
A Combined Molecular Dynamics and Hydropathic INTeraction (HINT) Approach to Investigate Protein Flexibility: The PPARγ Case Study


Abstract
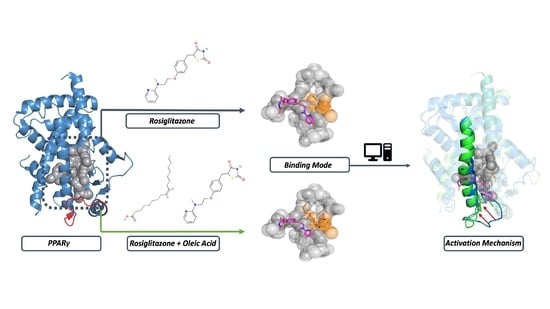
:
1. Introduction
2. Results and Discussion
2.1. Active Site Analysis
2.2. PPARγ Ligand Binding Mode
2.3. Molecular Dynamics
2.4. HINT Based Analysis
3. Materials and Methods
3.1. PDB Structure Analysis
3.2. Molecular Dynamics
3.3. HINT Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gund, P.; Halgren, T.A.; Smith, G.M. Chapter 27 Molecular Modeling as an Aid to Drug Design and Discovery. In Annual Reports in Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 1987; pp. 269–279. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Ahmad, S.; Tyagi, R.; Dahiya, V.; Yadav, M.K. Fundamentals of molecular modeling in drug design. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 125–155. [Google Scholar] [CrossRef]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef] [PubMed]
- van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. 1990, 29, 992–1023. [Google Scholar] [CrossRef]
- Ramanathan, A.; Savol, A.; Burger, V.; Chennubhotla, C.S.; Agarwal, P.K. Protein Conformational Populations and Functionally Relevant Substates. Accounts Chem. Res. 2013, 47, 149–156. [Google Scholar] [CrossRef]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [PubMed]
- Buldyrev, S.V. Application of Discrete Molecular Dynamics to Protein Folding and Aggregation. In Aspects of Physical Biology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 97–131. [Google Scholar] [CrossRef]
- Spyrakis, F.; BidonChanal, A.; Barril, X.; Luque, F.J. Protein Flexibility and Ligand Recognition: Challenges for Molecular Modeling. Curr. Top. Med. Chem. 2011, 11, 192–210. [Google Scholar] [CrossRef]
- Kellogg, G.E.; Semus, S.F.; Abraham, D.J. HINT: A new method of empirical hydrophobic field calculation for CoMFA. J. Comput. Mol. Des. 1991, 5, 545–552. [Google Scholar] [CrossRef]
- Agosta, F.; Kellogg, G.E.; Cozzini, P. From oncoproteins to spike proteins: The evaluation of intramolecular stability using hydropathic force field. J. Comput. Mol. Des. 2022, 36, 797–804. [Google Scholar] [CrossRef]
- Kellogg, G.E.; Abraham, D.J. Hydrophobicity: Is LogPo/w more than the sum of its parts? Eur. J. Med. Chem. 2000, 35, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Kellogg, G.E. Hydrophobicity-Shake Flasks, Protein Folding and Drug Discovery. Curr. Top. Med. Chem. 2010, 10, 67–83. [Google Scholar] [CrossRef]
- Cozzini, P.; Agosta, F.; Dolcetti, G.; Palù, A.D. A Computational Workflow to Predict Biological Target Mutations: The Spike Glycoprotein Case Study. Molecules 2023, 28, 7082. [Google Scholar] [CrossRef] [PubMed]
- Agosta, F.; Cozzini, P. Hint approach on Transthyretin folding/unfolding mechanism comprehension. Comput. Biol. Med. 2023, 155, 106667. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Bernal-Mizrachi, C.; Han, D.H.; Coleman, T.; Sambandam, N.; LaRiviere, L.L.; Holloszy, J.O.; Semenkovich, C.F.; Kelly, D.P. A potential link between muscle peroxisome proliferator- activated receptor-α signaling and obesity-related diabetes. Cell Metab. 2005, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative Analysis in Adult Rat Tissues and Regulation in Fasting and Refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Kamisaki, Y. Anti-inflammatory effect of PPARgamma agonists: Basics and clinical applications. Nihon Rinsho 2010, 68, 278–283. [Google Scholar]
- Imamura, K.; Lu, J.; Kaminishi, M. Roles of PPARs in carcinogenesis. Nihon Rinsho 2005, 63, 584–588. [Google Scholar] [PubMed]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular Recognition of Fatty Acids by Peroxisome Proliferator–Activated Receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Thorp, J.M.; Waring, W.S. Modification of Metabolism and Distribution of Lipids by Ethyl Chlorophenoxyisobutyrate. Nature 1962, 194, 948–949. [Google Scholar] [CrossRef]
- Stumvoll, M.; Häring, H.-U. Glitazones: Clinical effects and molecular mechanisms. Ann. Med. 2002, 34, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.S.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.-L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; Kojetin, D.J. An alternate binding site for PPARγ ligands. Nat. Commun. 2014, 5, 3571. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Brust, R.; Mosure, S.A.; Bass, J.; Munoz-Tello, P.; Lin, H.; Hughes, T.S.; Tang, M.; Ge, Q.; Kamenekca, T.M.; et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. eLife 2018, 7, e43320. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.P.; Sharma, R.; Jain, M.; Bandyopadhyay, D. Syntheses, biological evaluation of some novel substituted benzoic acid derivatives bearing hydrazone as linker. Res. Chem. Intermed. 2021, 47, 5061–5078. [Google Scholar] [CrossRef]
- Mishra, G.P.; Sharma, R. Identification of Potential PPAR γ Agonists as Hypoglycemic Agents: Molecular Docking Approach. Interdiscip. Sci. Comput. Life Sci. 2015, 8, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.; Henry, R.R. Thiazolidinedione safety. Expert Opin. Drug Saf. 2012, 11, 565–579. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-S.; Kim, E.S.; Koh, M.; Lee, S.-J.; Lim, D.; Yang, Y.R.; Jang, H.-J.; Seo, K.-A.; Min, S.-H.; Lee, I.H.; et al. A Novel Non-agonist Peroxisome Proliferator-activated Receptor γ (PPARγ) Ligand UHC1 Blocks PPARγ Phosphorylation by Cyclin-dependent Kinase 5 (CDK5) and Improves Insulin Sensitivity. J. Biol. Chem. 2014, 289, 26618–26629. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef]
- Bae, H.; Jang, J.Y.; Choi, S.-S.; Lee, J.-J.; Kim, H.; Jo, A.; Lee, K.-J.; Choi, J.H.; Suh, S.W.; Park, S.B. Mechanistic elucidation guided by covalent inhibitors for the development of anti-diabetic PPARγ ligands. Chem. Sci. 2016, 7, 5523–5529. [Google Scholar] [CrossRef]
- Jang, J.Y.; Koh, M.; Bae, H.; An, D.R.; Im, H.N.; Kim, H.S.; Yoon, J.Y.; Yoon, H.-J.; Han, B.W.; Park, S.B.; et al. Structural basis for differential activities of enantiomeric PPARγ agonists: Binding of S35 to the alternate site. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2017, 1865, 674–681. [Google Scholar] [CrossRef]
- Cozzini, P.; Fornabaio, M.; Marabotti, A.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Simple, Intuitive Calculations of Free Energy of Binding for Protein−Ligand Complexes. 1. Models without Explicit Constrained Water. J. Med. Chem. 2002, 45, 2469–2483. [Google Scholar] [CrossRef] [PubMed]
- Spyrakis, F.; Cozzini, P.; Bertoli, C.; Marabotti, A.; Kellogg, G.E.; Mozzarelli, A. Energetics of the protein-DNA-water interaction. BMC Struct. Biol. 2007, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Montanari, R.; Capelli, D.; Yamamoto, K.; Awaishima, H.; Nishikata, K.; Barendregt, A.; Heck, A.J.R.; Loiodice, F.; Altieri, F.; Paiardini, A.; et al. Insights into PPARγ Phosphorylation and Its Inhibition Mechanism. J. Med. Chem. 2020, 63, 4811–4823. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version~1.8; Schrödinger LLC.: New York, NY, USA, 2015. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. AmberTools. J. Chem. Inf. Model. 2023, 63, 6183–6191. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Kräutler, V.; van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Score |
|---|---|
| Volume | 1413.471 |
| Total SASA | 261.437 |
| Polar SASA | 96.962 |
| Apolar SASA | 164.475 |
| Hydrophobicity score | 48.681 |
| Polarity score | 17 |
| Charge score | 2 |
| Flexibility score | 0.431 |
| System | Total HINT Score (Average) | Hydrogen Bond (Average) | Electrostatic (Average) | Hydrophobic (Average) |
|---|---|---|---|---|
| PPARγ–Rosiglitazone (Run1) | 5.02 × 104 ±2.36 × 103 | 5.27 × 104 ±1.53 × 103 | 3.01 × 104 ±2.11 × 103 | 2.74 × 104 ±9.59 × 102 |
| PPARγ–Rosiglitazone (Run2) | 4.96 × 104 ±2.33 × 103 | 5.12 × 104 ±1.92 × 103 | 3.14 × 104 ±2.04 × 103 | 2.68 × 104 ±5.73 × 102 |
| PPARγ–Rosiglitazone (Run3) | 4.94 × 104 ±2.44 × 103 | 5.27 × 104 ±1.59 × 103 | 2.99 × 104 ±2.20 × 103 | 2.71 × 104 ±5.09 × 102 |
| PPARγ–Rosiglitazone–Oleic Acid (Run1) | 5.03 × 104 ±1.83 × 103 | 5.37 × 104 ±1.30 × 103 | 2.91 × 104 ±1.73 × 103 | 2.64 × 104 ±5.49 × 102 |
| PPARγ–Rosiglitazone–Oleic Acid (Run2) | 5.05 × 104 ±2.01 × 103 | 5.43 × 104 ±1.54 × 103 | 3.02 × 104 ±1.89 × 103 | 2.59 × 104 ±5.28 × 102 |
| PPARγ–Rosiglitazone–Oleic Acid (Run3) | 5.08 × 104 ±2.12 × 103 | 5.46 × 104 ±1.56 × 103 | 3.06 × 104 ±1.93 × 103 | 2.74 × 104 ±5.07 × 102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agosta, F.; Cozzini, P. A Combined Molecular Dynamics and Hydropathic INTeraction (HINT) Approach to Investigate Protein Flexibility: The PPARγ Case Study. Molecules 2024, 29, 2234. https://doi.org/10.3390/molecules29102234
Agosta F, Cozzini P. A Combined Molecular Dynamics and Hydropathic INTeraction (HINT) Approach to Investigate Protein Flexibility: The PPARγ Case Study. Molecules. 2024; 29(10):2234. https://doi.org/10.3390/molecules29102234
Chicago/Turabian StyleAgosta, Federica, and Pietro Cozzini. 2024. "A Combined Molecular Dynamics and Hydropathic INTeraction (HINT) Approach to Investigate Protein Flexibility: The PPARγ Case Study" Molecules 29, no. 10: 2234. https://doi.org/10.3390/molecules29102234
APA StyleAgosta, F., & Cozzini, P. (2024). A Combined Molecular Dynamics and Hydropathic INTeraction (HINT) Approach to Investigate Protein Flexibility: The PPARγ Case Study. Molecules, 29(10), 2234. https://doi.org/10.3390/molecules29102234