


Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase

 ,
,  , ,
, ,  and
and
Abstract
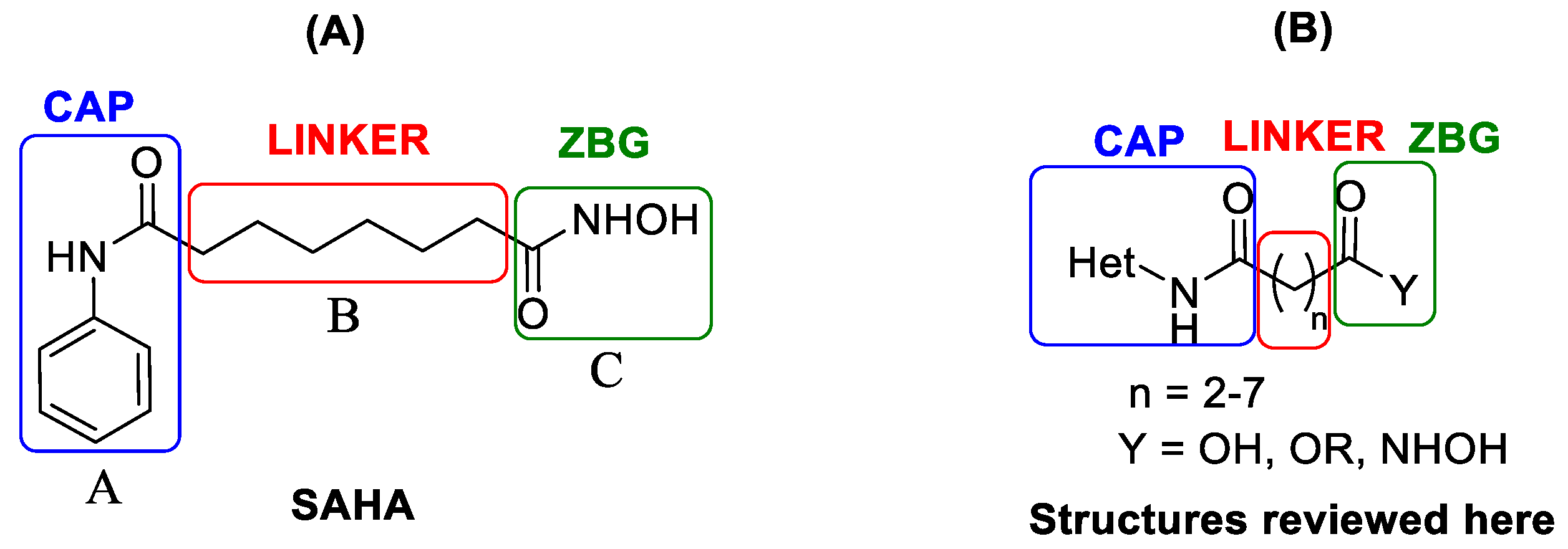
:1. Introduction
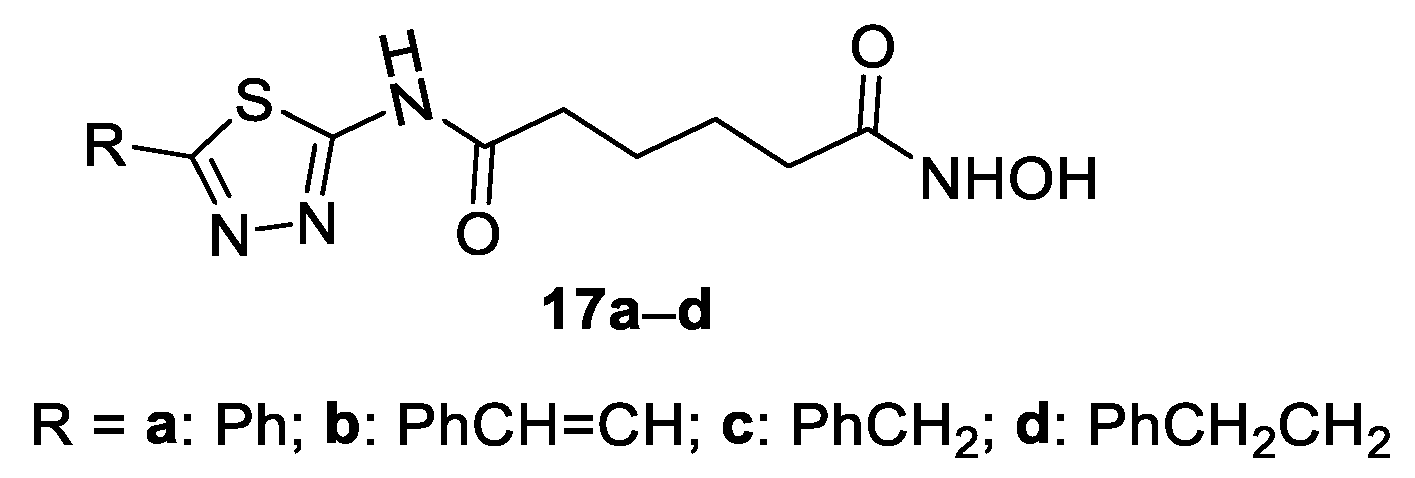
2. Two-Carbon Linker Chain
2-Amino-1,3,4-thiadiazoles in the CAP Group
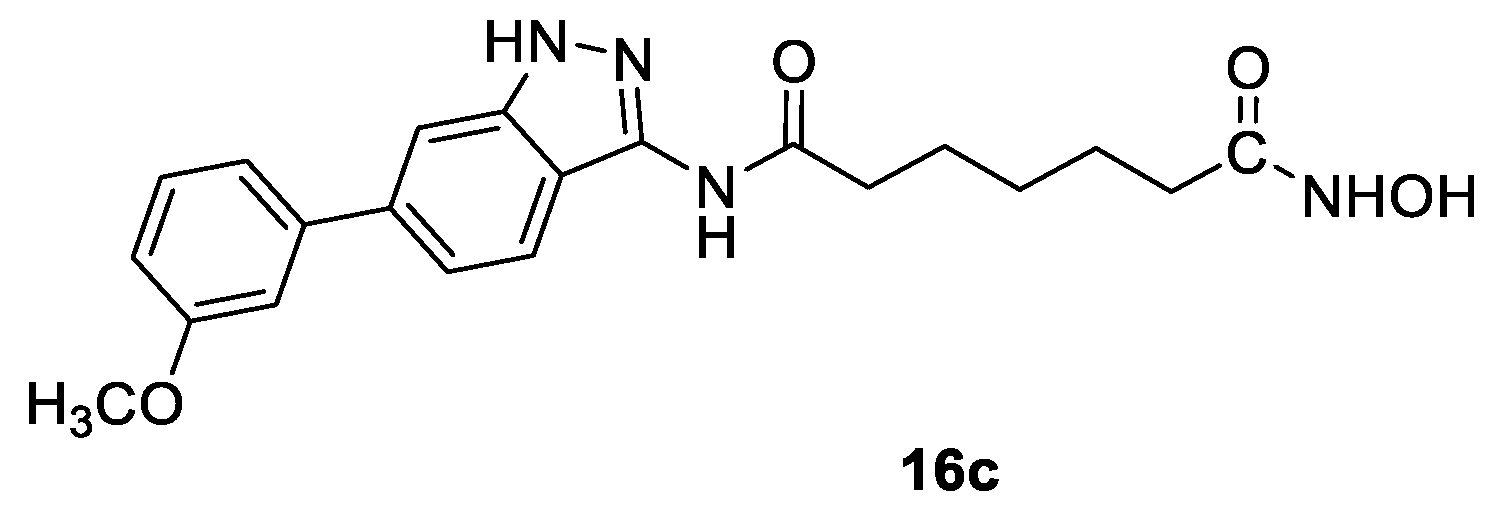
3. Three-Carbon Linker Chain
3.1. 2-Amino-1,3,4-thiadiazoles in the CAP GROUP
3.2. Indazoles in the CAP Group
4. Four-Carbon Linker Chain (4-C Spacer)
4.1. 2-Amino-1,3,4-thiadiazoles in the CAP Group
4.2. Indazoles in the CAP Group
4.3. Benzothiazoles in the CAP Group
4.4. 4-Anilinothieno [2,3-d]pyrimidine Derivatives in the CAP Group
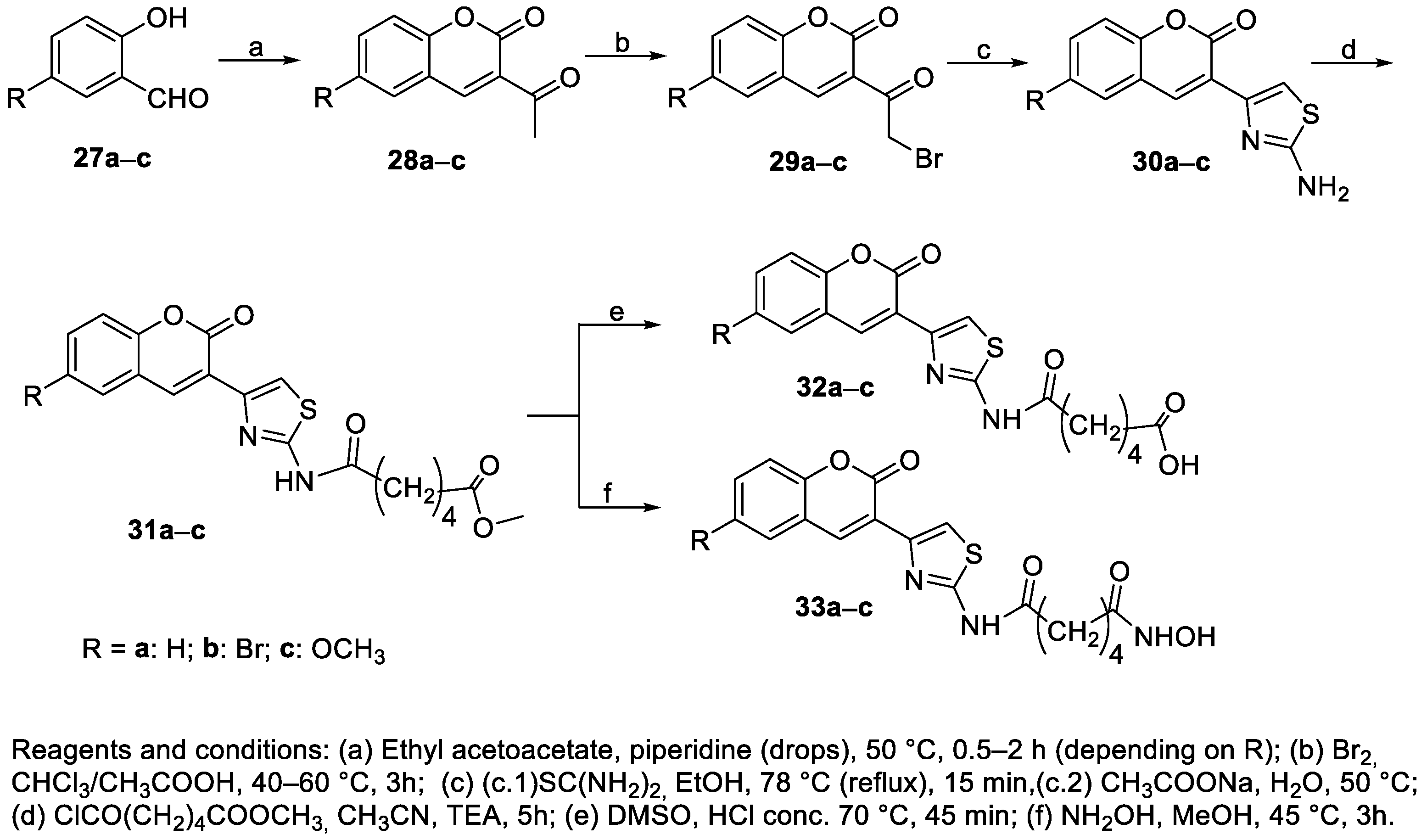
4.5. Thiazolyl-Coumarin Derivatives in the CAP Group
5. Five-Carbon Linker Chain (5-C Spacer)
5.1. 2-Amino-1,3,4-thiadiazoles in the CAP Group
5.2. 4-Anilinothieno [2,3-d]pyrimidine Derivatives in the CAP Group
5.3. Indazole Nucleus in the CAP Group
6. Six-Carbon Linker Chain (6-C Spacer)
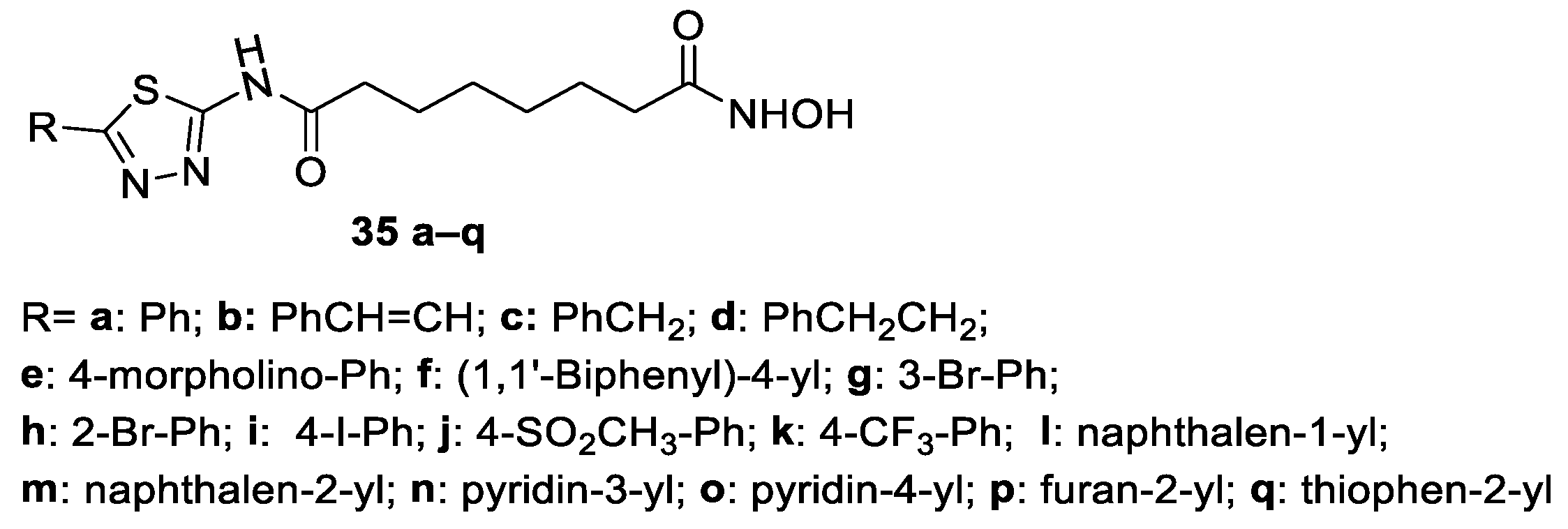
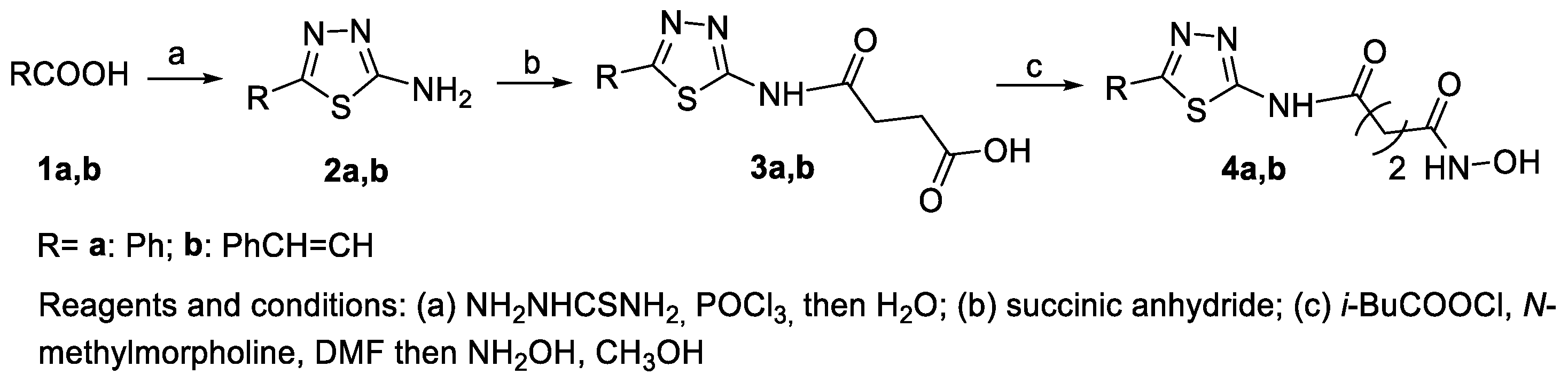
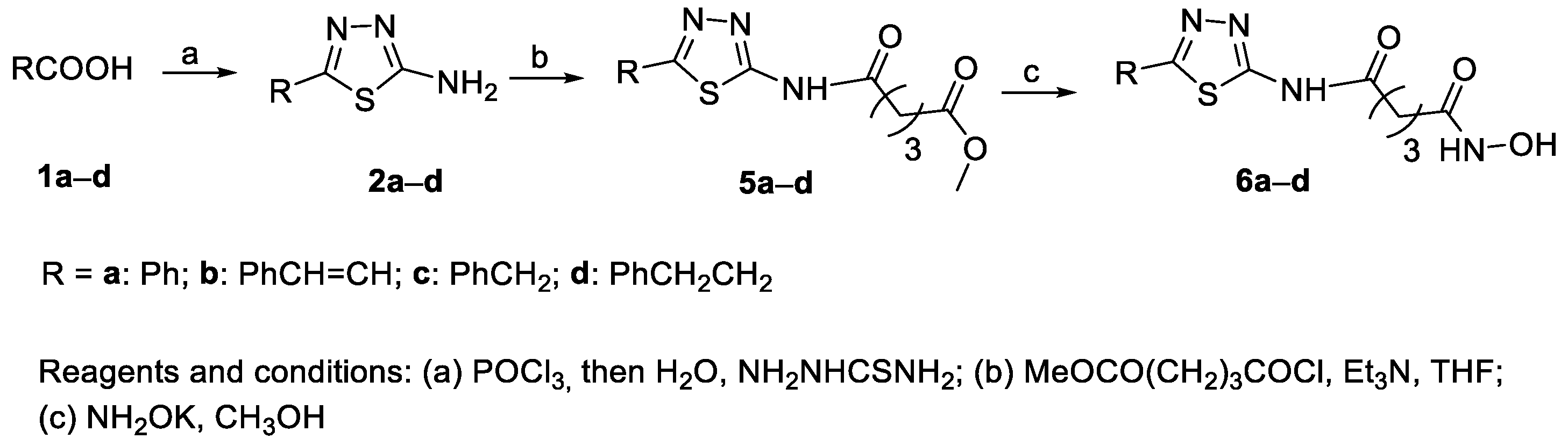
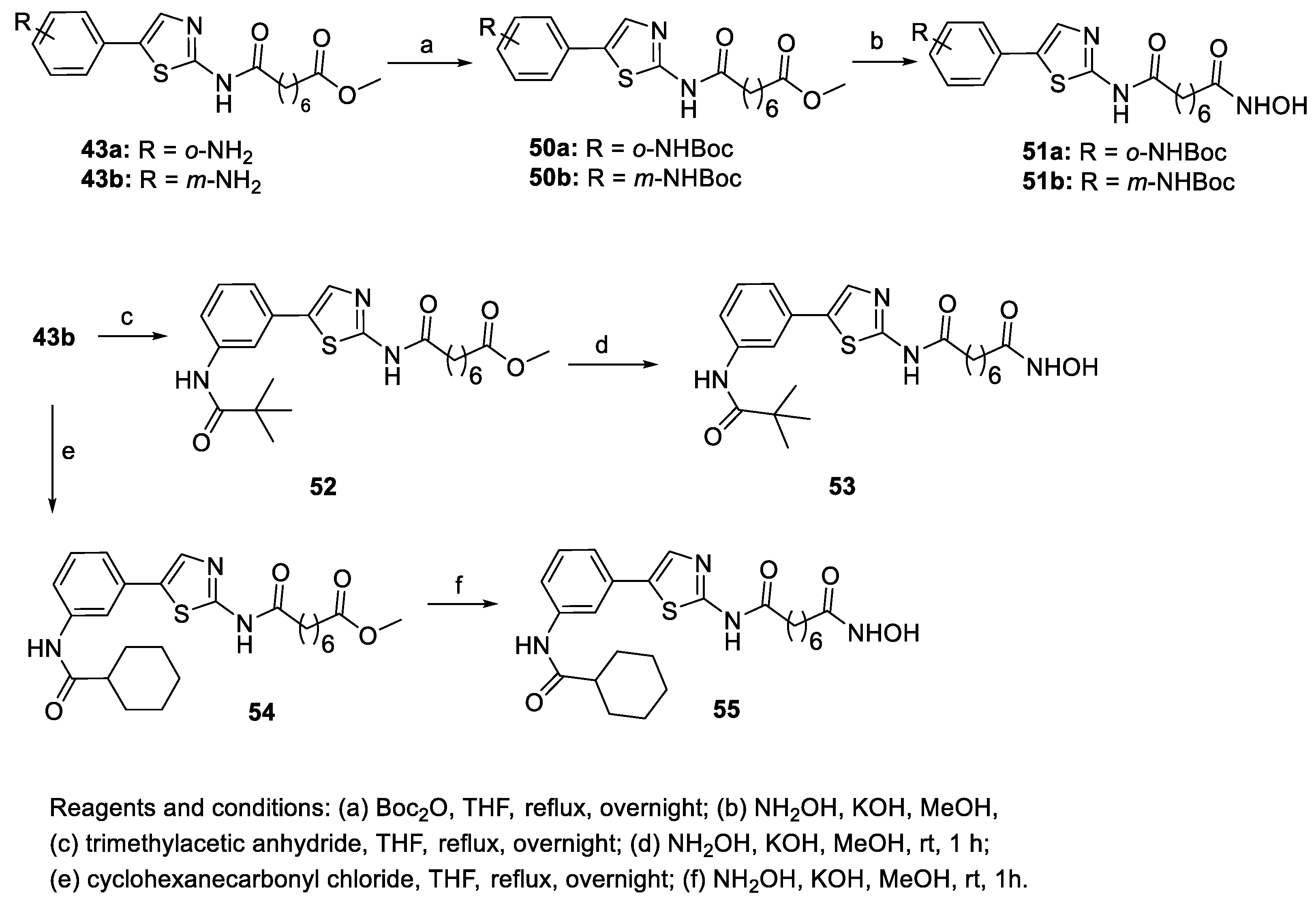
6.1. 2-Amino-1,3,4-thiadiazoles in the CAP Group
6.2. Thiazoles in the CAP Group
6.3. Pyrazole Nucleus in the CAP Group
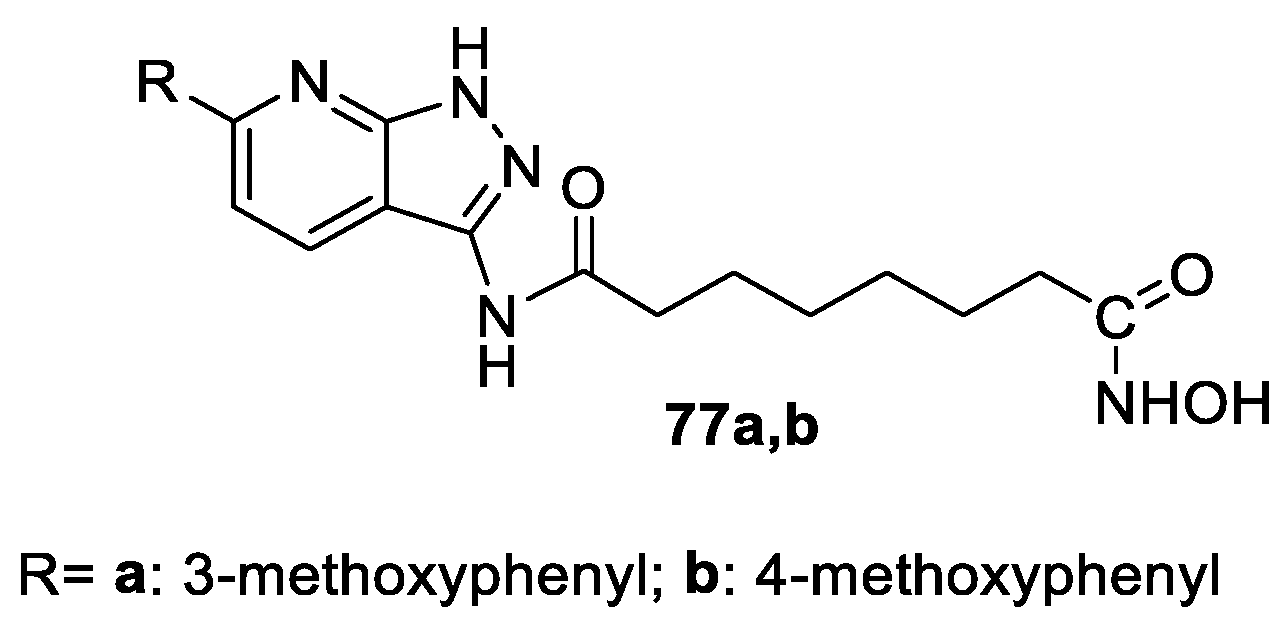
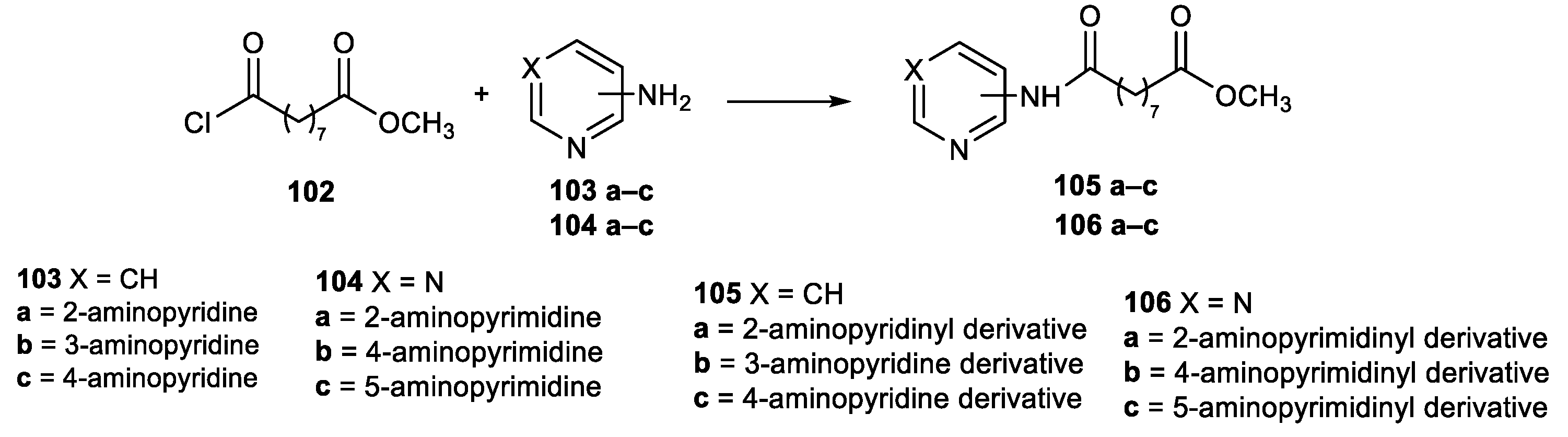
6.4. Pyridine and Pyrimidine Nucleus in the CAP Group
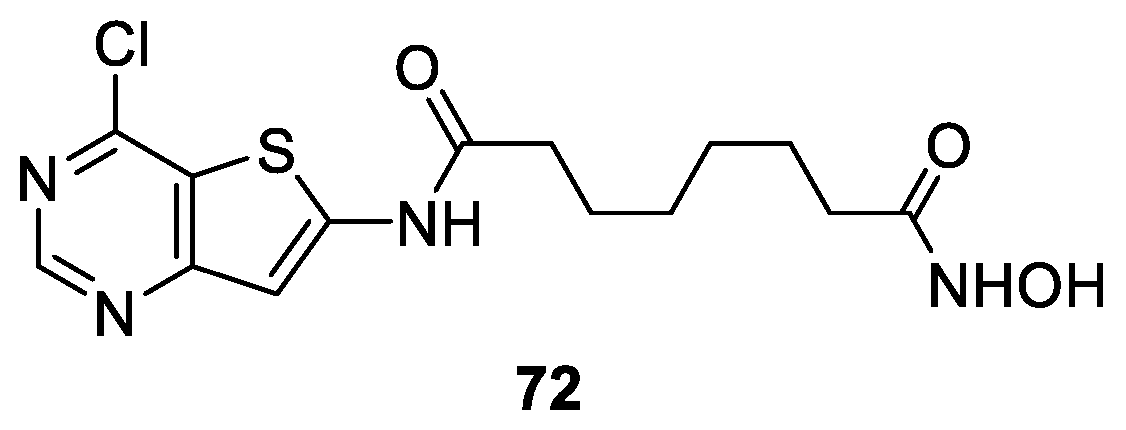
6.5. Thienopyrimidine Nucleus in the CAP Group
6.6. Indazole Nucleus in the CAP Group
6.7. Benzothiazole Moiety in the CAP Group
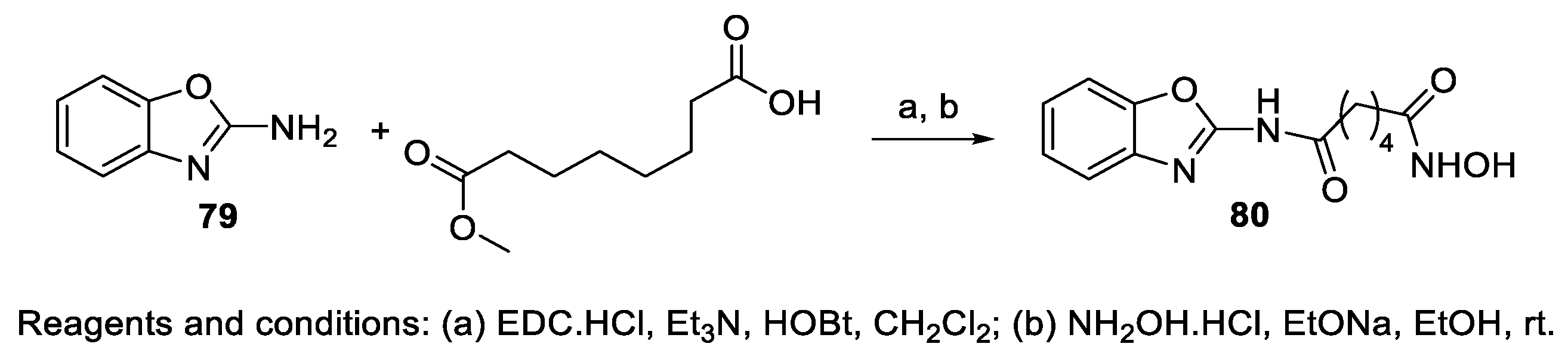
6.8. Benzoxazole Moiety in the CAP Group
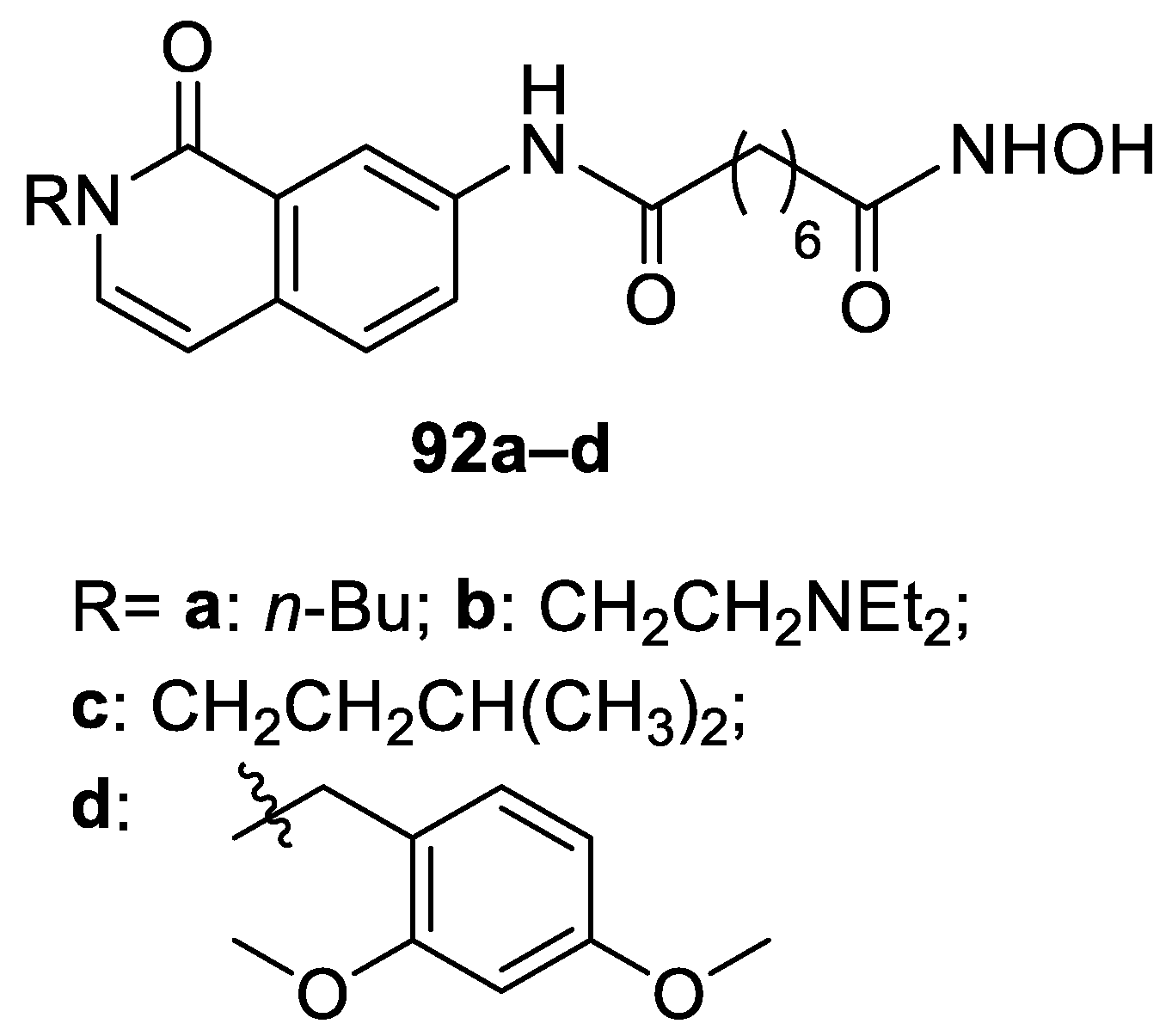
6.9. Isoquinoline Moiety in the CAP Group
6.10. Quinazoline Moiety in the CAP Group
7. Seven-Carbon Linker Chain (7-C Spacer)
Pyridine and Pyrimidine Moiety in the CAP Group
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and Co-regulates major cellular functions. Science 2009, 325, e834–e840. [Google Scholar] [CrossRef] [PubMed]
- Vaijayanthi, T.; Pandian, G.N.; Sugiyama, H. Chemical control system of epigenetics. Chem. Rec. 2018, 18, e1833–e1853. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone Deacetylase Inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659–671. [Google Scholar] [CrossRef]
- Langley, B.; D’Annibale, M.A.; Suh, K.; Ayoub, I.; Tolhurst, A.; Bastan, B.; Yang, L.; Ko, B.; Fisher, M.; Cho, S.; et al. Pulse Inhibition of Histone Deacetylases Induces Complete Resistance to Oxidative Death in Cortical Neurons without Toxicity and Reveals a Role for Cytoplasmic p21waf1/cip1 in Cell Cycle-Independent Neuroprotection. J. Neurosci. 2008, 28, 163–176. [Google Scholar] [CrossRef]
- Sinn, D.I.; Kim, S.J.; Chu, K.; Jung, K.H.; Lee, S.T.; Song, E.C.; Kim, J.M.; Park, D.K.; Kun Lee, S.; Kim, M.; et al. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol. Dis. 2007, 26, 464–472. [Google Scholar] [CrossRef]
- Petri, S.; Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Crow, J.P.; Beal, M.F. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2006, 22, 40–49. [Google Scholar] [CrossRef]
- Hahnen, E.; Hauke, J.; Trankle, C.; Eyupoglu, I.Y.; Wirth, B.; Blumcke, I. Histone deacetylase inhibitors: Possible implications for neurodegenerative disorders. Expert Opin. Investig. Drugs 2008, 17, 169–184. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. Mechanisms and molecular probes of sirtuins. Chem. Biol. 2008, 15, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Bae, S.-C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Matore, B.W.; Banjare, P.; Guria, T.; Roy, P.P.; Singh, J. Oxadiazole derivatives: Histone deacetylase inhibitors in anticancer therapy and drug discovery. Eur. J. Med. Chem. Rep. 2022, 5, 100058. [Google Scholar] [CrossRef]
- Moinul, M.; Amin, S.A.; Khatun, S.; Das, S.; Jha, T.; Gayen, S. A detail survey and analysis of selectivity criteria for indole-based histone deacetylase 8 (HDAC8) inhibitors. J. Mol. Struct. 2023, 1271, 133967. [Google Scholar] [CrossRef]
- Da´sko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef]
- Melesina, J.; Simoben, C.V.; Praetorius, L.; Bülbül, E.F.; Robaa, D.; Sippl, W. Strategies To Design Selective Histone Deacetylase Inhibitors. ChemMedChem 2021, 16, 1336–1359. [Google Scholar] [CrossRef]
- Ru, J.; Wang, Y.; Li, Z.; Wang, J.; Ren, C.; Zhang, J. Technologies of targeting histone deacetylase in drug discovery: Current progress and emerging prospects. Eur. J. Med. Chem. 2023, 261, 115800. [Google Scholar] [CrossRef]
- Jung, M.; Hoffmann, K.; Brosch, G.; Loidl, P. Analogues of trichosTatin a and trapoxin B as histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 1997, 7, 1655–1658. [Google Scholar] [CrossRef]
- Jung, M.; Brosch, G.; Kolle, D.; Scherf, H.; Gerhauser, C.; Loidl, P. Amide Analogues of Trichostatin A as Inhibitors of Histone Deacetylase and Inducers of Terminal Cell Differentiation. J. Med. Chem. 1999, 42, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Guan, P.; Sun, F.; Hou, X.; Wang, F.; Yi, F.; Xu, W.; Fang, H. Design, synthesis and preliminary bioactivity studies of 1,3,4-thiadiazole hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem 2012, 20, 3865. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, J.; He, F.; Gao, L.; Wen, Y.; Gao, L.; Wang, P.; Kang, D.; Hu, L. Design, synthesis and biological evaluation of novel indazole-based derivatives as potent HDAC inhibitors via fragment-based virtual screening. Eur. J. Med. Chem. 2020, 192, 112189. [Google Scholar] [CrossRef] [PubMed]
- Oanh, D.T.K.; Hai, H.V.; Park, S.H.; Kim, H.-J.; Han, B.-W.; Kim, H.-S.; Hong, J.-T.; Han, S.-B.; Hue, V.T.M.; Nam, N.-H. Benzothiazole-containing hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorg. Med. Chem. Lett. 2011, 21, 7509–7512. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, L.; Ji, X.; Wu, X.; Su, M.; Sheng, L.; Zang, Y.; Li, J.; Liu, H. Design, synthesis and biological evaluation of 4-anilinothieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem. 2014, 22, 6146–6155. [Google Scholar] [CrossRef]
- Pardo-Jiménez, V.; Navarrete-Encina, P.; Díaz-Araya, G. Synthesis and Biological Evaluation of Novel Thiazolyl-Coumarin Derivatives as Potent Histone Deacetylase Inhibitors with Antifibrotic Activity. Molecules 2019, 24, 739. [Google Scholar] [CrossRef]
- Guan, P.; Wang, L.; Hou, X.; Wan, Y.; Xu, W.; Tang, W.; Fang, H. Improved antiproliferative activity of 1,3,4-thiadiazole-containing histone deacetylase (HDAC) inhibitors by introduction of the heteroaromatic surface recognition motif. Bioorg. Med. Chem. 2014, 22, 5766–5775. [Google Scholar] [CrossRef]
- Nam, N.-H.; Huong, T.L.; Dung, D.T.M.; Dung, P.T.P.; Oanh, D.T.K.; Park, S.H.; Kim, K.; Han, B.W.; Yun, J.; Kang, J.S.; et al. Synthesis, bioevaluation and docking study of 5-substitutedphenyl-1,3,4-thiadiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. J. Enzym. Inhib. Med. Chem. 2014, 29, 611–618. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.M.; Savoy, D.N.; Billadeau, D.D.; Kim, K.H. Chemistry, Biology, and QSAR Studies of Substituted Biaryl Hydroxamates and Mercaptoacetamides as HDAC Inhibitors—Nanomolar-Potency Inhibitors of Pancreatic Cancer Cell Growth. ChemMedChem 2008, 3, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Brett, C.; Langley, B.C. Functional Differences in Epigenetic Modulators Superiority of Mercaptoacetamide-Based Histone Deacetylase Inhibitors Relative to Hydroxamates in Cortical Neuron Neuroprotection Studies. J. Med. Chem. 2007, 50, 3054–3061. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Velaparthi, S.; Pieffet, G.; Pennington, C.; Mahesh, A.; Holzle, D.L.; Brunsteiner, M.; van Breemen, R.; Blond, S.Y.; Petukhov, P.A. Binding Ensemble Profiling with Photoaffinity Labeling (BEProFL) Approach: Mapping the Binding Poses of HDAC8 Inhibitors. J. Med. Chem. 2009, 52, 7003–7013. [Google Scholar] [CrossRef] [PubMed]
- Glaser, K.B.; Li, J.; Pease, L.J.; Staver, M.J.; Marcotte, P.A.; Guo, J.; Frey, R.R.; Garland, R.B.; Heyman, H.R.; Wada, C.K.; et al. Differential protein acetylation induced by novel histone deacetylase inhibitors. Biochem. Biophys. Res. Commun. 2004, 325, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Neelarapu, R.; Holzle, D.L.; Velaparthi, S.; Bai, H.; Brunsteiner, M.; Blond, S.Y.; Petukhov, P.A. Design, Synthesis, Docking, and Biological Evaluation of Novel Diazide-Containing Isoxazole-and Pyrazole-Based Histone Deacetylase Probes. J. Med. Chem. 2011, 54, 4350–4364. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.S.; Neelarapu, R.; Madriaga, A.; Bai, H.; Mendonca, E.; Abdelkarim, H.; van Breemen, R.B.; Blond, S.Y.; Petukhov, P.A. Novel histone deacetylase 8 ligands without a zinc chelating group: Exploring an ‘upside-down’ binding pose. Bioorg. Med. Chem. Lett. 2012, 22, 6621–6627. [Google Scholar] [CrossRef]
- Albrow, V.E.; Grimley, R.L.; Clulow, J.; Rose, C.R.; Sun, J.; Warmus, J.S.; Tate, E.W.; Jonesd, L.H.; Storer, R.I. Design and development of histone deacetylase (HDAC) chemical probes for cell-based profiling. Mol. BioSyst. 2016, 12, 1781–1789. [Google Scholar] [CrossRef]
- Li, Y.; Luo, X.; Guo, Q.; Nie, Y.; Wang, T.; Zhang, C.; Huang, Z.; Wang, X.; Liu, Y.; Chen, Y.; et al. Discovery of N1-(4-((7-Cyclopentyl-6-(dimethylcarbamoyl)-7 H-pyrrolo[2,3- d]pyrimidin-2-yl)amino)phenyl)- N8-hydroxyoctanediamide as a Novel Inhibitor Targeting Cyclin-dependent Kinase 4/9 (CDK4/9) and Histone Deacetlyase1 (HDAC1) against Malignant Cancer. J. Med. Chem. 2018, 61, 3166–3192. [Google Scholar] [CrossRef]
- Remiszewski, S.W.; Sambucetti, L.C.; Atadja, P.; Bair, K.W.; Cornell, W.D.; Green, M.A.; Howell, K.L.; Jung, M.; Kwon, P.; Trogani, N.; et al. Inhibitors of Human Histone Deacetylase: Synthesis and Enzyme and Cellular Activity of Straight Chain Hydroxamates. J. Med. Chem. 2002, 45, 753–757. [Google Scholar] [CrossRef]
- Wang, J.; Su, M.; Li, T.; Gao, A.; Yang, W.; Sheng, L.; Zang, Y.; Li, J.; Liu, H. Design, synthesis and biological evaluation of thienopyrimidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. Eur. J. Med. Chem. 2017, 128, 293–299. [Google Scholar] [CrossRef]
- Mantzourani, C.; Gkikas, D.; Kokotos, A.; Nummela, P.; Theodoropoulou, M.A.; Wu, K.-C.; Fairlie, D.P.; Politis, P.K.; Ristimäki, A.; Kokotos, G. Synthesis of benzoxazole-based vorinostat analogs and their antiproliferative activity. Bioorg. Chem. 2021, 114, 105132. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, L.; Wang, Y.; Wu, X.; Li, T.; Yang, N.; Su, M.; Sheng, L.; Zheng, M.; Zang, Y.; et al. Design, synthesis and biological evaluation of isoquinoline-based derivatives as novel histone deacetylase inhibitors. Bioorg. Med. Chem. 2015, 23, 5881–5890. [Google Scholar] [CrossRef] [PubMed]
- Salmi-Smail, C.; Fabre, A.; Dequiedt, F.; Restouin, A.; Castellano, R.; Garbit, S.; Roche, P.; Morelli, X.; Brunel, J.M.; Collette, Y. Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J. Med. Chem. 2010, 53, 3038–3047. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Boga, C.; Micheletti, G. Design and Synthesis of Organic Molecules as Antineoplastic Agents. Molecules 2020, 25, 2808. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
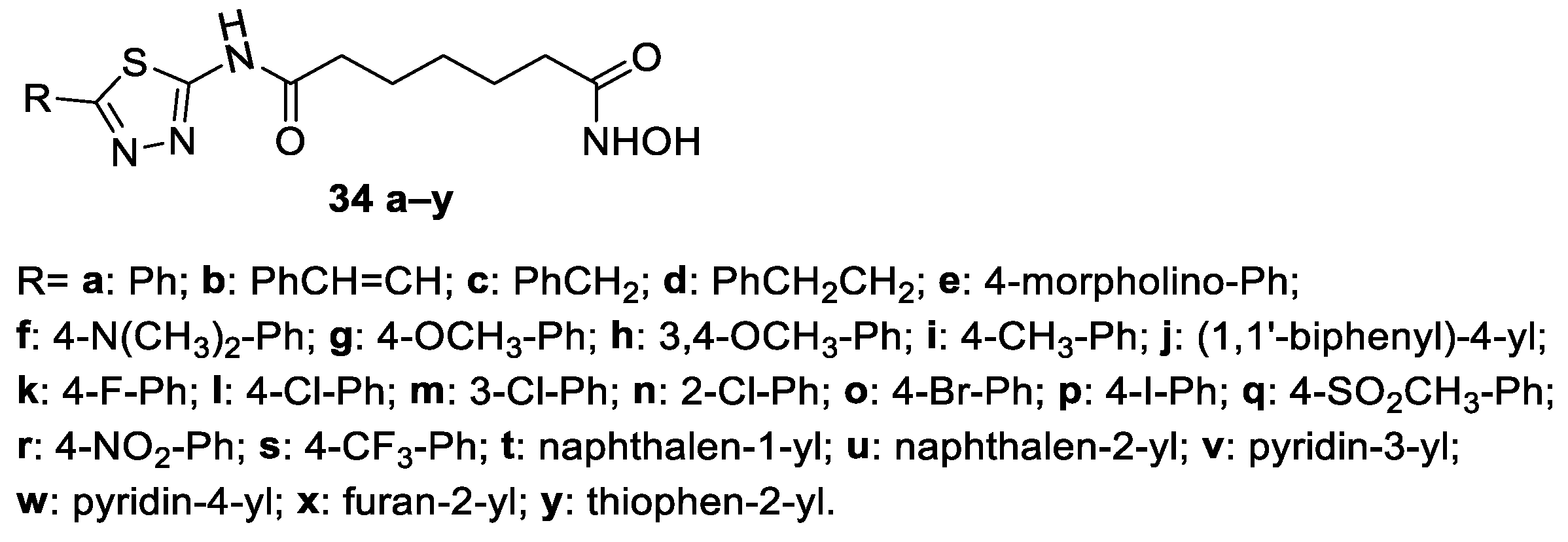{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
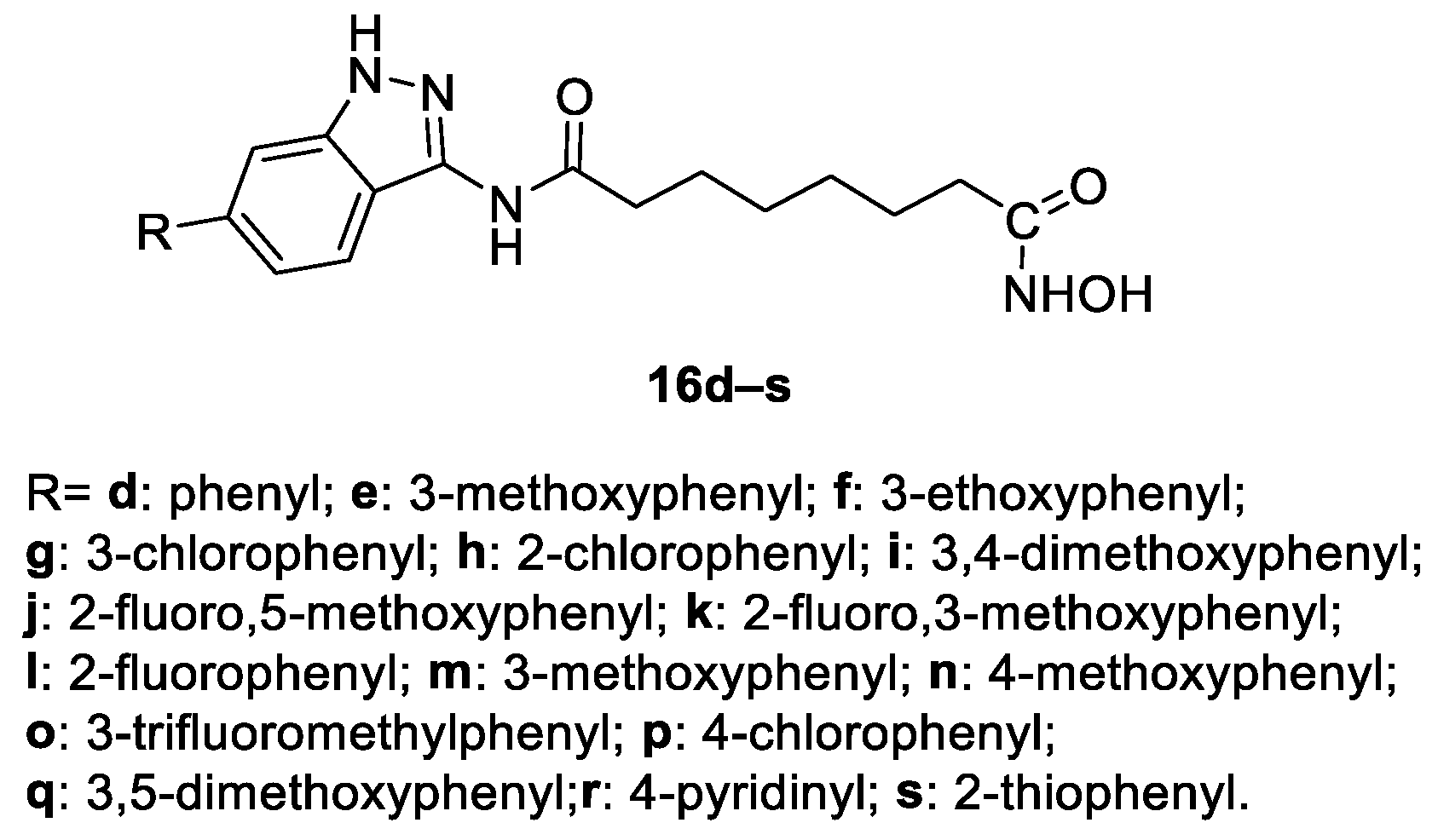{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HDAC IC50 (μM) |
|---|---|
| 6a | >5 |
| 6b | 0.16 ± 0.03 |
| 6c | 1.87 ± 0.41 |
| 6d | 2.71 ± 0.25 |
| SAHA | 0.15 ± 0.02 |
| Compound | HDAC IC50 (μM) |
|---|---|
| 17a | 1.03 ± 0.04 |
| 17b | 1.70 ± 0.40 |
| 17c | 1.12 ± 0.01 |
| 17d | 3.49 ± 0.04 |
| SAHA | 0.15 ± 0.02 |
| Compound | HDAC IC50 (μM) |
|---|---|
| 34a | 0.089 ± 0.005 |
| 34b | >5 |
| 34c | 0.22 ± 0.04 |
| 34d | 0.33 ± 0.05 |
| SAHA | 0.15 ± 0.02 |
| Compound | R | HDAC1 IC50 (nM) | HDAC3 IC50 (nM) | HDAC6 IC50 (nM) |
|---|---|---|---|---|
| 26c | 3-Cl, 4-F | 35.89 ± 16.34 | 37.67 ± 1.61 | 23.99 ± 0.72 |
| 26d | 3-CF3, 4-Cl | 40.84 ± 8.23 | 48.26 ± 1.78 | 30.00 ± 1.14 |
| 26e | H | 11.77 ± 0.50 | 20.77 ± 0.64 | 26.99 ± 4.95 |
| 26f | 4-CH3 | 14.01 ± 1.32 | 9.33 ± 0.10 | 19.68 ± 1.96 |
| 26g | 3-CH3, 4-CH3 | 29.82 ± 11.51 | 14.74 ± 0.03 | 16.87 ± 3.02 |
| SAHA | 93.34 ± 2.78 | 158.17 ± 6.66 | 78.98 ± 13.19 |
| Compound | HDAC IC50 (μM) |
|---|---|
| 35a | 0.27 ± 0.004 |
| 34b | 3.21 ± 0.10 |
| 34c | 0.26 ± 0.05 |
| 34d | 0.32 ± 0.05 |
| SAHA | 0.15 ± 0.02 |
| Compound | HDAC3 IC50 ± SD (nM) | HDAC8 IC50 ± SD (nM) |
|---|---|---|
| SAHA | 27 ± 1.0 | 440 ± 21 |
| 61 | 128 ± 9.8 | 17 ± 3 |
| 67 | 432 ± 52 | 487 ± 80 |
| 68a | 44 ± 5.8 | 76 ± 5.0 |
| 68b | 59 ± 1.0 | 82 ± 9.0 |
| 68c | 22 ± 1.3 | 28 ± 3.0 |
| 68d | 191 ± 18 | 147 ± 15 |
| Comp. | Alkyl Chain Linker n | HDAC1 IC50 (nM) | HDAC2 IC50 (nM) | HDAC8 IC50 (nM) | HCT-116 IC50 (μM) | MCF-7 IC50 (μM) | HeLa IC50 (μM) |
|---|---|---|---|---|---|---|---|
| SAHA | 13 | 70 | 44 | ||||
| 16a | n = 3 | 76 | 168 | 54 | >50 | 41.5 | >50 |
| 16b | n = 4 | 13 | 62 | 41 | 23.5 | 4.4 | 5.8 |
| 16c | n = 5 | 2.6 | 6.3 | 4.5 | 10.6 | 7.4 | 20.1 |
| 16d | n = 6 | 1.9 | 3.9 | 3.0 | 4.9 | 0.8 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheletti, G.; Boga, C.; Drius, G.; Bordoni, S.; Calonghi, N. Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase. Molecules 2024, 29, 238. https://doi.org/10.3390/molecules29010238
Micheletti G, Boga C, Drius G, Bordoni S, Calonghi N. Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase. Molecules. 2024; 29(1):238. https://doi.org/10.3390/molecules29010238
Chicago/Turabian StyleMicheletti, Gabriele, Carla Boga, Giacomo Drius, Silvia Bordoni, and Natalia Calonghi. 2024. "Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase" Molecules 29, no. 1: 238. https://doi.org/10.3390/molecules29010238
APA StyleMicheletti, G., Boga, C., Drius, G., Bordoni, S., & Calonghi, N. (2024). Suberoylanilide Hydroxamic Acid Analogs with Heteroaryl Amide Group and Different Chain Length: Synthesis and Effect on Histone Deacetylase. Molecules, 29(1), 238. https://doi.org/10.3390/molecules29010238