
Identification of New Substrates and Inhibitors of Human CYP2A7

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Cloning of a Fission Yeast Expression Strain for CYP2A7-WT
2.2. Activities of CYP2A6*1, CYP2A7*1 and CYP2A7-WT towards Different Luminogenic Substrates
2.3. Insights from Molecular Modeling into Differences between CYP2A7-WT and CYP2A7*1
2.4. Lack of CYP2A7 Activity towards Three Known CYP2A6 Substrates
2.5. Metabolism of Diclofenac by CYP2A6 and CYP2A7
2.6. Identification of CYP2A7 Inhibitors
3. Materials and Methods
3.1. Materials
3.2. Fission Yeast Media and General Techniques
3.3. Construction of Expression Plasmids and Fission Yeast Strains
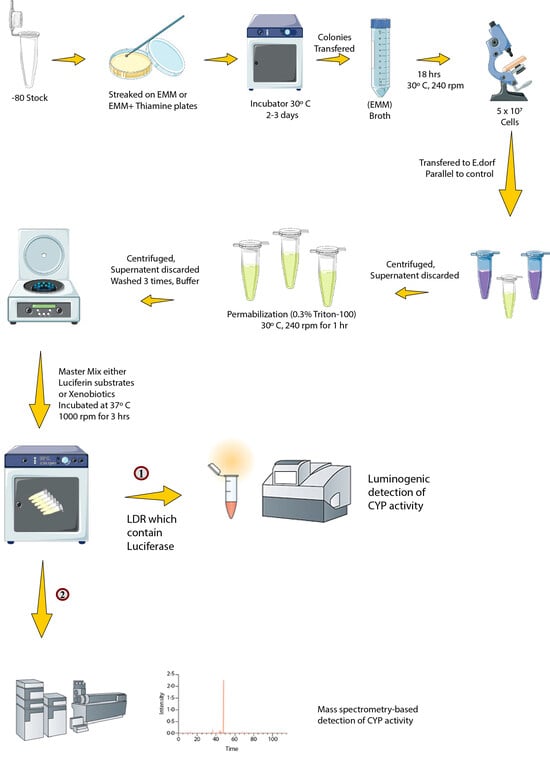
3.4. Biotransformation with Enzyme Bags
3.5. Bioluminescence Detection
3.6. Fluorescence Detection of Coumarin 7-Hydroxylation and Deethylation of 7-Ethoxycoumarin
3.7. Analysis of Nicotine Metabolites
3.8. Analysis of Hydroxydiclofenac on HPLC/microTOF-QII
3.9. Analysis of Hydroxydiclofenac on HPLC/micro-QE
3.10. Statistical Analysis
3.11. Protein Modeling and Molecular Dynamics (MD) Simulations
3.12. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bernhardt, R.; Urlacher, V.B. Cytochromes P450 as promising catalysts for biotechnological application: Chances and limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [Google Scholar] [CrossRef] [PubMed]
- Durairaj, P.; Fan, L.; Du, W.; Ahmad, S.; Mebrahtu, D.; Sharma, S.; Ashraf, R.A.; Liu, J.; Liu, Q.; Bureik, M. Functional expression and activity screening of all human cytochrome P450 enzymes in fission yeast. FEBS Lett. 2019, 593, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Machalz, D.; Pach, S.; Bermudez, M.; Bureik, M.; Wolber, G. Structural insights into understudied human cytochrome P450 enzymes. Drug Discov. Today 2021, 26, 2456–2464. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Walsky, R.L.; Obach, R.S. Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Weldemichael, D.M.; Zhou, K.; Su, S.-j.; Zhao, L.; Marchisio, M.A.; Bureik, M. Futile cycling by human microsomal cytochrome P450 enzymes within intact fission yeast cells. Arch. Biochem. Biophys. 2021, 701, 108791. [Google Scholar] [CrossRef] [PubMed]
- Lauschke, V.M.; Zhou, Y.; Ingelman-Sundberg, M. Pharmacogenomics Beyond Single Common Genetic Variants: The Way Forward. Annu. Rev. Pharmacol. Toxicol. 2024, 64, 33–51. [Google Scholar] [CrossRef]
- Cao, X.; Durairaj, P.; Yang, F.; Bureik, M. A comprehensive overview of common polymorphic variants that cause missense mutations in human CYPs and UGTs. Biomed. Pharmacother. 2019, 111, 983–992. [Google Scholar] [CrossRef]
- Cali, J.J.; Ma, D.; Sobol, M.; Simpson, D.J.; Frackman, S.; Good, T.D.; Daily, W.J.; Liu, D. Luminogenic cytochrome P450 assays. Expert Opin. Drug Metab. Toxicol. 2006, 2, 629–645. [Google Scholar] [CrossRef]
- Sharma, S.; Liu, J.; Zhang, X.; Sharma, S.S.; Sorensen, E.J.; Bureik, M. New luciferin-based probe substrates for human CYP26A1. Biochem. Biophys. Rep. 2020, 24, 100861. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Machalz, D.; Wolber, G.; Sorensen, E.J.; Bureik, M. New proluciferin substrates for human CYP4 family enzymes. Appl. Biochem. Biotechnol. 2021, 193, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, X.; Wang, Y.; Huang, H.; Sharma, S.; Sharma, S.S.; Wolf, C.A.; Liu, S.; Wolber, G.; Sorensen, E.J.; et al. Exploring the Chemical Space of Proluciferins as Probe Substrates for Human Cytochrome P450 Enzymes. Appl. Biochem. Biotechnol. 2023, 195, 1042–1058. [Google Scholar] [CrossRef]
- Cali, J.J.; Ma, D.; Wood, M.G.; Meisenheimer, P.L.; Klaubert, D.H. Bioluminescent assays for ADME evaluation: Dialing in CYP selectivity with luminogenic substrates. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1115–1130. [Google Scholar] [CrossRef]
- Hritz, J.; de Ruiter, A.; Oostenbrink, C. Impact of plasticity and flexibility on docking results for cytochrome P450 2D6: A combined approach of molecular dynamics and ligand docking. J. Med. Chem. 2008, 51, 7469–7477. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: Their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Sørensen, J.; Hensley, N.; Wong, C.; Zhu, C.; Perison, T.; Amaro, R.E. POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput. 2017, 13, 4584–4592. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yamagishi, S.; Yamamoto, H.; Yamamoto, T.; Kuroiwa, Y.; Yokoi, T. Deficient cotinine formation from nicotine is attributed to the whole deletion of the CYP2A6 gene in humans. Clin. Pharmacol. Ther. 2000, 67, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Uno, T.; Obe, Y.; Ogura, C.; Goto, T.; Yamamoto, K.; Nakamura, M.; Kanamaru, K.; Yamagata, H.; Imaishi, H. Metabolism of 7-ethoxycoumarin, safrole, flavanone and hydroxyflavanone by cytochrome P450 2A6 variants. Biopharm. Drug Dispos. 2013, 34, 87–97. [Google Scholar] [CrossRef]
- Loose, D.S.; Kan, P.B.; Hirst, M.A.; Marcus, R.A.; Feldman, D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J. Clin. Investig. 1983, 71, 1495–1499. [Google Scholar] [CrossRef]
- Yan, Q.; Machalz, D.; Zollner, A.; Sorensen, E.J.; Wolber, G.; Bureik, M. Efficient substrate screening and inhibitor testing of human CYP4Z1 using permeabilized recombinant fission yeast. Biochem. Pharmacol. 2017, 146, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, A.S.; Hausler, A.; Schieweck, K.; Lang, M.; Bowman, R. Highly selective inhibition of estrogen biosynthesis by CGS 20267, a new non-steroidal aromatase inhibitor. J Steroid Biochem. Mol. Biol. 1990, 37, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Lacy, C.F.; Armstrong, L.L.; Goldman, M.P.; Lance, L.L. Cytochrome P450 Enzymes: Substrates, Inhibitors, and Inducers; LexiComp Inc.: Hudson, OH, USA, 2007. [Google Scholar]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Alfa, C.; Cold Spring Harbor Laboratory. Experiments with Fission Yeast: A Laboratory Course Manual; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1993; p. ix. 186p. [Google Scholar]
- Maundrell, K. Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 1993, 123, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Drăgan, C.A.; Peters, F.T.; Bour, P.; Schwaninger, A.E.; Schaan, S.M.; Neunzig, I.; Widjaja, M.; Zapp, J.; Kraemer, T.; Maurer, H.H.; et al. Convenient gram-scale metabolite synthesis by engineered fission yeast strains expressing functional human P450 systems. Appl. Biochem. Biotechnol. 2011, 163, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Maundrell, K. nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem. 1990, 265, 10857–10864. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.K.; Denton, T.T.; Cerny, M.A.; Zhang, X.; Johnson, E.F.; Cashman, J.R. Synthetic inhibitors of cytochrome P-450 2A6: Inhibitory activity, difference spectra, mechanism of inhibition, and protein cocrystallization. J. Med. Chem. 2006, 49, 6987–7001. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins Struct. Funct. Bioinform. 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossváry, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference SNP a | Minor Allele Frequency | Residue Change b |
|---|---|---|
| rs3869579 | 0.47770050 | Cys311Arg |
| rs4142867 | 0.47727665 | Glu169Asp |
| rs12460590 | 0.33779851 | Gly479Val |
| rs4079366 | 0.24078928 | Arg274His |
| Enzyme | Luminescence (RLU) | ||||||
|---|---|---|---|---|---|---|---|
| Luciferin-H | Luciferin-ME | Luciferin-1A2 | Luciferin-BE | Luciferin-2FBE | Luciferin-3FBE | Luciferin-4FBE | |
| CYP2A7*1 | 318 ± 60 | 276 ± 63 | 79 ± 11 | 16,216 ± 369 | 17,900 ± 1470 | 22,110 ± 1060 | 29,790 ± 8760 |
| CYP2A7-WT | 263 ± 72 | 439 ± 113 ** | 63 ± 67 | 6312 ± 699 * | 18,610 ± 3160 | 11,720 ± 3560 **** | 29,580 ± 5550 |
| Compound | Structure | % Inhibition of CYP2A6*1 | % Inhibition of CYP2A7*1 | % Inhibition of CYP2A7-WT |
|---|---|---|---|---|
| Ketoconazole |  | 70 ± 11 **** | 74 ± 4 **** | 58 ± 17 **** |
| 1-Benzyl-imidazole |  | 64 ± 12 **** | 55 ± 11 **** | 80 ± 21 **** |
| Letrozole |  | 64 ± 10 **** | 78 ± 7 **** | 72 ± 13 **** |
| Strain | Expressed Protein(s) | Parental Strain | Genotype | References |
|---|---|---|---|---|
| CAD62 | CPR | NCYC2036 | h- ura4-D18 leu1::pCAD1-CPR | [27] |
| RAJ122 | CPR, CYP2A6*1 | CAD62 | h- ura4-D18 leu1::pCAD1-CPR/ pREP1-CYP2A6*1 | [2] |
| RAJ127 | CPR, CYP2A7*1 | CAD62 | h- ura4-D18 leu1::pCAD1-CPR/ pREP1-CYP2A7*1 | [2] |
| AA01 | CPR, CYP2A7-WT | CAD62 | h- ura4-D18 leu1::pCAD1-CPR/ pREP1-CYP2A7-WT | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, R.A.; Liu, S.; Wolf, C.A.; Wolber, G.; Bureik, M. Identification of New Substrates and Inhibitors of Human CYP2A7. Molecules 2024, 29, 2191. https://doi.org/10.3390/molecules29102191
Ashraf RA, Liu S, Wolf CA, Wolber G, Bureik M. Identification of New Substrates and Inhibitors of Human CYP2A7. Molecules. 2024; 29(10):2191. https://doi.org/10.3390/molecules29102191
Chicago/Turabian StyleAshraf, Rana Azeem, Sijie Liu, Clemens Alexander Wolf, Gerhard Wolber, and Matthias Bureik. 2024. "Identification of New Substrates and Inhibitors of Human CYP2A7" Molecules 29, no. 10: 2191. https://doi.org/10.3390/molecules29102191
APA StyleAshraf, R. A., Liu, S., Wolf, C. A., Wolber, G., & Bureik, M. (2024). Identification of New Substrates and Inhibitors of Human CYP2A7. Molecules, 29(10), 2191. https://doi.org/10.3390/molecules29102191