
Rational Design of Palladium(II) Indenyl and Allyl Complexes Bearing Phosphine and Isocyanide Ancillary Ligands with Promising Antitumor Activity

 , ,
, ,
Abstract
:
1. Introduction
2. Results
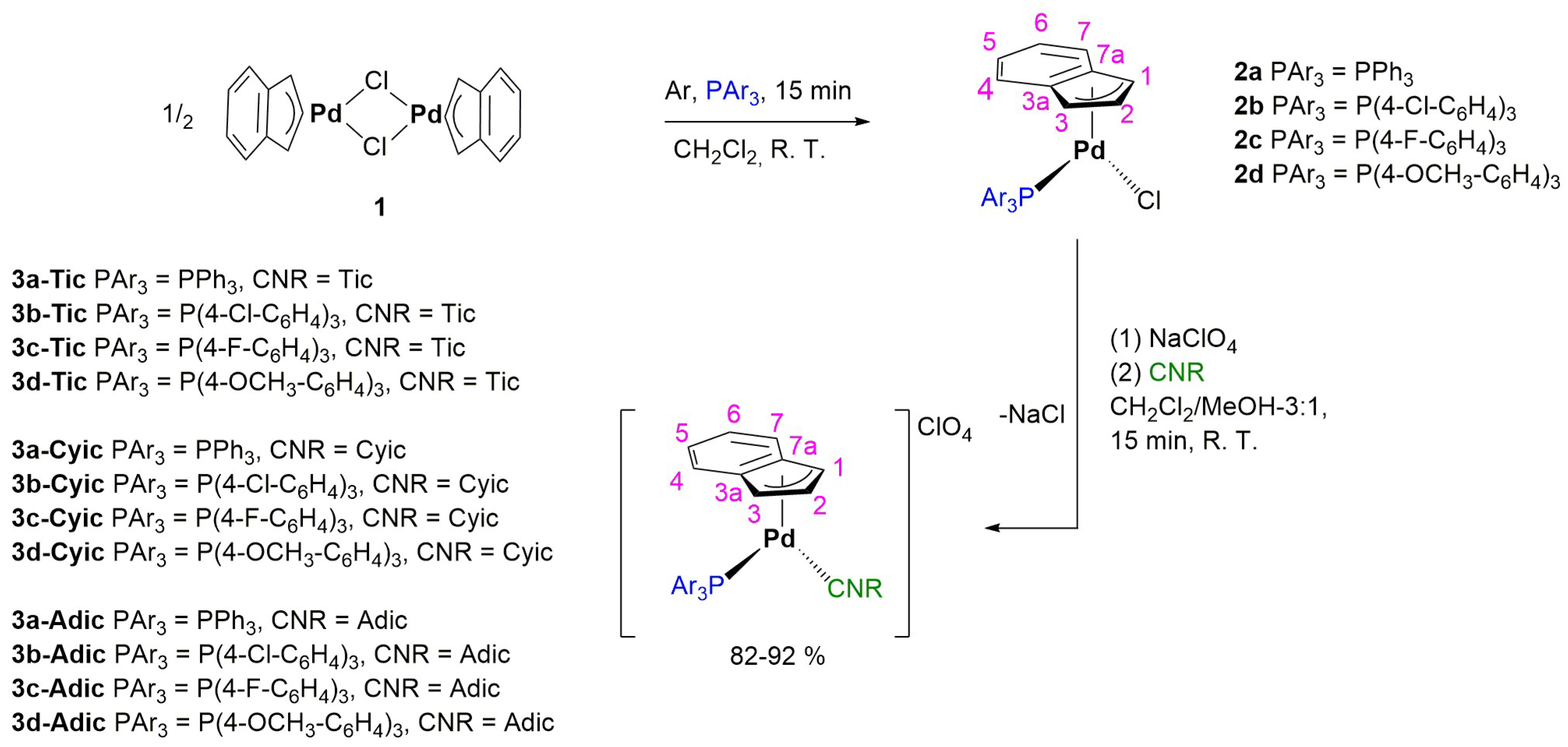
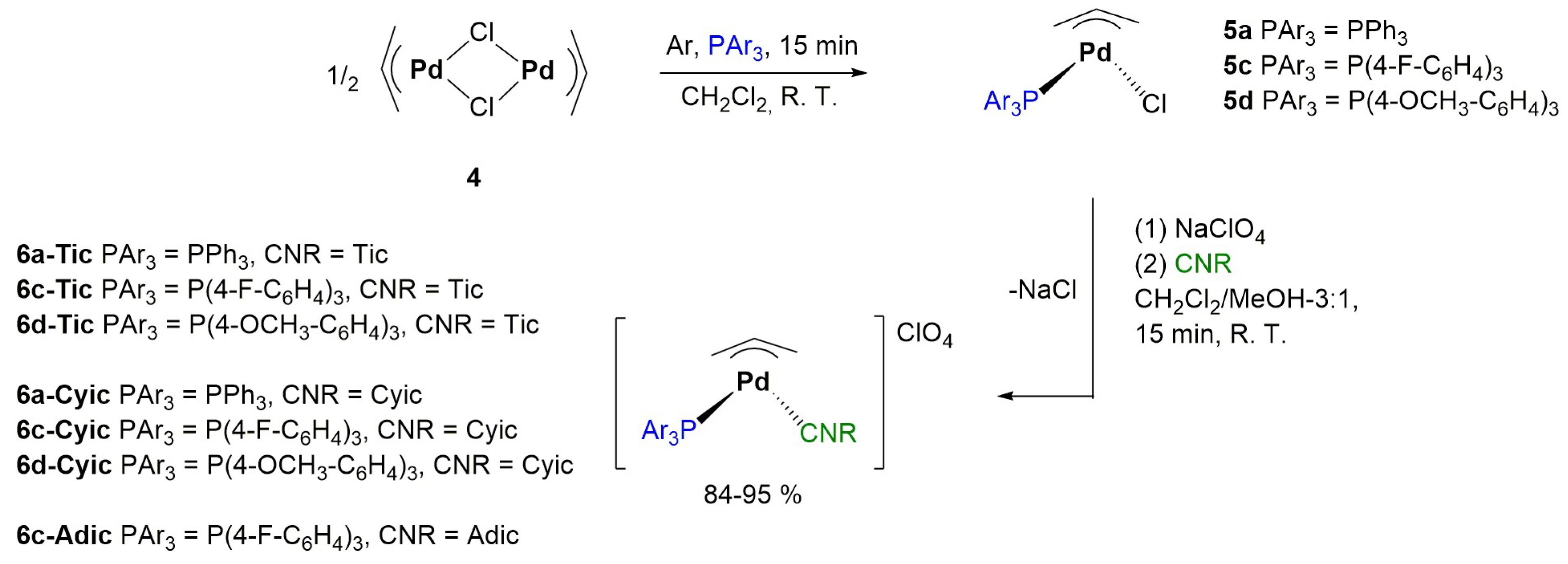
2.1. Synthesis and Characterization of the Complexes
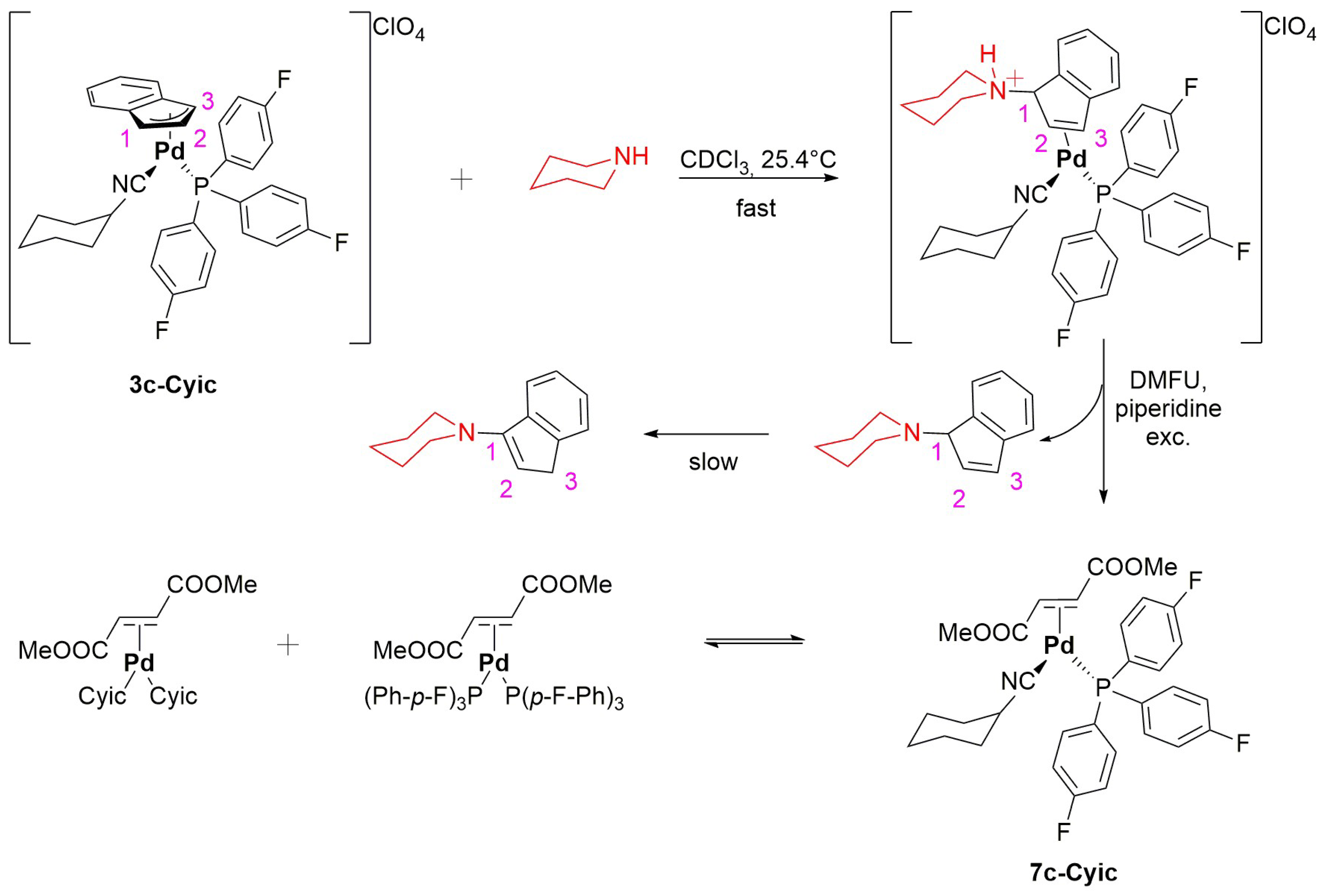
2.2. Reactivity of Indenyl Complexes
2.3. Anti-Cancer Activity of Indenyl and Allyl Complexes
- (a)
- All the tested complexes exhibit high cytotoxicity towards all three ovarian cancer cell lines. This cytotoxicity is comparable with that of cisplatin and, in many cases, better when we consider the cisplatin-resistant A2780cis cell line.
- (b)
- With a few exceptions, the activities of all the compounds are practically the same towards the A2780 and A2780cis cell lines. This evidence seems to suggest a mechanism of action different from that of classical platinum-based drugs.
- (c)
- Importantly, the high cytotoxicity of all the compounds was even maintained towards the OVCAR-5 cell line, which is classified as one of the more aggressive according to the High-Grade Serous Ovarian Cancer (HGSOC) classification [58].
- (d)
- The type of phosphine or isocyanide coordinated on the metal centre does not significantly or recurringly influence the cytotoxicity of the complexes. This fact seems to indicate that it is the general structure of these kinds of compounds that determines their biological activity.
- (e)
- Generally speaking, the allyl derivatives are more active than the corresponding indenyl complexes towards the A2780cis and OVCAR-5 cell lines, whereas towards A2780 cells, their behaviour remains basically the same.
- (f)
- The IC50 values with respect to the MRC-5 cell line point out the significant cytotoxicity of our compounds even towards normal cells, although generally being less marked. In particular, in the case of Pd(II) allyl derivatives, the IC50 values obtained for the three ovarian cancer cell lines are of 1–2 orders of magnitude lower than those obtained against MRC-5 fibroblasts, thus suggesting a certain degree of selectivity.
- (g)
- Finally, we tested the potential of our compounds even against another type of cancer cells. We turned our attention to the MDA-MB-231 line, consisting of highly invasive and poorly differentiated cells of triple-negative breast cancer. Our complexes have exhibited an activity significantly higher than that of cisplatin by at least one order of magnitude, with the indenyl derivatives seeming in this case to be slightly more effective than the allyl ones. The only exception is represented by complex 6c-Adic, which showed an IC50 value over two orders of magnitude lower than cisplatin and 40 times lower than that recorded for the MRC-5 cell line.
3. Materials and Methods
3.1. Solvents and Reagents
3.2. Instruments
3.3. Synthesis of Cationic Pd(II)-Indenyl Complexes Bearing One Phosphine and One Isocyanide Serving as Ancillary Ligands
General Procedure
3.4. Synthesis of Cationic Pd(II)-Allyl Complexes Bearing One Phosphine and One Isocyanide as Ancillary Ligands
General Procedure
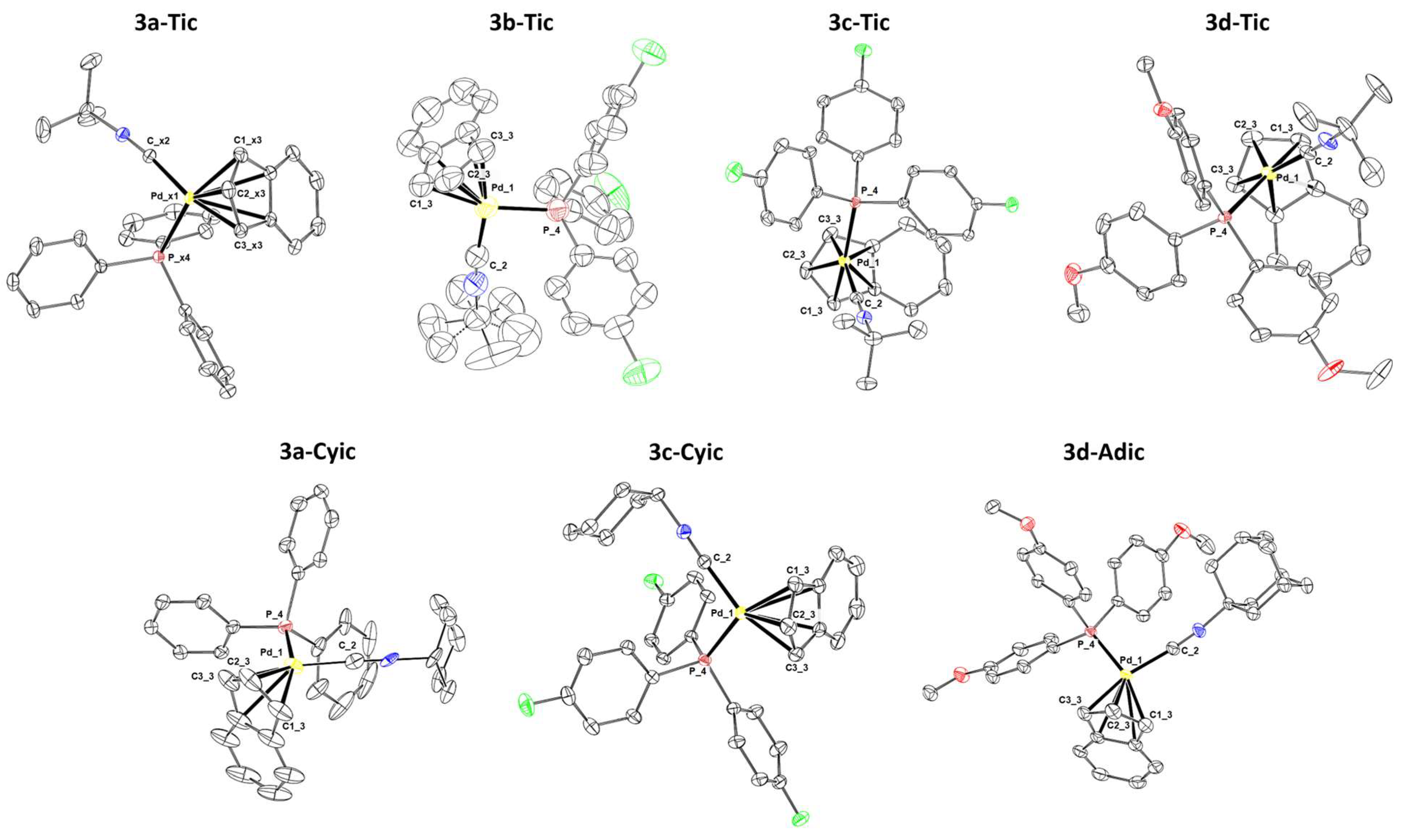
3.5. Crystal Structure Determination
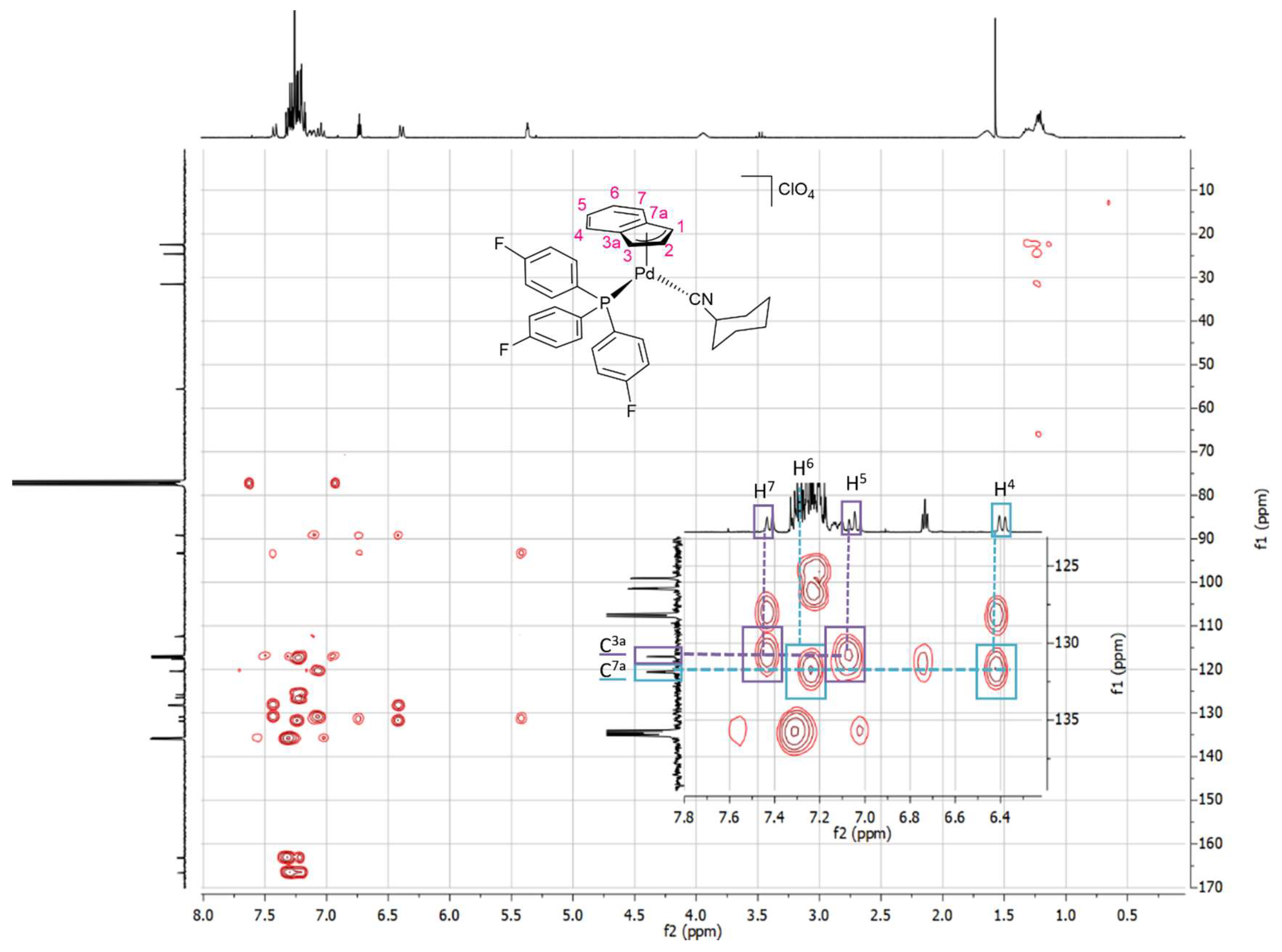
3.6. NMR Studies of Indenyl Amination
3.7. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peña, Q.; Wang, A.; Zaremba, O.; Shi, Y.; Scheeren, H.W.; Metselaar, J.M.; Kießling, F.; Pallares, R.M.; Wuttke, S.; Lammers, T. Metallodrugs in Cancer Nanomedicine. Chem. Soc. Rev. 2022, 51, 2544–2582. [Google Scholar] [CrossRef] [PubMed]
- Nafees, M.; Hanif, M.; Yang, P. Beyond Cisplatin: New Frontiers in Metallodrugs for Hard-to-Treat Triple Negative Breast Cancer. Coord. Chem. Rev. 2024, 499, 215507. [Google Scholar] [CrossRef]
- González-Ballesteros, M.; Mejía, C.; Ruiz-Azuara, L. Metallodrugs: An Approach against Invasion and Metastasis in Cancer Treatment. FEBS Open Bio 2022, 12, 880–899. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Sun, H. Metalloproteomics for Unveiling the Mechanism of Action of Metallodrugs. Inorg. Chem. 2019, 58, 13673–13685. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Pitto-Barry, A.; Sadler, P.J. Exploration of the Medical Periodic Table: Towards New Targets. Chem. Commun. 2013, 49, 5106. [Google Scholar] [CrossRef]
- Boros, E.; Dyson, P.J.; Gasser, G. Classification of Metal-Based Drugs According to Their Mechanisms of Action. Chem 2020, 6, 41–60. [Google Scholar] [CrossRef]
- Buchwald, S.L.; Milstein, D. Key Concepts in Ligand Design: An Introduction. In Ligand Design in Metal Chemistry: Reactivity and Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2016; pp. 1–12. [Google Scholar]
- Crabtree, R.H. NHC Ligands versus Cyclopentadienyls and Phosphines as Spectator Ligands in Organometallic Catalysis. J. Organomet. Chem. 2005, 690, 5451–5457. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric Effects of Phosphorus Ligands in Organometallic Chemistry and Homogeneous Catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Tolman, C.A. Electron Donor-Acceptor Properties of Phosphorus Ligands. Substituent Additivity. J. Am. Chem. Soc. 1970, 92, 2953–2956. [Google Scholar] [CrossRef]
- Niemeyer, Z.L.; Milo, A.; Hickey, D.P.; Sigman, M.S. Parameterization of Phosphine Ligands Reveals Mechanistic Pathways and Predicts Reaction Outcomes. Nat. Chem. 2016, 8, 610–617. [Google Scholar] [CrossRef]
- Jover, J.; Fey, N.; Harvey, J.N.; Lloyd-Jones, G.C.; Orpen, A.G.; Owen-Smith, G.J.J.; Murray, P.; Hose, D.R.J.; Osborne, R.; Purdie, M. Expansion of the Ligand Knowledge Base for Chelating P,P-Donor Ligands (LKB-PP). Organometallics 2012, 31, 5302–5306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Gensch, T.; Murray, B.; Niemeyer, Z.L.; Sigman, M.S.; Biscoe, M.R. Enantiodivergent Pd-Catalyzed C–C Bond Formation Enabled through Ligand Parameterization. Science 2018, 362, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Michelin, R.A.; Pombeiro, A.J.L.; Guedes da Silva, M.F.C. Aminocarbene Complexes Derived from Nucleophilic Addition to Isocyanide Ligands. Coord. Chem. Rev. 2001, 218, 75–112. [Google Scholar] [CrossRef]
- Pombeiro, A.J.; da Silva, M.F.C.G.; Michelin, R.A. Aminocarbyne Complexes Derived from Isocyanides Activated towards Electrophilic Addition. Coord. Chem. Rev. 2001, 218, 43–74. [Google Scholar] [CrossRef]
- Boyarskiy, V.P.; Bokach, N.A.; Luzyanin, K.V.; Kukushkin, V.Y. Metal-Mediated and Metal-Catalyzed Reactions of Isocyanides. Chem. Rev. 2015, 115, 2698–2779. [Google Scholar] [CrossRef]
- Knorn, M.; Lutsker, E.; Reiser, O. Isonitriles as Supporting and Non-Innocent Ligands in Metal Catalysis. Chem. Soc. Rev. 2020, 49, 7730–7752. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Tron, G.C.; Purghè, B.; Grosa, G.; Aprile, S. Metabolic Fate of the Isocyanide Moiety: Are Isocyanides Pharmacophore Groups Neglected by Medicinal Chemists? Chem. Res. Toxicol. 2020, 33, 955–966. [Google Scholar] [CrossRef]
- Schäfer, R.J.B.; Monaco, M.R.; Li, M.; Tirla, A.; Rivera-Fuentes, P.; Wennemers, H. The Bioorthogonal Isonitrile–Chlorooxime Ligation. J. Am. Chem. Soc. 2019, 141, 18644–18648. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Gazzarrini, S.; He, S.; Chen, L.; Li, J.; Xing, L.; Li, C.; Chen, L.; Neochoritis, C.G.; et al. Isocyanides as In-fluenza A Virus Subtype H5N1 Wild-Type M2 Channel Inhibitors. ChemMedChem 2015, 10, 1837–1845. [Google Scholar] [CrossRef]
- Scattolin, T.; Moro, G.; Serena, A.; Guadagnin Pattaro, A.; Rizzolio, F.; Canzonieri, V.; Demitri, N.; Bortolamiol, E.; Moretto, L.M.; Visentin, F. Synthesis, Characterization, and Anticancer Activity of Ferrocenyl Complexes Bearing Different Organopalladium Fragments. Appl. Organomet. Chem. 2022, 36, e6629. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Caligiuri, I.; Rizzolio, F.; Demitri, N.; Visentin, F. Synthesis and Comparative Study of the Anticancer Activity of H3-Allyl Palladium(II) Complexes Bearing N-Heterocyclic Carbenes as Ancillary Ligands. Polyhedron 2020, 186, 114607. [Google Scholar] [CrossRef]
- Bortolamiol, E.; Visentin, F.; Scattolin, T. Recent Advances in Bioconjugated Transition Metal Complexes for Cancer Therapy. Appl. Sci. 2023, 13, 5561. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Visentin, F.; Palazzolo, S.; Caligiuri, I.; Perin, T.; Canzonieri, V.; Demitri, N.; Rizzolio, F.; Togni, A. Palladium(II)-η3-Allyl Complexes Bearing N-Trifluoromethyl N-Heterocyclic Carbenes: A New Generation of Anticancer Agents That Restrain the Growth of High-Grade Serous Ovarian Cancer Tumoroids. Chem.—Eur. J. 2020, 26, 11868–11876. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Rizzolio, F.; Demitri, N.; Visentin, F. Allyl Palladium Complexes Bearing Carbohydrate-based N -heterocyclic Carbenes: Anticancer Agents for Selective and Potent In Vitro Cytotoxicity. Appl. Organomet. Chem. 2020, 34, e5876. [Google Scholar] [CrossRef]
- Scattolin, T.; Caligiuri, I.; Canovese, L.; Demitri, N.; Gambari, R.; Lampronti, I.; Rizzolio, F.; Santo, C.; Visentin, F. Synthesis of New Allyl Palladium Complexes Bearing Purine-Based NHC Ligands with Antiproliferative and Proapoptotic Activities on Human Ovarian Cancer Cell Lines. Dalton Trans. 2018, 47, 13616–13630. [Google Scholar] [CrossRef]
- Scattolin, T.; Caligiuri, I.; Mouawad, N.; El Boustani, M.; Demitri, N.; Rizzolio, F.; Visentin, F. Synthesis and In-Depth Studies on the Anticancer Activity of Novel Palladacyclopentadienyl Complexes Stabilized by N-Heterocyclic Carbene Ligands. Eur. J. Med. Chem. 2019, 179, 325–334. [Google Scholar] [CrossRef]
- Scattolin, T.; Andreetta, G.; Mauceri, M.; Rizzolio, F.; Demitri, N.; Canzonieri, V.; Visentin, F. Imidazo[1,5-a]Pyridine-3-Ylidenes and Dipyridoimidazolinylidenes as Ancillary Ligands in Palladium Allyl Complexes with Potent In Vitro Anticancer Activity. J. Organomet. Chem. 2021, 952, 122014. [Google Scholar] [CrossRef]
- Scattolin, T.; Voloshkin, V.A.; Visentin, F.; Nolan, S.P. A Critical Review of Palladium Organometallic Anticancer Agents. Cell Rep. Phys. Sci. 2021, 2, 100446. [Google Scholar] [CrossRef]
- Bortolamiol, E.; Fama, F.; Zhang, Z.; Demitri, N.; Cavallo, L.; Caligiuri, I.; Rizzolio, F.; Scattolin, T.; Visentin, F. Cationic Palladium (II)-Indenyl Complexes Bearing Phosphines as Ancillary Ligands: Synthesis, and Study of Indenyl Amination and Anticancer Activity. Dalton Trans. 2022, 51, 11135–11151. [Google Scholar] [CrossRef]
- Bortolamiol, E.; Isetta, G.; Caligiuri, I.; Demitri, N.; Paganelli, S.; Rizzolio, F.; Scattolin, T.; Visentin, F. Biological and Catalytic Applications of Pd(II)-Indenyl Complexes Bearing Phosphine and N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2023, 26, e202300084. [Google Scholar] [CrossRef]
- Scattolin, T.; Pessotto, I.; Cavarzerani, E.; Canzonieri, V.; Orian, L.; Demitri, N.; Schmidt, C.; Casini, A.; Bortolamiol, E.; Visentin, F.; et al. Indenyl and Allyl Palladate Complexes Bearing N-Heterocyclic Carbene Ligands: An Easily Accessible Class of New Anticancer Drug Candidates. Eur. J. Inorg. Chem. 2022, 2022, e202200103. [Google Scholar] [CrossRef]
- Viciu, M.S.; Germaneau, R.F.; Navarro, O.; Stevens, E.D.; Nolan, S.P. Activation and Reactivity of (NHC)Pd(Allyl)Cl (NHC = N-Heterocyclic Carbene) Complexes in Cross-Coupling Reactions. Organometallics 2002, 21, 5470–5472. [Google Scholar] [CrossRef]
- Viciu, M.S.; Germaneau, R.F.; Nolan, S.P. Well-Defined, Air-Stable (NHC)PD(Allyl)CL (NHC = N-Heterocyclic Carbene) Catalysts for the Arylation of Ketones. Org. Lett. 2002, 4, 4053–4056. [Google Scholar] [CrossRef] [PubMed]
- Navarro, O.; Oonishi, Y.; Kelly, R.A.; Stevens, E.D.; Briel, O.; Nolan, S.P. General and Efficient Methodology for the Suzuki–Miyaura Reaction in Technical Grade 2-Propanol. J. Organomet. Chem. 2004, 689, 3722–3727. [Google Scholar] [CrossRef]
- Navarro, O.; Kaur, H.; Mahjoor, P.; Nolan, S.P. Cross-Coupling and Dehalogenation Reactions Catalyzed by (N-Heterocyclic Carbene)Pd(Allyl)Cl Complexes. J. Org. Chem. 2004, 69, 3173–3180. [Google Scholar] [CrossRef]
- Marion, N.; Navarro, O.; Mei, J.; Stevens, E.D.; Scott, N.M.; Nolan, S.P. Modified (NHC)Pd(Allyl)Cl (NHC = N-Heterocyclic Carbene) Complexes for Room-Temperature Suzuki−Miyaura and Buchwald−Hartwig Reactions. J. Am. Chem. Soc. 2006, 128, 4101–4111. [Google Scholar] [CrossRef] [PubMed]
- Chartoire, A.; Claver, C.; Corpet, M.; Krinsky, J.L.; Mayen, J.; Nelson, D.J.; Nolan, S.P.; Peñafiel, I.; Woodward, R.L.; Meadows, R.E. Recyclable NHC Catalyst for the Development of a Generalized Approach to Continuous Buchwald–Hartwig Reaction and Workup. Org. Process Res. Dev. 2016, 20, 551–557. [Google Scholar] [CrossRef]
- Lei, P.; Meng, G.; Szostak, M. General Method for the Suzuki–Miyaura Cross-Coupling of Amides Using Commercially Available, Air- and Moisture-Stable Palladium/NHC (NHC = N-Heterocyclic Carbene) Complexes. ACS Catal. 2017, 7, 1960–1965. [Google Scholar] [CrossRef]
- Lei, P.; Ling, Y.; An, J.; Nolan, S.P.; Szostak, M. 2-Methyltetrahydrofuran (2-METHF): A Green Solvent for PD−NHC-Catalyzed Amide and Ester Suzuki-Miyaura Cross-Coupling by N−C/O−C Cleavage. Adv. Synth. Catal. 2019, 361, 5654–5660. [Google Scholar] [CrossRef]
- Melvin, P.R.; Nova, A.; Balcells, D.; Dai, W.; Hazari, N.; Hruszkewycz, D.P.; Shah, H.P.; Tudge, M.T. Design of a Versatile and Improved Precatalyst Scaffold for Palladium-Catalyzed Cross-Coupling: (η3-1-tBu-Indenyl)2(Μ-CL)2PD2. ACS Catal. 2015, 5, 3680–3688. [Google Scholar] [CrossRef]
- Espinosa, M.R.; Doppiu, A.; Hazari, N. Differences in the Performance of Allyl Based Palladium Precatalysts for Suzuki-Miyaura Reactions. Adv. Synth. Catal. 2020, 362, 5062–5078. [Google Scholar] [CrossRef] [PubMed]
- Canovese, L.; Visentin, F.; Levi, C.; Dolmella, A. Synthesis, Characterization, Dynamics and Reactivity toward Amination of η 3-Allyl Palladium Complexes Bearing Mixed Ancillary Ligands. Evaluation of the Electronic Characteristics of the Ligands from Kinetic Data. Dalton Trans. 2011, 40, 966–981. [Google Scholar] [CrossRef] [PubMed]
- Sui-Seng, C.; Enright, G.D.; Zargarian, D. New Routes to η 1- and (η 3↔η 5)-Indenylpalladium Complexes: Synthesis, Characterization, and Reactivities. Organometallics 2004, 23, 1236–1246. [Google Scholar] [CrossRef]
- Sui-Seng, C.; Enright, G.D.; Zargarian, D. New Palladium(II)−(η 3/5- or η 1-Indenyl) and Dipalladium(I)−(μ,η 3-Indenyl) Complexes. J. Am. Chem. Soc. 2006, 128, 6508–6519. [Google Scholar] [CrossRef] [PubMed]
- Canovese, L.; Visentin, F.; Santo, C.; Bertolasi, V. Insertion of Isocyanides across the Pd-C Bond of Phosphinoquinoline Allyl Palladium Complexes Bearing H1- and H3-Coordinated Allyl Groups. A Synthetic and Mechanistic Study. Organometallics 2014, 33, 1700–1709. [Google Scholar] [CrossRef]
- Singleton, E.; Oosthuizen, H.E. Metal Isocyanide Complexes. Adv. Organomet. Chem. 1983, 22, 209–310. [Google Scholar] [CrossRef]
- Westcott, S.A.; Kakkar, A.K.; Stringer, G.; Taylor, N.J.; Marder, T.B. Flexible Coordination of Indenyl Ligands in Sandwich Complexes of Transition Metals. Molecular Structures of [(η-C9R7)2M] (M = Fe, R = H, Me; M = Co, Ni, R = H): Direct Measurement of the Degree of Slip-Fold Distortion as a Function of d-Electron Count. J. Organomet. Chem. 1990, 394, 777–794. [Google Scholar] [CrossRef]
- Baker, R.T.; Tulip, T.H. Synthesis, Molecular Structure, Solution Dynamics, and Reactivity of (.Eta.-C5H5)2M(.Mu.-PR2)2Rh(.Eta.-Indenyl) (M = Zr, Hf; R = Et, Ph). Organometallics 1986, 5, 839–845. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Scattolin, T.; Santo, C.; Bertolasi, V. Synthesis, Characterization and a Reactivity Study of Some Allyl Palladium Complexes Bearing Bidentate Hemi-Labile Carbene or Mixed Carbene/PPh3 Ligands. Polyhedron 2016, 119, 377–386. [Google Scholar] [CrossRef]
- Scrivanti, A.; Carturan, G.; Crociani, B. Synthesis, Characterization and Solution Behavior of (.Eta.3-Allyl)(Carbene)Palladium(II) Complexes. Organometallics 1983, 2, 1612–1617. [Google Scholar] [CrossRef]
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian Cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of Ovarian Cancer: A Review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed]
- Scattolin, T.; Bortolamiol, E.; Palazzolo, S.; Caligiuri, I.; Perin, T.; Canzonieri, V.; Demitri, N.; Rizzolio, F.; Cavallo, L.; Dereli, B.; et al. The Anticancer Activity of an Air-Stable Pd(I)-NHC (NHC = N-Heterocyclic Carbene) Dimer. Chem. Commun. 2020, 56, 12238–12241. [Google Scholar] [CrossRef]
- Scattolin, T.; Moro, G.; Rizzolio, F.; Santo, C.; Moretto, L.M.; Visentin, F. Improved Synthesis, Anticancer Activity and Electrochemical Characterization of Unusual Zwitterionic Palladium Compounds with a Ten-Term Coordinative Ring. ChemistrySelect 2019, 4, 10911–10919. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating Cell Lines as Tumour Models by Comparison of Genomic Profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P.; Liu, Y.; Scattolin, T.; Gobbo, A.; Beliš, M.; Van Hecke, K.; Cazin, C.S.J. A Simple Synthetic Route to Well-Defined [PD(NHC)CL(1-TBU-indenyl)] Pre-catalysts for Cross-Coupling Reactions. Eur. J. Inorg. Chem. 2021, 2022, e202100840. [Google Scholar] [CrossRef]
- Hartley, F.R.; Jones, S.R. Metal π-Allyl Chemistry III. The Preparation of π-Allylpalladium Complexes from Palladium(II) Salts. J. Organomet. Chem. 1974, 66, 465–473. [Google Scholar] [CrossRef]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the Crystallography Beamlines at Elettra. Eur. Phys. J. Plus 2015, 130, 43. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Baslé, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Mármol, J.J.; Berrisford, J.M. The CCP4 Suite: Integrative Software for Macromolecular Crystallography. Acta Crystallogr. Sect. Struct. Biol. 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How Good Are My Data and What Is the Resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. checkCIF Validation ALERTS: What They Mean and How to Respond. Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Schrodinger, L. The PyMOL Molecular Graphics System. 2015. Available online: https://pymol.org/2/ (accessed on 1 January 2024).
- Zargarian, D. Group 10 Metal Indenyl Complexes. Coord. Chem. Rev. 2002, 233–234, 157–176. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | ||||
|---|---|---|---|---|---|
| A2780 | A2780cis | OVCAR-5 | MDA-MB-231 | MRC-5 | |
| cisplatin | 0.30 ± 0.02 | 7.2 ± 0.8 | 0.9 ± 0.1 | 11.5 ± 0.8 | 4.5 ± 0.1 |
| 3a-Tic | 0.70 ± 0.09 | 1.5 ± 0.2 | 2.4 ± 0.3 | 0.32 ± 0.05 | 3.1 ± 0.2 |
| 3b-Tic | 0.43 ± 0.08 | 0.3 ± 0.1 | 0.44 ± 0.03 | 0.36 ± 0.08 | 3.38 ± 0.09 |
| 3c-Tic | 0.6 ± 0.1 | 1.8 ± 0.3 | 1.60 ± 0.06 | 1.3 ± 0.2 | 3.1 ± 0.2 |
| 3d-Tic | 0.32 ± 0.02 | 1.6 ± 0.8 | 3.3 ± 0.2 | 0.46 ± 0.09 | 4.11 ± 0.05 |
| 3a-Cyic | 0.29 ± 0.06 | 1.87 ± 0.08 | 2.6 ± 0.5 | 0.65 ± 0.05 | 3.0 ± 0.1 |
| 3b-Cyic | 0.45 ± 0.05 | 0.8 ± 0.2 | 0.43 ± 0.08 | 0.33 ± 0.05 | 3.4 ± 0.1 |
| 3c-Cyic | 0.69 ± 0.01 | 0.97 ± 0.03 | 1.13 ± 0.07 | 1.4 ± 0.2 | 5.3 ± 0.5 |
| 3d-Cyic | 0.28 ± 0.03 | 2.8 ± 0.1 | 3.0 ± 0.4 | 1.1 ± 0.4 | 3.8 ± 0.1 |
| 3a-Adic | 0.40 ± 0.01 | 2.1 ± 0.3 | 0.33 ± 0.07 | 1.2 ± 0.5 | 3.2 ± 0.1 |
| 3b-Adic | 0.21 ± 0.08 | 0.73 ± 0.07 | 0.26 ± 0.04 | 2.0 ± 0.5 | 2.8 ± 0.3 |
| 3c-Adic | 0.12 ± 0.02 | 1.9 ± 0.1 | 0.26 ± 0.06 | 0.9 ± 0.5 | 3.2 ± 0.2 |
| 3d-Adic | 0.32 ± 0.05 | 1.27 ± 0.09 | 1.14 ± 0.07 | 0.36 ± 0.03 | 2.5 ± 0.4 |
| 6a-Tic | 0.10 ± 0.02 | 0.30 ± 0.06 | 0.37 ± 0.07 | 1.2 ± 0.2 | 10 ± 2 |
| 6c-Tic | 0.14 ± 0.03 | 0.55 ± 0.09 | 0.34 ± 0.03 | 3.9 ± 0.7 | 4.7 ± 0.8 |
| 6d-Tic | 0.33 ± 0.03 | 0.41 ± 0.07 | 0.35 ± 0.07 | 1.21 ± 0.09 | 3.4 ± 0.2 |
| 6aCyic | 0.27 ± 0.08 | 0.3 ± 0.1 | 0.241 ± 0.007 | 2.9 ± 0.4 | 3.6 ± 0.2 |
| 6c-Cyic | 0.23 ± 0.03 | 0.41 ± 0.03 | 0.26 ± 0.02 | 2.5 ± 0.2 | 4.2 ± 0.5 |
| 6a-Cyic | 0.20 ± 0.04 | 0.28 ± 0.04 | 0.26 ± 0.03 | 1.3 ± 0.4 | 3.7 ± 0.2 |
| 6c-Adic | 0.27 ± 0.05 | 0.4 ± 0.2 | 0.29 ± 0.06 | 0.071 ± 0.007 | 2.9 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortolamiol, E.; Botter, E.; Cavarzerani, E.; Mauceri, M.; Demitri, N.; Rizzolio, F.; Visentin, F.; Scattolin, T. Rational Design of Palladium(II) Indenyl and Allyl Complexes Bearing Phosphine and Isocyanide Ancillary Ligands with Promising Antitumor Activity. Molecules 2024, 29, 345. https://doi.org/10.3390/molecules29020345
Bortolamiol E, Botter E, Cavarzerani E, Mauceri M, Demitri N, Rizzolio F, Visentin F, Scattolin T. Rational Design of Palladium(II) Indenyl and Allyl Complexes Bearing Phosphine and Isocyanide Ancillary Ligands with Promising Antitumor Activity. Molecules. 2024; 29(2):345. https://doi.org/10.3390/molecules29020345
Chicago/Turabian StyleBortolamiol, Enrica, Eleonora Botter, Enrico Cavarzerani, Matteo Mauceri, Nicola Demitri, Flavio Rizzolio, Fabiano Visentin, and Thomas Scattolin. 2024. "Rational Design of Palladium(II) Indenyl and Allyl Complexes Bearing Phosphine and Isocyanide Ancillary Ligands with Promising Antitumor Activity" Molecules 29, no. 2: 345. https://doi.org/10.3390/molecules29020345
APA StyleBortolamiol, E., Botter, E., Cavarzerani, E., Mauceri, M., Demitri, N., Rizzolio, F., Visentin, F., & Scattolin, T. (2024). Rational Design of Palladium(II) Indenyl and Allyl Complexes Bearing Phosphine and Isocyanide Ancillary Ligands with Promising Antitumor Activity. Molecules, 29(2), 345. https://doi.org/10.3390/molecules29020345