fold, reached between 5 and 10× IC
50 of eugenol, followed by a decrease back to values comparable to the control at 20 and 30× IC
50 of eugenol after 24 h of treatment (
Figure 1B). After 72 h of treatment, a dose-dependent increase of the Feret ratio was observed till 1.7 fold at 2× IC
50 (
Figure 1C).
Altogether, these results indicate that eugenol induced rounding of Lmm promastigotes bodies, similar to amphotericin B.
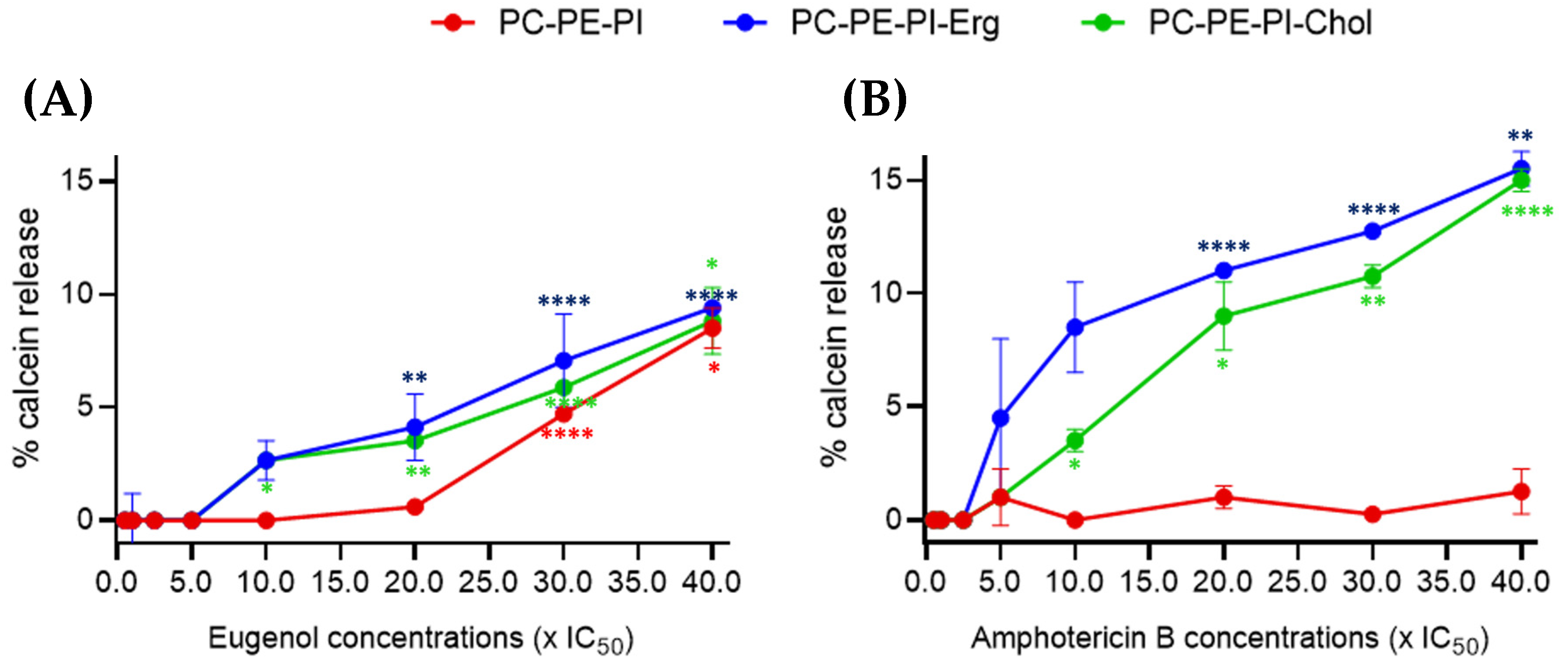
2.2. Eugenol Induces Milder Membrane Permeability in LUVs Than Amphotericin B
To then evaluate the potential effect of eugenol on membranes, we examined changes in the permeability of large unilamellar vesicles (LUVs) mimicking
Lmm membranes. Three LUVs compositions were tested. All were composed of phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) but differential sterol content: PC:PE:PI, PC:PE:PI:Cholesterol, or PC:PE:PI:Ergosterol. Each model was treated with increasing concentrations of eugenol and amphotericin B and the calcein leakage was followed (
Figure 2).
Between 0.5 and 5× IC
50, eugenol showed no effect on LUVs, regardless of the liposome composition. At 10× IC
50, a low calcein leakage of 2.7% was observed in ergosterol- and cholesterol-containing LUVs. At 30× IC
50, a calcein release of 4.7% was observed in sterol-free LUVs. At 40× IC
50, ~9% of calcein was released for all three liposome membrane compositions (
Figure 2A).
Amphotericin B was used as a reference drug since its interactions with ergosterol in
Lmm plasma membranes are well known [
4,
19,
20]. No effect was observed until 2.5× IC
50. A ~4% calcein leakage was observed starting from 5× IC
50 for ergosterol-containing LUVs and from 10× IC
50 for cholesterol-containing LUVs, while, as expected, no leakage was observed for the LUVs without sterol even at the highest-tested concentration. A gradual increase was observed for both sterol-containing compositions until 40× IC
50 (
Figure 2B).
All in all, these data show that eugenol increased LUV permeability at two-times-higher IC50 concentrations than amphotericin B. Furthermore, there was seemingly a slight difference in effect between sterol- and nonsterol-containing LUVs treated with eugenol, though no difference between ergosterol- and cholesterol-containing LUVs, suggesting a different mechanism of action than amphotericin B.
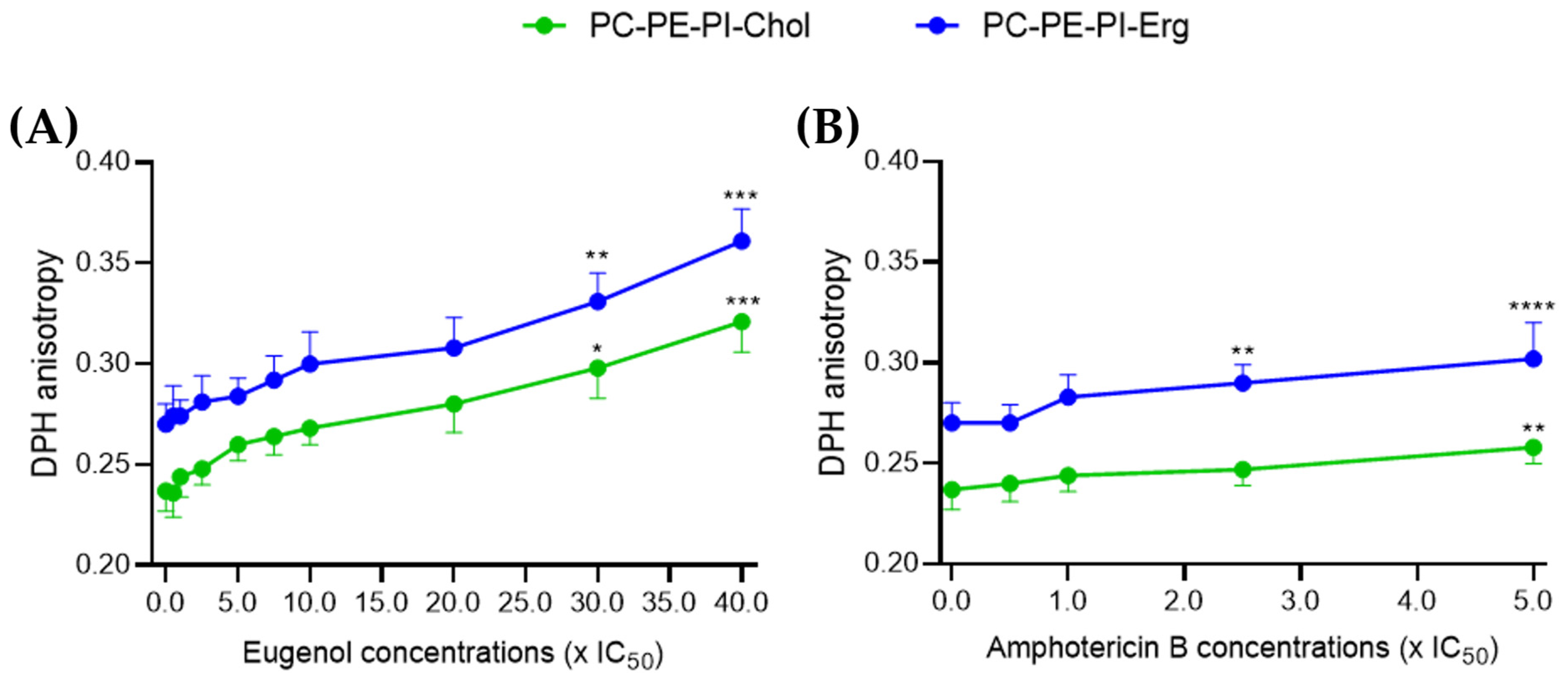
2.3. Eugenol Similarly Decreases Membrane Fluidity in Cholesterol and Ergosterol LUVs
Then, using the same eugenol and amphotericin B concentrations as in the permeability assay, we evaluated the membrane fluidity through the measurement of DPH anisotropy. An increase in DPH anisotropy indicates a decrease in membrane fluidity [
21]. Only the models containing sterols were kept as there was no significant effect on permeability after treatment in the sterol-free model for amphotericin B.
Between 0.5 and 20× IC
50, eugenol showed no significant effect on DPH anisotropy compared to the control. However, starting from 30× IC
50, eugenol induced a 30 to 35% dose-dependent decrease in membrane fluidity in both ergosterol- and cholesterol-containing LUVs (
Figure 3A).
A significant 7% decrease in membrane fluidity was observed after treatment with amphotericin B at 2.5× IC
50 and 12% at 5× IC
50 in ergosterol-containing LUVs. This is in agreement with previous results that a colloidal suspension containing 45% amphotericin B was shown to decrease the plasma membrane fluidity of
L. amazonensis promastigotes by increasing EPR spin labeling in a dose-dependent manner [
22]. For cholesterol-containing LUVs, only a 9% decrease in fluidity was observed at 5× IC
50 (
Figure 3B).
Altogether, these results on LUVs indicate that, like amphotericin B, eugenol was able to decrease the membrane fluidity, whether in the presence of ergosterol or cholesterol. However, concentrations of eugenol 15 times higher than those used for amphotericin B are required to show a significant decrease in membrane fluidity.
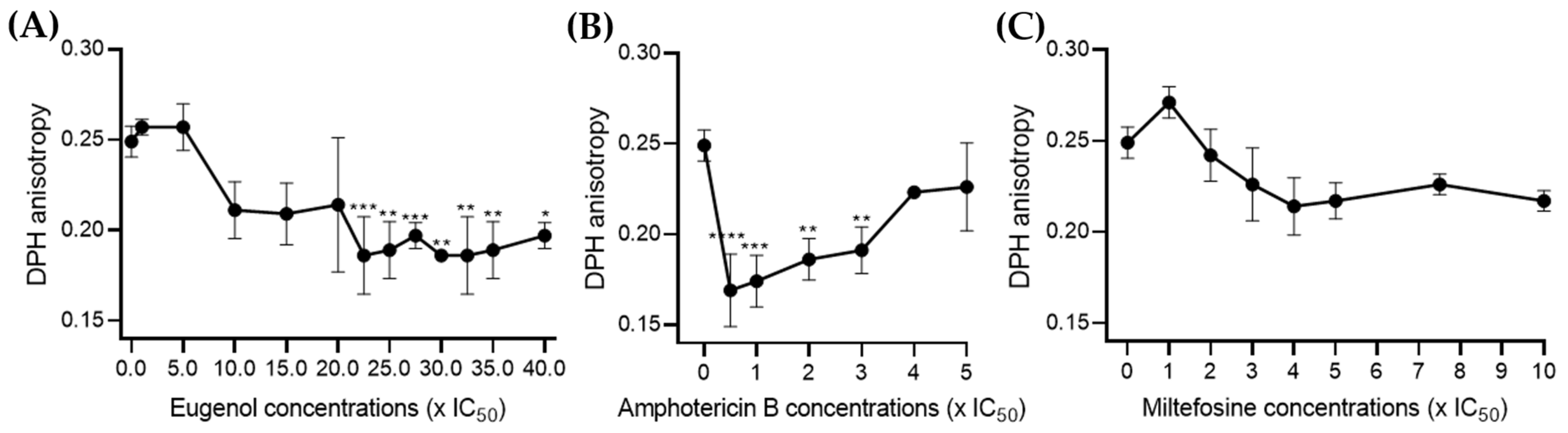
2.4. Eugenol Increases the Fluidity of Lmm Membranes at Higher Concentrations Than Amphotericin B
To enhance the biological relevance of our results and ensure that the differences only in concentrations but not in activity between eugenol and amphotericin B did not result from the simple composition of LUVs, we directly measured DPH anisotropy on the
Lmm membranes [
23,
24].
For eugenol, no significant effect was observed below 20× IC
50. Between 22.5 and 40× IC
50, eugenol increased membrane fluidity by 20–30%, which is contrary to its activity in LUVs (
Figure 4A). Amphotericin B also displayed an increase in membrane fluidity below 3× IC
50, while the DPH value increased dose dependently from 0.5 to 5× IC
50 (
Figure 4B). In contrast, miltefosine showed no significant effect on membrane DPH at any of the concentrations tested (
Figure 4C).
In sum, the above data indicate that eugenol induced similar changes to membrane permeability and fluidity than amphotericin B, though alterations were seen at higher concentrations and were less sterol-dependent, which could suggest a different mechanism of action.
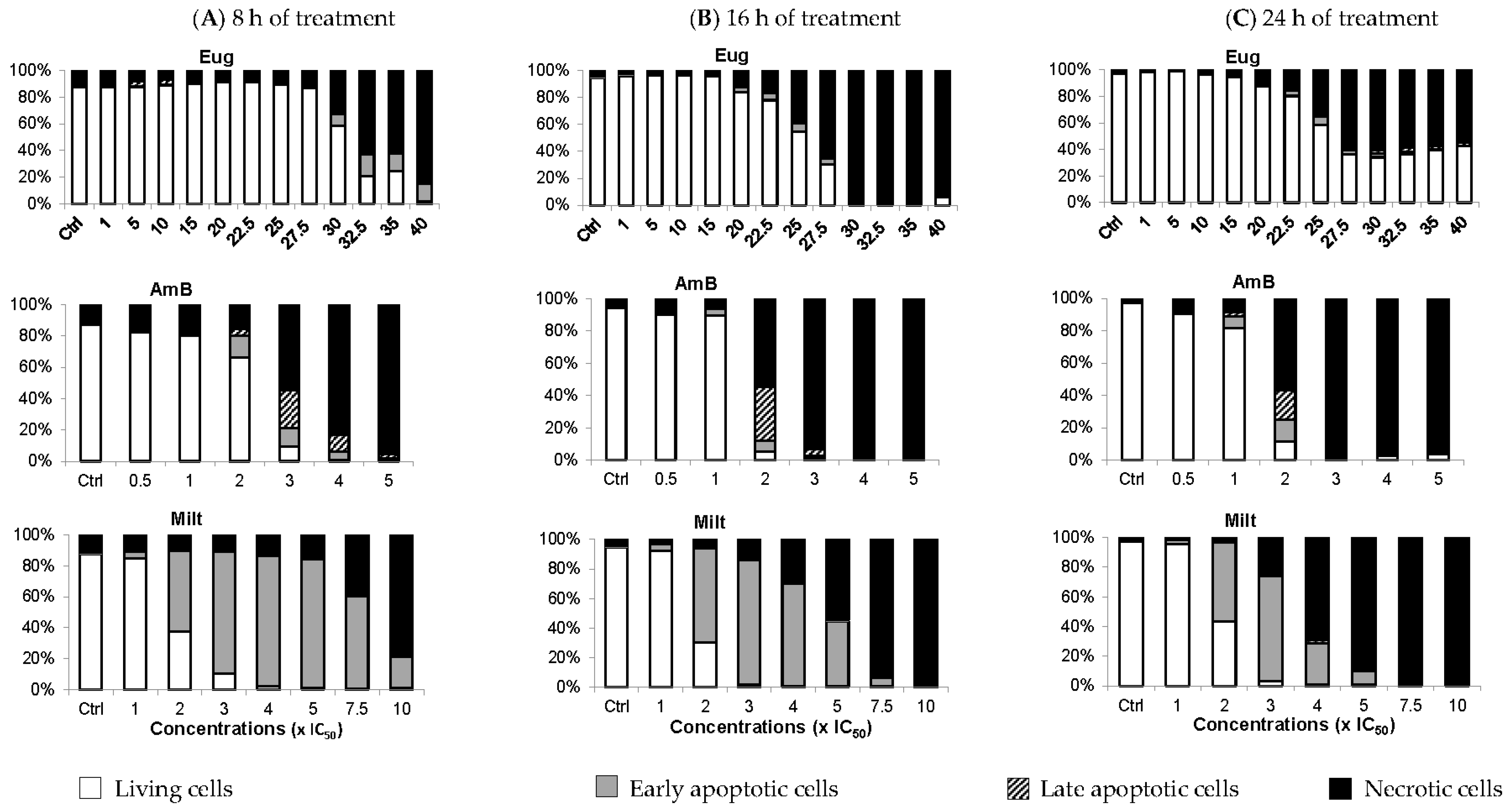
2.5. Eugenol Exerts Its Leishmanicidal Effect with Kinetic and Cell-Death Mechanisms Different from Those of Amphotericin B and Miltefosine
To further assess the mode of action of eugenol, we studied the cell-death mechanisms induced by this compound in comparison with amphotericin B and miltefosine.
Calcein-acetoxymethyl ester (AM)/propidium iodide (PI) double labelling can be used on
Leishmania to distinguish between living, early apoptotic, late apoptotic, and necrotic cells. Contrary to mammalian cells, calcein-AM has greater difficulty entering into viable
Leishmania cells with intact membranes, resulting in low-to-no fluorescence (calcein−/PI−) [
25,
26]. During apoptosis, calcein-AM enters the cells where it is cleaved by esterases into a fluorescent molecule. However, at the early apoptosis stage, membrane modifications with the maintenance of the membrane integrity allow calcein-AM entry but prevent PI entry (calcein+/PI−). During late apoptosis, membrane degradation creates pores in the plasma membrane allowing PI to enter the cells (calcein+/PI+). Finally, in necrotic cells, complete membrane loss leads to PI being solely observed (calcein−/PI+) [
25].
In
Figure 5, between 1 and 15× IC
50, eugenol did not induce apoptosis whether the treatment lasted 8 h, 16 h, or 24 h. Only 3.5% of cells were in the late apoptotic stage at 5 and 10× IC
50, after 8 h of treatment. At 30 and 32.5× IC
50, 9.7% and 16.7% of promastigotes were in early apoptosis, respectively, after 8 h of treatment. After 16 h of treatment at these concentrations, over 95% of cells were necrotic, while after 24 h of treatment, the living cell population rose to 35%, with ~5% early and late apoptotic cells combined. The lower percentage of necrotic cells after 24 h of treatment compared to living cells could be due to more cells evolving from a necrotic state to being completely deteriorated, and thus not detected using our double labeling. Alternatively, the
Lmm doubling time enabled more living cells to multiply, increasing their proportion, or cells started adjusting to the presence of eugenol, which led to more cells in the early apoptotic phase than in the necrotic phase.
In contrast to eugenol, amphotericin B induced early and late apoptotic cells starting at 1 or 2× IC
50 according to the duration of treatment. However, it mainly caused necrosis at higher concentrations. Miltefosine was also added as it was already shown to induce apoptosis in
L. tropica at 10 µM and
L. major promastigotes at 22 µM after 18 h, 24 h, and 36 h of incubation by flow-cytometry analysis of annexin V [
27]. In the case of miltefosine,
Lmm cells were in their early apoptosis phase starting at 2× IC
50 for all three treatment durations. Regarding necrosis, the concentration of miltefosine required to induce at least 40% of necrotic cells is inversely proportional to the incubation time.
In conclusion, eugenol induced apoptosis when applied for 8 h at a high concentration (≥30× IC50). After 24 h of treatment at these concentrations, eugenol likely induced necrosis, a phenomenon that can be linked to membrane disruption via increased membrane permeability and fluidity. At lower concentrations of eugenol, cells were mainly living and membrane integrity was maintained. We, therefore, focused on evaluating the effect of eugenol on metabolic activity to characterize intracellular events that could be responsible for the antileishmanial activity of the compound.
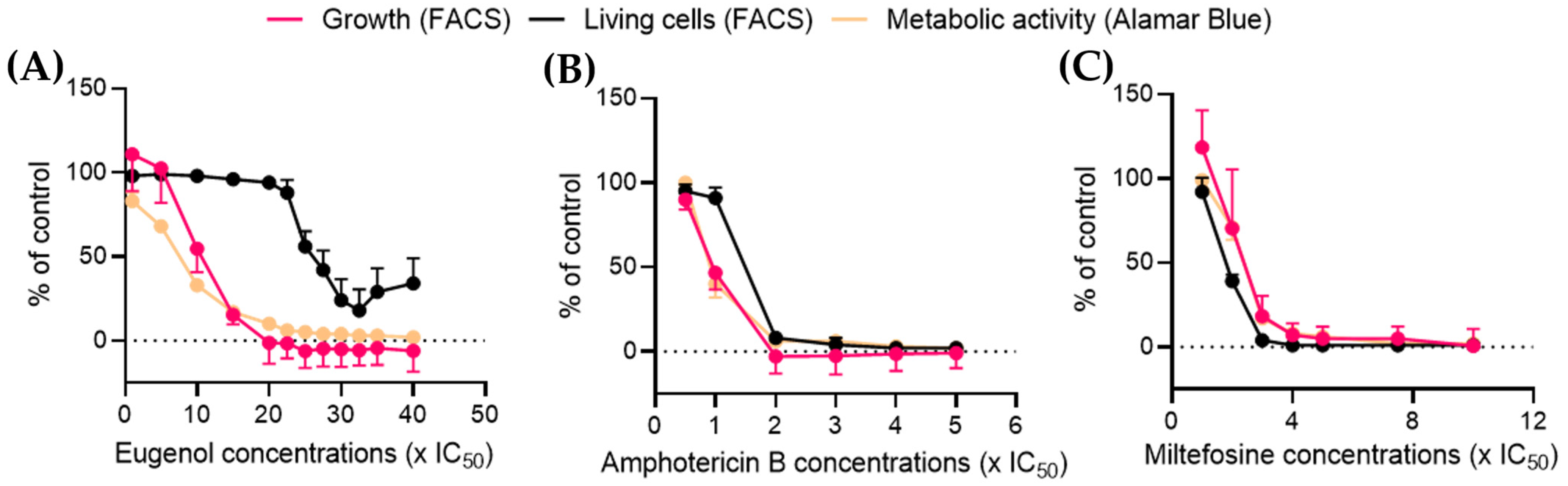
2.6. Eugenol Drastically Reduces Metabolic Activity at Low Concentrations While Maintaining a High Percentage of Living Cells
Alamar Blue is a reagent containing resazurin, a blue dye. Resazurin enters the cell where it is reduced to the highly fluorescent pink-molecule resofurin by enzymes and cofactors such as NADH oxidoreductase or cytochrome
c oxidase [
28,
29,
30] without impairing mitochondrial respiration. It is used to follow cell viability and metabolic function [
31], as it is sensitive to changes in energy or respiratory metabolism [
32,
33]. Furthermore, NADH/NAD
+ balance and related enzymes have been shown to be crucial in
Leishmania survival [
34]. The metabolic activity data obtained by the Alamar Blue assay were then plotted in comparison with the percentage of living cells obtained by FACS (see
Figure 5).
After 24 h of treatment at 10× IC
50, eugenol reduced the metabolic activity by more than half while simultaneously inducing a 40% loss of
Lmm division. In contrast, the calcein-AM/PI double labeling showed 95% living parasites at this concentration. The percentage of living cells was reduced by half only at concentrations higher than 25× IC
50 and reached a plateau at 27.5× IC
50 (
Figure 6A). Amphotericin B and miltefosine both induced a sharp decline in the living-cell population coupled with a dose-dependent reduction of cell growth and metabolic activity (
Figure 6B,C).
These results indicate that contrary to amphotericin B and miltefosine, eugenol induces a leishmanostatic effect at concentrations below 15× IC50, which is characterized by a reduction of metabolic activity and cell growth while maintaining living cell populations.
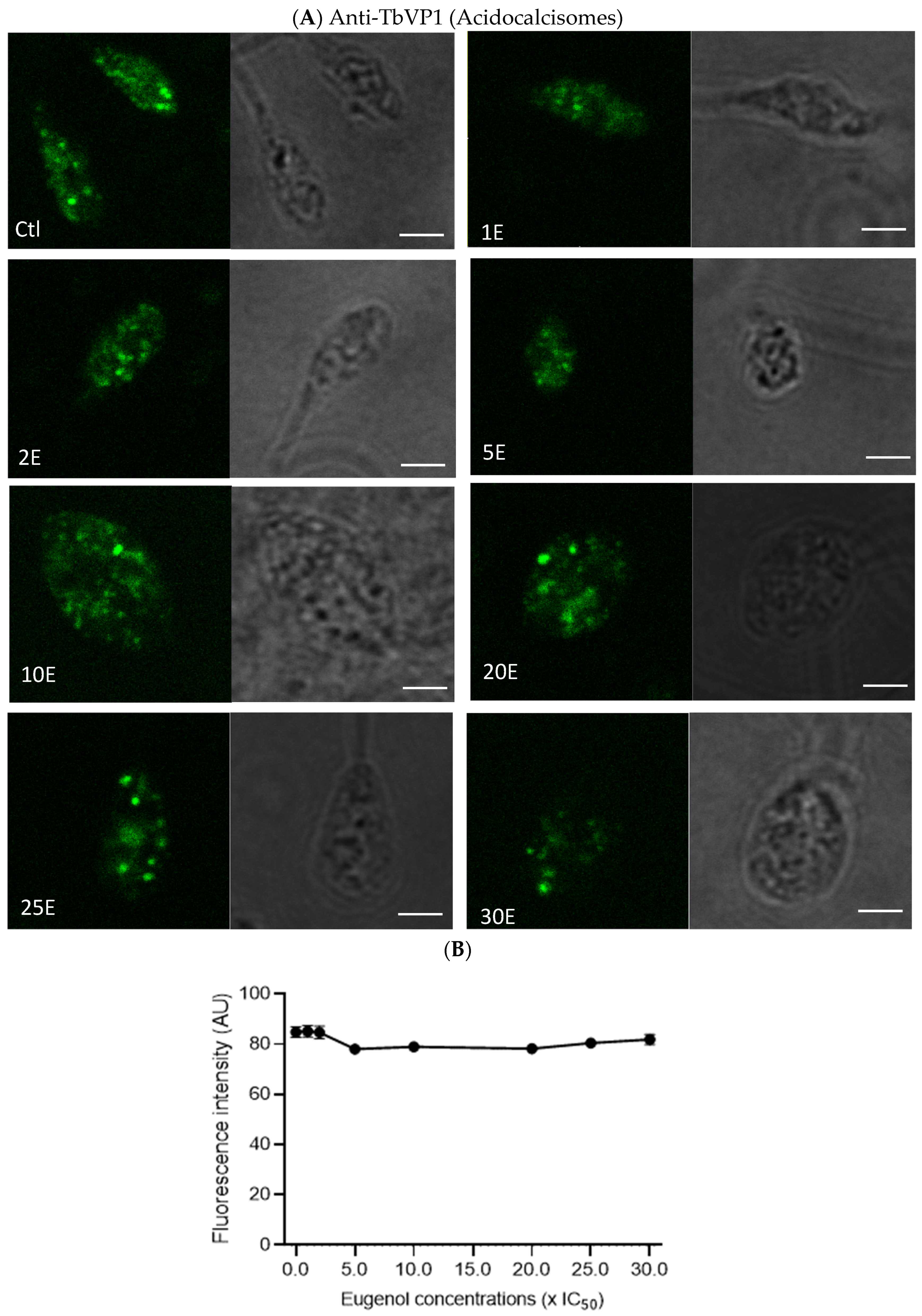
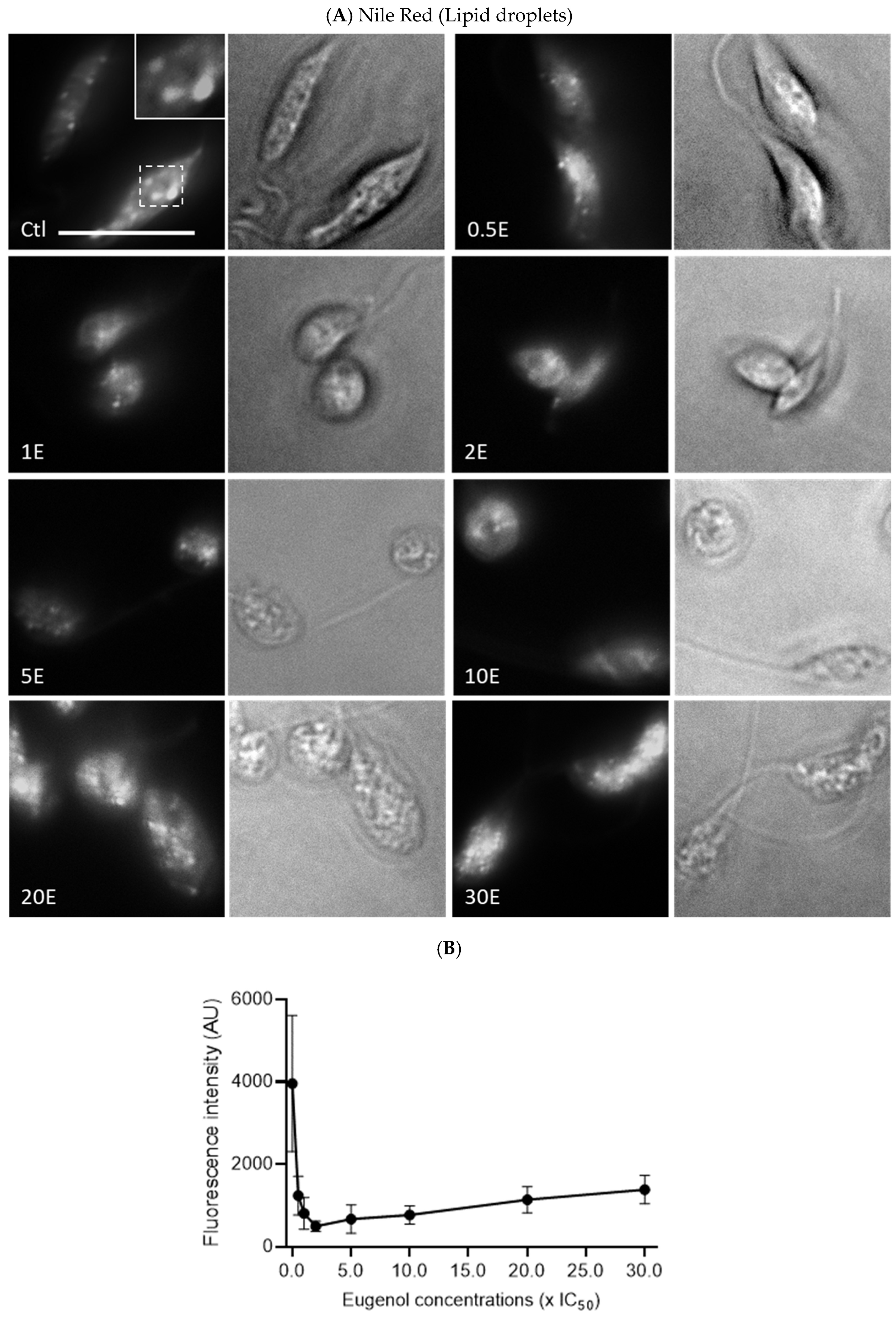
2.7. Eugenol Reduces Lipid Inclusions but Does Not Impair Acidocalcisomes
Since the metabolic activity of
Lmm measured by the Alamar Blue assay was reduced in the presence of eugenol, we evaluated whether this observation could be linked to an alteration of polyphosphate storage and osmoregulation in acidocalcisomes and/or lipid metabolism via lipid droplets [
35,
36]. Acidocalcisomes and lipid droplets were therefore assessed after 24 h of treatment with increasing concentrations of eugenol by fluorescence microscopy upon anti-TbVP1 and Nile Red labeling, respectively.
In
Figure 7A,B, no obvious effect on acidocalcisomes was noticed after 24 h or 72 h. This suggests that the overall content and number of acidocalcisomes was not impacted by eugenol.
In contrast, lipid droplets decreased significantly and dose dependently between 0.5× IC
50 and 2–5× IC
50, nearly eightfold compared to the untreated promastigotes, before re-increasing (
Figure 8A,B).
3. Discussion
The effect of eugenol on Lmm promastigotes was studied to gain a better understanding of its mode of action. Eugenol-induced rounding of promastigotes after 24 and 72 h of treatment at concentrations around IC50. An increase in membrane permeability and a decrease in fluidity in LUVs were observed at concentrations above 20× IC50, while an increase in the fluidity of Lmm membranes was observed at the same concentrations. The results showed that eugenol mainly induced necrosis rather than apoptosis (whether early or late) as a cell-death mechanism, at the various concentrations and incubation periods tested. Further investigation revealed that according to the Alamar Blue assay, metabolic activity related to NADH/NAD+ and cytochrome C oxidase was drastically reduced starting at 5× IC50. Lipid droplets were also reduced between 0.5–5× IC50 of eugenol while the abundance of acidocalcisomes was not affected. Throughout our experiments, we chose to test eugenol at increasing concentrations up to the ones allowing for a significant effect on the activities studied, while amphotericin B was tested at lower concentrations for which it was already known to have a significant effect. This allowed for the measurement of the difference in the extent and efficacy of eugenol compared to known reference drugs.
The change in promastigotes morphology from elongated to round and swollen was previously observed in the shape of
L. donovani promastigotes after treatment with a eugenol-rich
Syzygium aromaticum oil at 100 µg/mL and eugenol emulsions [
17,
37] as well as several other antileishmanial compounds.
Feret ratios obtained after eugenol treatment of
Lmm indicate a reduction of body length, describing the shift from elongated to rounded promastigotes. Although the morphology was affected by concentrations near the IC
50 of eugenol, membrane disruption was neither the cause nor the end result at these concentrations. Eugenol only showed a 10% increase in LUVs permeability at concentrations above 30× IC
50. These observations concur with those of a previous publication showing that, at a MIC concentration (or 100×
Lmm IC
50), eugenol was found to induce 12.5% leakage in
Saccharomyces cerevisiae membrane-like LUVs [
38]. LUVs composition differs between
S. cerevisiae and
Lmm models, though, in both cases, weak LUV membrane permeability was observed at high concentrations (>20× IC
50). Nonetheless, amphotericin B is known to target
Lmm membranes and showed a comparable increase in the permeability to eugenol, although at concentrations approximately 30 times lower than eugenol (0.25× IC
50 instead of 7.5× IC
50). This indicates that, although the permeabilizing effect is present with high doses of eugenol, the extent of the disruption might be comparable to that of amphotericin B. Furthermore, a closer investigation of the impact of eugenol on the membranes highlighted a different mode of action than that of amphotericin B. Indeed, the activity of eugenol does not appear dependent on the presence of sterols (ergosterol or cholesterol) in LUVs, while amphotericin B is known (and shown) to preferentially target ergosterol-containing LUVs [
19].
Similar to permeability, membrane fluidity is only significantly affected by eugenol at concentrations above 20× IC
50. In this regard, loss of membrane integrity mainly occurs at high concentrations of the compound and leads to necrosis. The high concentrations needed to observe membrane disruption suggest the role of other membrane components than the lipids included in the LUVs. Furthermore, the opposite effects on the membrane fluidity of LUVs versus
Lmm membranes were observed. This could be due to differences in the components present in artificial membranes and membranes from parasites, and especially the presence of membrane proteins. Furthermore, differences in experimental conditions such as the duration of incubation (30 min for LUVs experiments and 24 h for
Lmm plasma membranes) could also explain the differences in results since the compounds could be differently inserted in the membrane because of a lesser or greater duration of contact with the membranes, thus modifying the DPH location [
39].
Intracellularly, nuclear DNA fragmentation might have occurred while membranes remained intact, as previously observed for
L. donovani promastigotes treated with 4 mM H
2O
2 for 6 h [
40]. We here showed that acidocalcisomes revealed by the pyrophosphatase immunolabeling were not significantly affected by eugenol concentrations. Nonetheless, eugenol may still target Ca
2+. Indeed, an eugenol-rich
S. aromaticum oil caused depolarization of the mitochondrial membrane [
37], while an eugenol-rich essential oil from
Ocimum gratissimum induced mitochondrial swelling after 72 h of treatment [
41]. SQ109 is a drug inhibiting
Leishmania donovani proliferation and was shown to cause deregulation of Ca
2+ in mitochondria, via disruption of the mitochondrial membrane potential and acidocalcisomes, by their alkalinization [
42]. These findings agree with the Alamar Blue assay results as cytochrome c oxidase is involved in mitochondrial energetic mechanisms.
This study also shows for the first time that eugenol dose-dependently decreases the concentration of intracellular lipid inclusions between 0.5 and 5× IC
50. Miltefosine was shown to cause an overall depletion of lipid metabolites after 7.5 h of treatment, which was suggested to be caused by leakage and, thus, impairment of cellular membranes [
43]. However, we observe a sharp decline in lipid droplets at concentrations for which membrane permeability and fluidity are not or very mildly affected. Lipids have a variety of functions in
Leishmania sp. and some are linked to the structural needs of the parasites while others are linked to the metabolic needs as energy storage for the cells [
36]. This could be due to the conversion and subsequent use of lipids to fulfill energetic needs in stress conditions induced by exposure to eugenol.
4. Materials and Methods
4.1. Chemicals and Reagents
Eugenol, amphotericin B, calcein, sephadex G-50, LH-20, ergosterol (Erg-), pentamidine isethionate, propidium iodide (PI), and 1,6-diphenyl-1,3,5-hexatriene (DPH) were obtained from Sigma-Aldrich (Bornem, Belgium). Miltefosine was obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Calcein-AM included in a live/dead Viability/Cytotoxicity Kit for mammalian cells, Alamar blue and goat anti-Guinea Pig IgG Secondary Antibody, Alexa Fluor™ 488 were obtained from Thermo Fisher Scientific (Merelbeke, Belgium). The 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), l-α-phosphatidylinositol (PI—Liver, Bovine), and cholesterol (Chol—Ovine Wool) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). All organic solvents used were from VWR (Leuven, Belgium).
4.2. Scanning Electron Microscopy
L. mexicana mexicana promastigotes at the density of 105 cells/mL were treated for 72 h with 0.5, 1, or 2× IC50 of eugenol (IC50 = 16.59 µM), or IC50 of amphotericin B (IC50 = 0.25 µM). After centrifugation at 600× g for 10 min, parasites were washed 2 times with phosphate-buffered saline (PBS, pH 7.4) and once with cacodylate buffer 0.1 M (pH 7.4). They were then immobilized on poly-L-lysine-coated coverslips for 5 min at room temperature. After washing 3 times with cacodylate 0.1 M (pH 7.4) to remove the excess of free-floating Leishmania, the coverslips were incubated in 1% glutaraldehyde in cacodylate 0.1 M (pH 7.4) to crosslink the fixed parasites on the poly-L-lysine coating. The parasites were postfixed with 1% osmium tetroxide in a cacodylate buffer for 2 h at 4 °C and washed in water to eliminate traces of the remaining osmium tetroxide, then dehydrated in a graded series of ethanol, critical-point dried, and coated with 10 nm of gold. Samples were observed with a CM12 Philips electron microscope at 80 kV.
4.3. Preparation of Large Unilamellar Vesicles (LUVs) Mimicking Leishmania Plasma Membrane
Based on the lipid profile of the isolated surface membrane of
L. donovani promastigotes reported by Wassef et al. [
44], LUVs composed of PC:PE:PI:Erg (10:26:12:15) were prepared using the extrusion technique with the Avanti Mini Extruder (Avanti Polar Lipids, Inc., Alabaster, AB, USA) [
45]. Briefly, lipids were dissolved in chloroform to obtain a stock solution of 25 mg/mL and then mixed in the required ratio. The lipid film, which was formed after evaporating the solvent under a vacuum with a Buchi Rotavapor R-200, rotary evaporator (Büchi Labortechnik AG, Flawil, Switzerland), was hydrated by Tris 10 mM and NaCl 159 mM at pH 7.4. The suspension was submitted to five cycles of freezing–thawing to obtain multilamellar vesicles (MLVs). The MLVs were extruded at 45 °C, 19 times through two polycarbonate Nucleopore Track Etch membrane filters (pore size = 100 nm) (Whatman Nucleopore, Corning Costar Corp., Badhoevedorp, The Netherlands) to obtain LUVs. Their size (average ± 150 nm) and the polydispersity index (PDI < 0.2) were controlled via a dynamic light-scattering technique using Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). The phospholipid content was measured by phosphorus assay [
46] and then adjusted to a final concentration of 5 μM.
To study the selective role of ergosterol, it was omitted in the preparation of some liposomes or replaced by cholesterol.
4.4. Calcein Release Assay
The leakage of calcein entrapped inside LUVs at the self-quenching concentration induced by a permeabilizing agent can be measured by fluorescence increase as calcein is diluted. For this permeability study, calcein, after purifying using column chromatography with sephadex LH-20, was diluted to the required concentration (73 mM) in the 10 mM Tris-HCl buffer solution at pH 7.4. This solution was then used for the hydration of the lipid film. The unencapsulated calcein was removed using the minicolumn centrifugation technique with sephadex G-50. Calcein-filled LUVs were incubated with eugenol or the reference drug amphotericin B, for 15 min at concentrations ranging from 0.5× IC
50 to 40× IC
50. Triton X 0.1% was used to induce 100% of the calcein leakage. After measuring the fluorescence intensity (excitation wavelength of 472 nm, emission wavelength of 512 nm) using a Perkin Elmer LS 30 Fluorescence Spectrophotometer (Perkin-Elmer Ltd., Beaconsfield, UK), the percentage of calcein released was calculated using the formula:
Ft: fluorescence signal measured in the presence of compound;
Fctrl: fluorescence signal measured for LUVs control;
Ftot: fluorescence signal measured in the presence of Triton X-100;
Results are means of three independent experiments in triplicates.
4.5. DPH Fluorescence Anisotropy Assay
Fluorescence anisotropy values of DPH (r) can be used to probe the effect of a compound on membrane fluidity as the probe locates in the hydrophobic core of the lipid bilayer of the membrane and is sensitive to changes in the lipid acyl chain physical properties [
47].
In LUVs- During the LUVs preparation process, DPH 2 mM in tetrahydrofuran was mixed with lipids at the molar ratio of 1:200 (DPH: lipid) before evaporating the solvents. The following steps were done as previously described in the section “Preparation of LUV mimicking Leishmania plasma membrane”.
DPH-containing LUVs in a Tris HCl/NaCl buffer were then incubated with eugenol or amphotericin B for 30 min in the dark at the previously prepared concentrations. DPH was excited at 365 nm, and emission was read at 425 nm using an LS55 luminescence spectrometer connected to a circulating water bath to maintain the temperature at 28 °C [
48].
DPH fluorescence anisotropy was determined using the equation:
IVV and IVH: the fluorescence intensities with the excitation (wavelength of 365 nm) and emission (wavelength of 425 nm) polarization filters in vertical (v) and horizontal (h) orientations, respectively.
G: an inherent factor to the spectrophotometer used.
In
Lmm- Samples were prepared as described by Xu et al. [
49]. Briefly, parasites at the density of 10
7 cells/mL were treated for 24 h with eugenol (in the range of 1–40 × IC
50) with IC
50 24 h = 5× IC
50 72 h, or reference drugs amphotericin B (in the range of 0.5–5× IC
50) with IC
50 24 h = IC
50 72 h, and miltefosine (in the range of 1–10× IC
50) with IC
50 24 h = 25.09 µM = 2× IC
50 72 h, then washed once with PBS, and resuspended in 1 mL of PBS. Then, 2 mM DPH in tetrahydrofuran were added to a final concentration of 0.5 μM and incubated in the dark for 20 min at 28 °C (the concentration of tetrahydrofuran in
Leishmania suspension was 0.025%).
DPH parameters are the same as previously described and Equation (2) was used to determine DPH anisotropy.
Results of the DPH anisotropy for both LUVs and Lmm are means of three independent experiments in triplicates.
4.6. Flow-Cytometry Analysis
Lmm promastigotes at the density of 107 cells/mL were treated for 8 h, 16 h, and 24 h with eugenol in the range of 1–40× IC50 (IC50 8, 16 and 24 h = 5× IC50 72 h), or by two reference drugs, amphotericin B in the range of 0.5–5× IC50 (IC50 8, 16 and 24 h = IC50 72 h), and miltefosine between 1 and 10× IC50 (IC50 24 h = 2× IC50 72 h). Treated parasites were collected by centrifugation at 600× g for 10 min. After washing once with PBS, Lmm promastigotes were resuspended in 1 mL of PBS-containing calcein-AM 0.1 μM and PI 3 μM, then incubated for 15 min at room temperature in the dark. A total of 50,000 events per sample were acquired in a FACSCalibur cytometer using 488 nm excitation. Green fluorescence emission for calcein (530/30 bandpass) and red fluorescence emission for PI (610/20 bandpass) were measured.
4.7. Alamar Blue Assay
Using Alamar blue to analyze the effect of eugenol on the metabolic activity of L. mexicana mexicana promastigotes, parasites at the density of 107 cells/mL were incubated with eugenol, amphotericin B, or miltefosine at the same range of concentrations as the flow cytometry experiment. After 20 h of incubation at 28 °C, Alamar blue (diluted 1:1 in PBS) was added and parasites were further incubated for 4 h at a final concentration of 4.5%. Fluorescence was measured on a spectrofluorimeter (SpectraMax-Molecular Devices, Berkshire, UK) (excitation wavelength of 530 nm, emission wavelength of 590 nm). The percentage of metabolic activity was calculated by comparing fluorescence values to those reported for the nontreated group.
4.8. Nile Red Labelling and Image Quantification
Lmm promastigotes at 10
7 cells/mL were treated for 24 h with eugenol at concentrations between 0.5 and 30× IC
50 (IC
50 24 h = 5× IC
50 72 h). The parasites were then centrifuged at 600×
g for 10 min. The pellet was washed twice with PBS 1X, then fixed in 4% PFA. Then, 10 µL of the sample were mixed with 30 µL of a 10 µg/mL Nile Red solution in acetone and incubated for 10 min. The mixture was observed in a Zeiss LSM980 microscope. Quantification of
Lmm morphology was obtained by measuring 4–6 images from two independent experiments for the Feret maximum diameter (accounting for length) and the Feret minimum diameter (accounting for width). The Feret ratio of
Lmm was then calculated as follows:
Similarly, quantification of the Nile Red fluorescence was done by measuring the mean green intensity in 4–6 images per concentration of eugenol after manually drawing the Lmm contour using confocal images in black and white. The results were obtained from 2 independent experiments.
4.9. Anti-TbVP1 Labelling and Image Quantification
Lmm promastigotes at the density of 10
7 cells/mL were treated for 24 h with eugenol at 1–30× IC
50 (IC
50 24 h = 5× IC
50 72 h). Treated parasites were centrifuged at 600×
g for 10 min. After washing twice with PBS,
Lmm promastigotes were resuspended in 1 mL of PBS. Cells at 3 × 10
7 Lmm/mL were then transferred to a 24-well poly-l-lysine-coated plate. After several washes in PBS 1X, they were fixed with 4% formaldehyde in 0.1 M phosphate buffer for 20 min at room temperature. Permeabilization was done in 0.1% Triton-X100 for 5 min at room temperature. After several washes in Q-PBS (1% bovine serum albumin, lysine at 1 mg/mL, 0.01% saponin, and 0.025% azide in PBS 1X), the cells were incubated with anti-TbVP1 primary antibodies [
50] for 1 h and then with fluorescent Alexa Fluor 488 goat antiguinea pig secondary antibodies for 1 h. The coverslips were mounted in prolong gold and left to polymerize for 24 h. The images were taken on a Zeiss LSM980 microscope. Quantification was obtained by measuring the mean green intensity of
Lmm in 4–6 images per concentration of eugenol after manually drawing the
Lmm contour using confocal images in black and white. The results were obtained from 1 experiment.
4.10. Statistical Analyses
All statistical tests were performed on GraphPad Prism 9.4.1. One-way ANOVA with Dunnett’s post tests to compare the control versus compound-treated LUVs or Leishmania promastigotes, * p < 0.05; ** p < 0.01, *** p < 0.001, and **** p < 0.0001.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







