

Radical Reactions in Organic Synthesis: Exploring in-, on-, and with-Water Methods


Abstract
:1. Organic Reactions in Aqueous Media
2. Free Radicals in Water
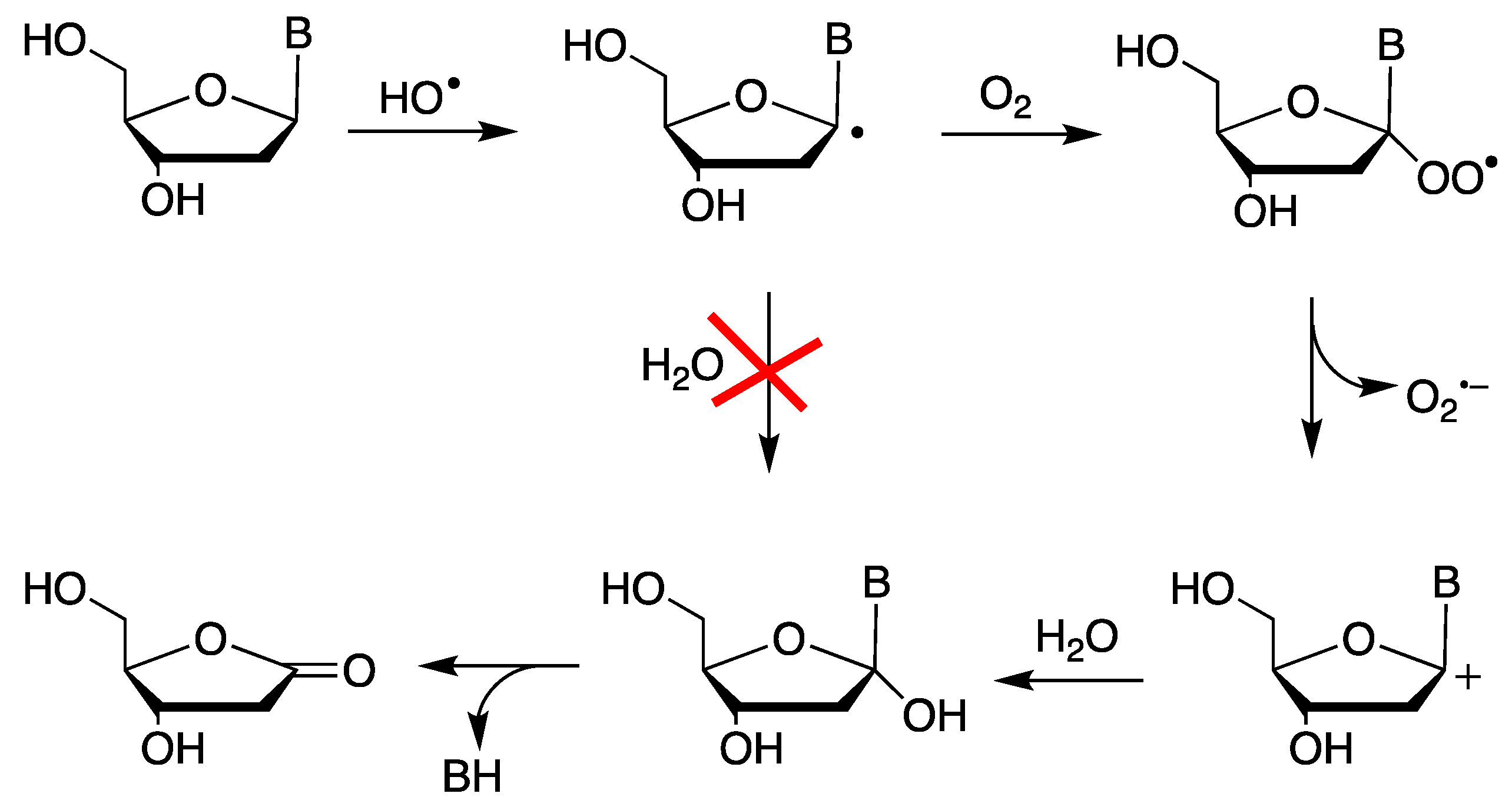
2.1. Neutral Carbon-Centered Radicals Do Not React with Water
2.2. Polar Effects and the Increase in Reaction Rates
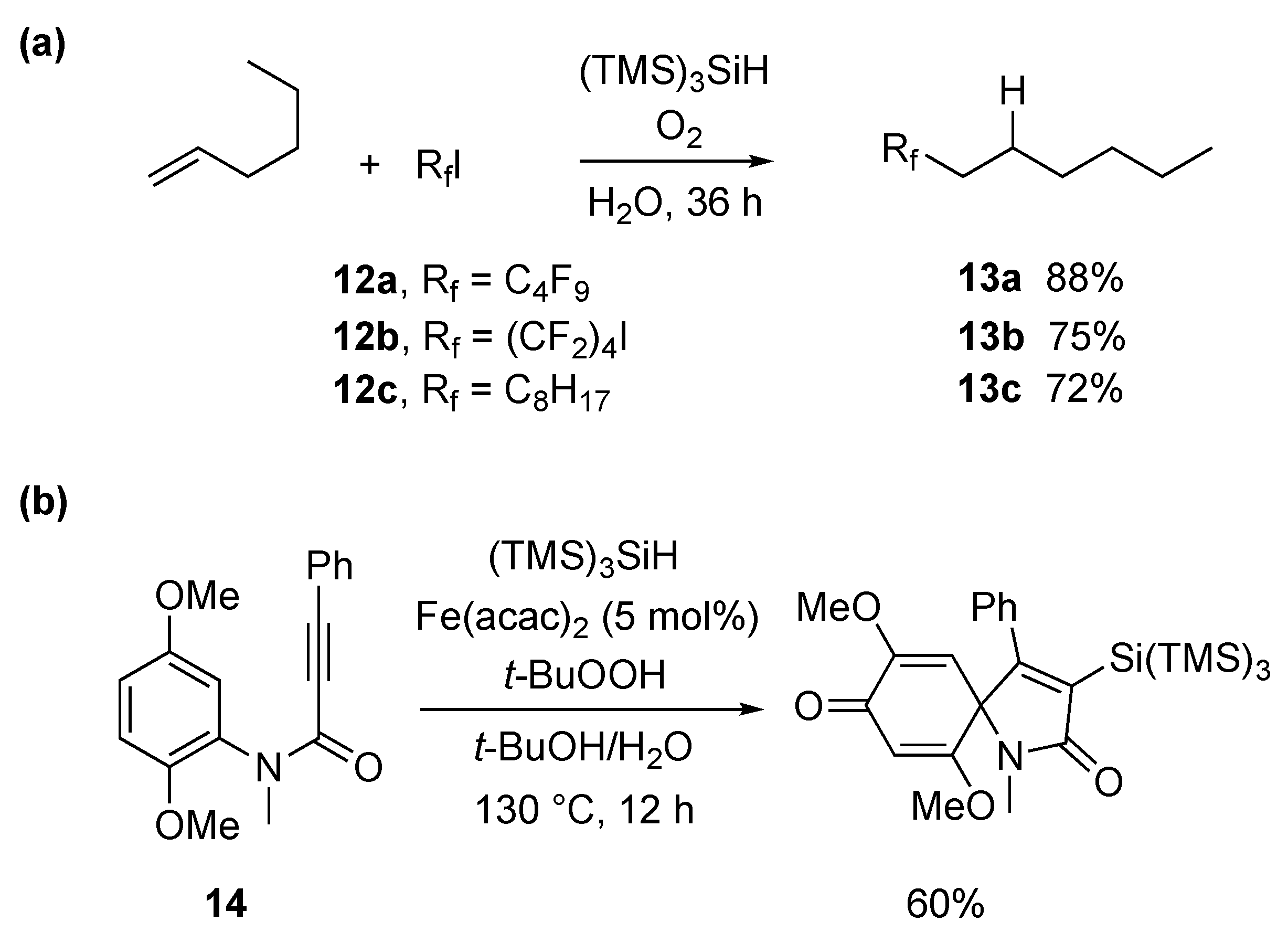
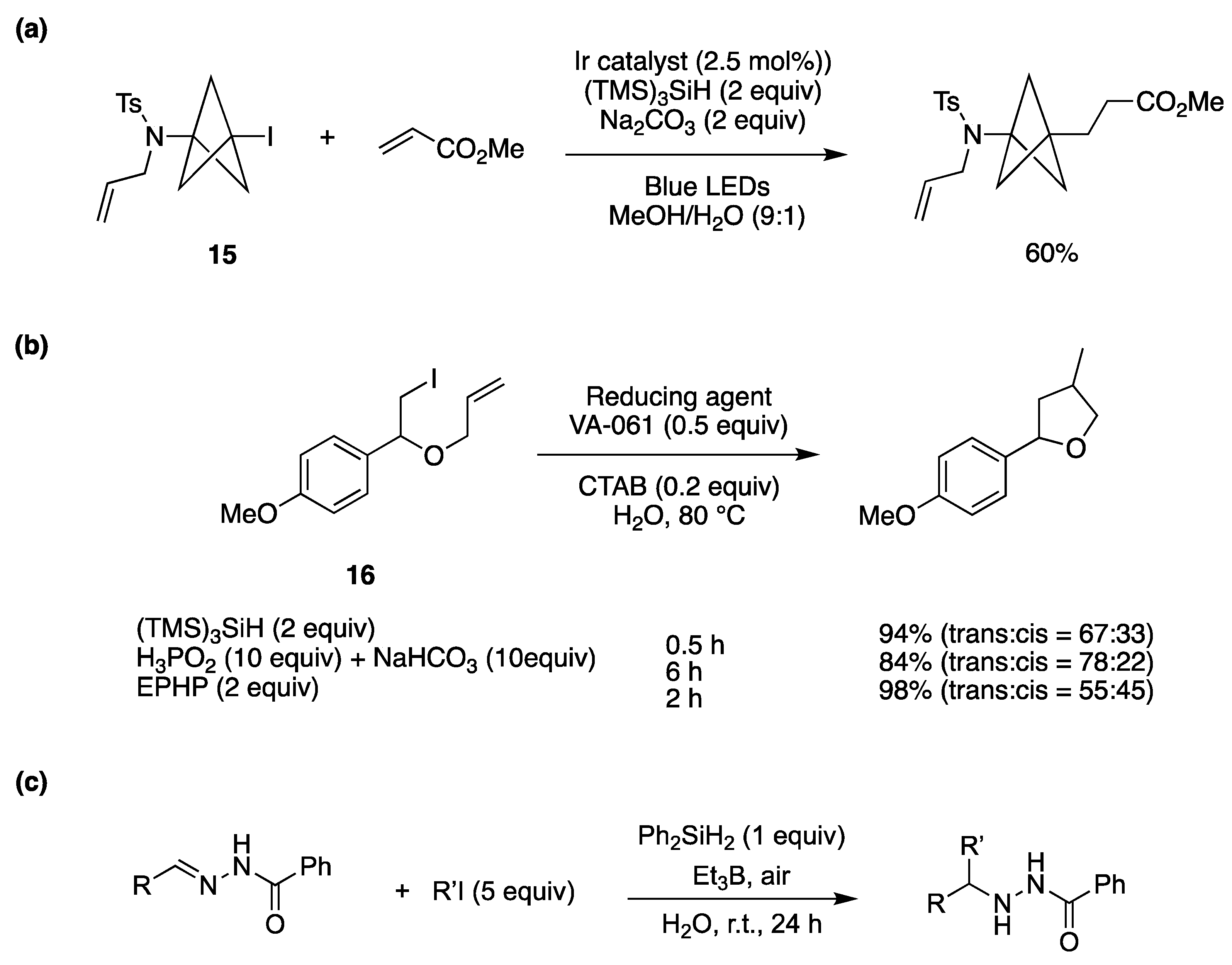
3. Synthetic Approaches Using Radical Intermediates in Water and Aqueous Media
4. Radical Reactions Occurring “on Water” and in a Heterogeneous Phase
5. Reaction of Carbon-Centered Radicals with Water-Activated by Lewis Acids
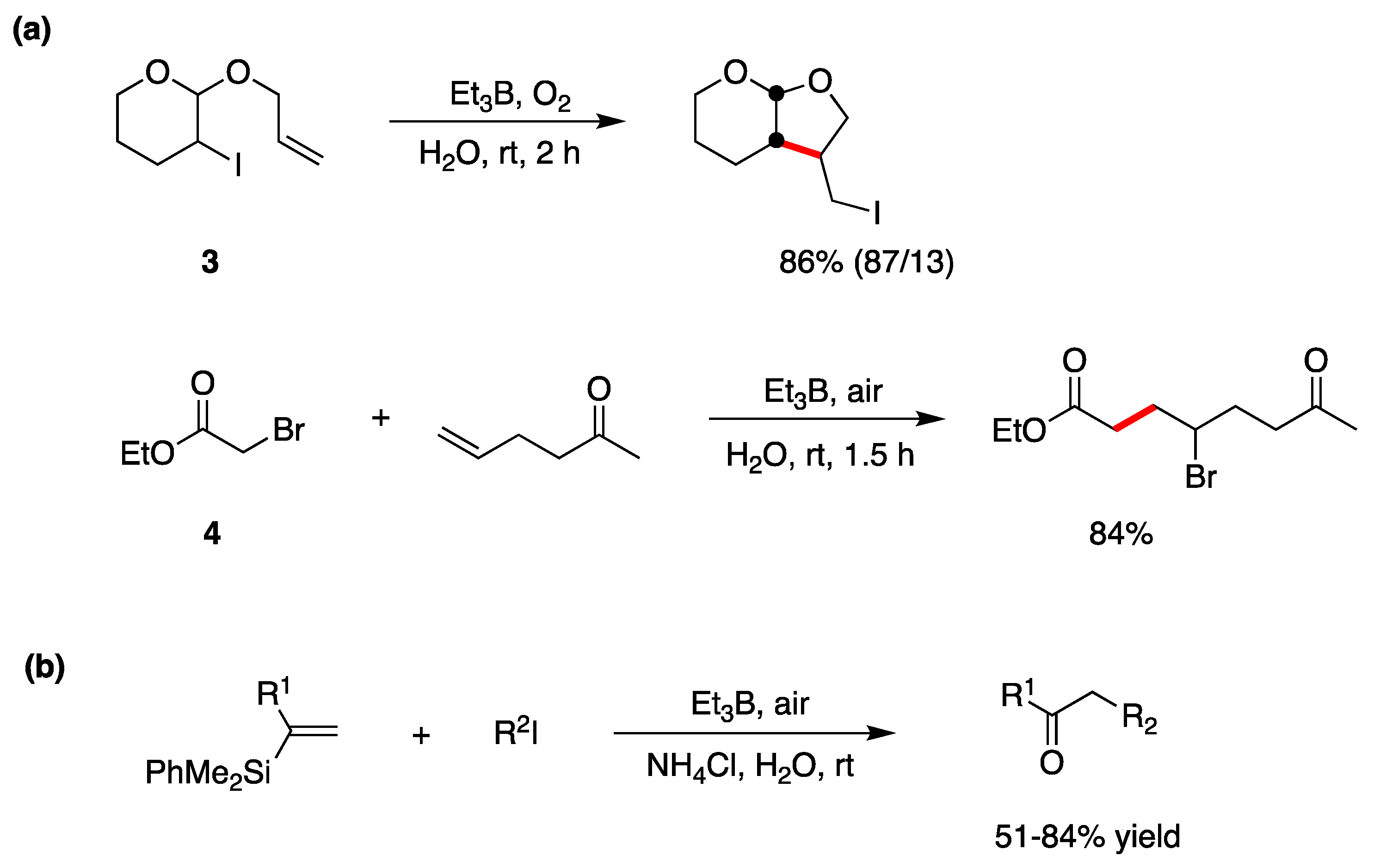
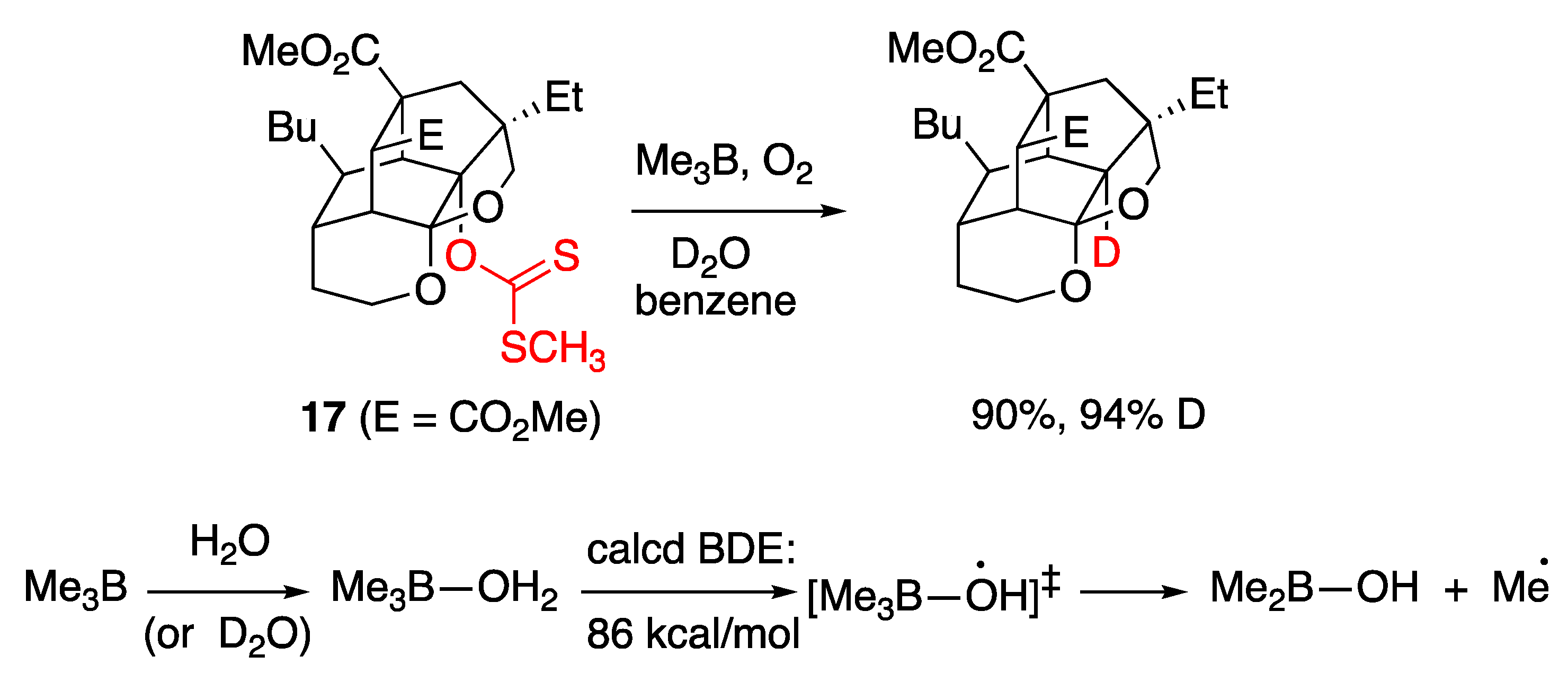
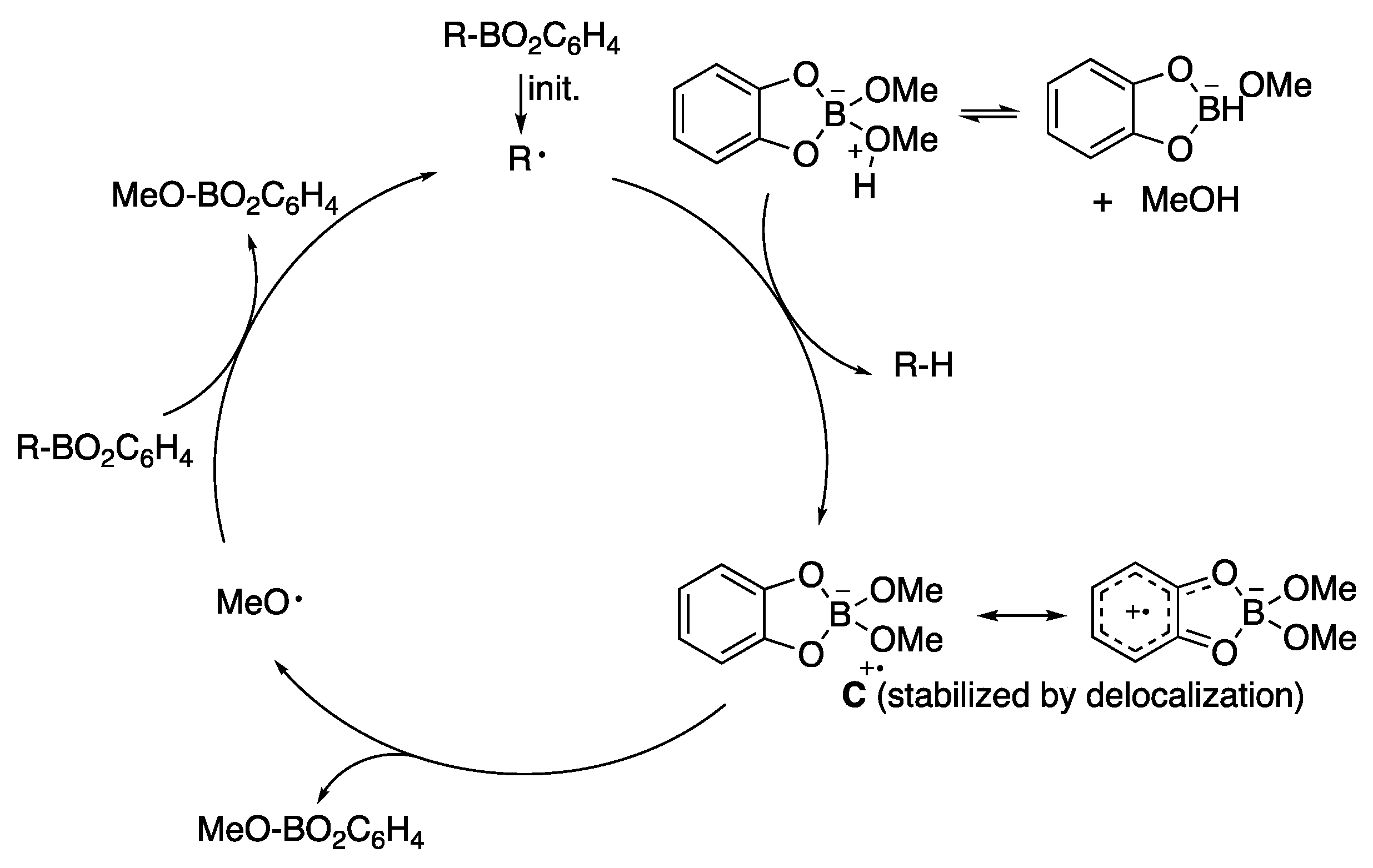
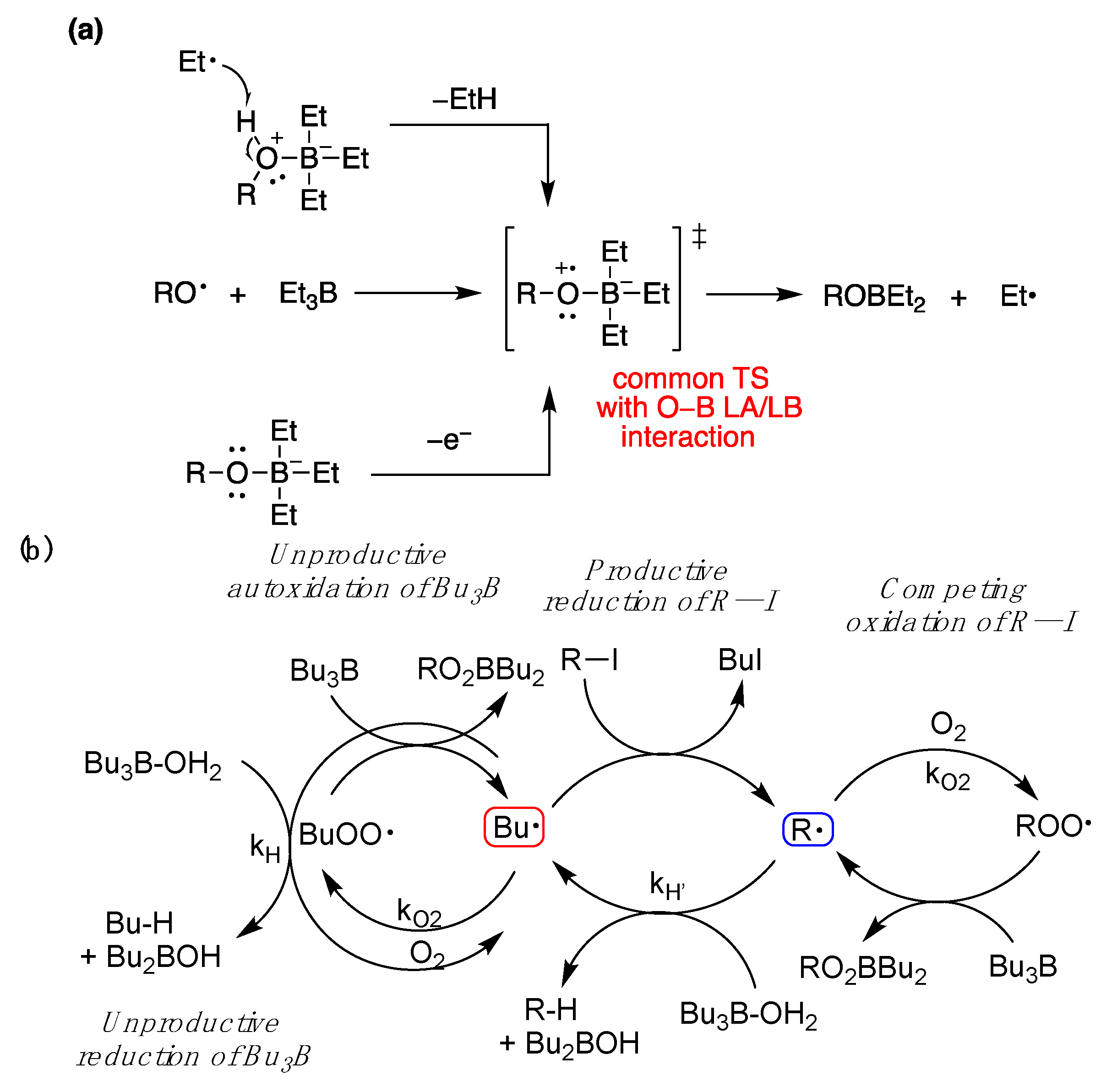
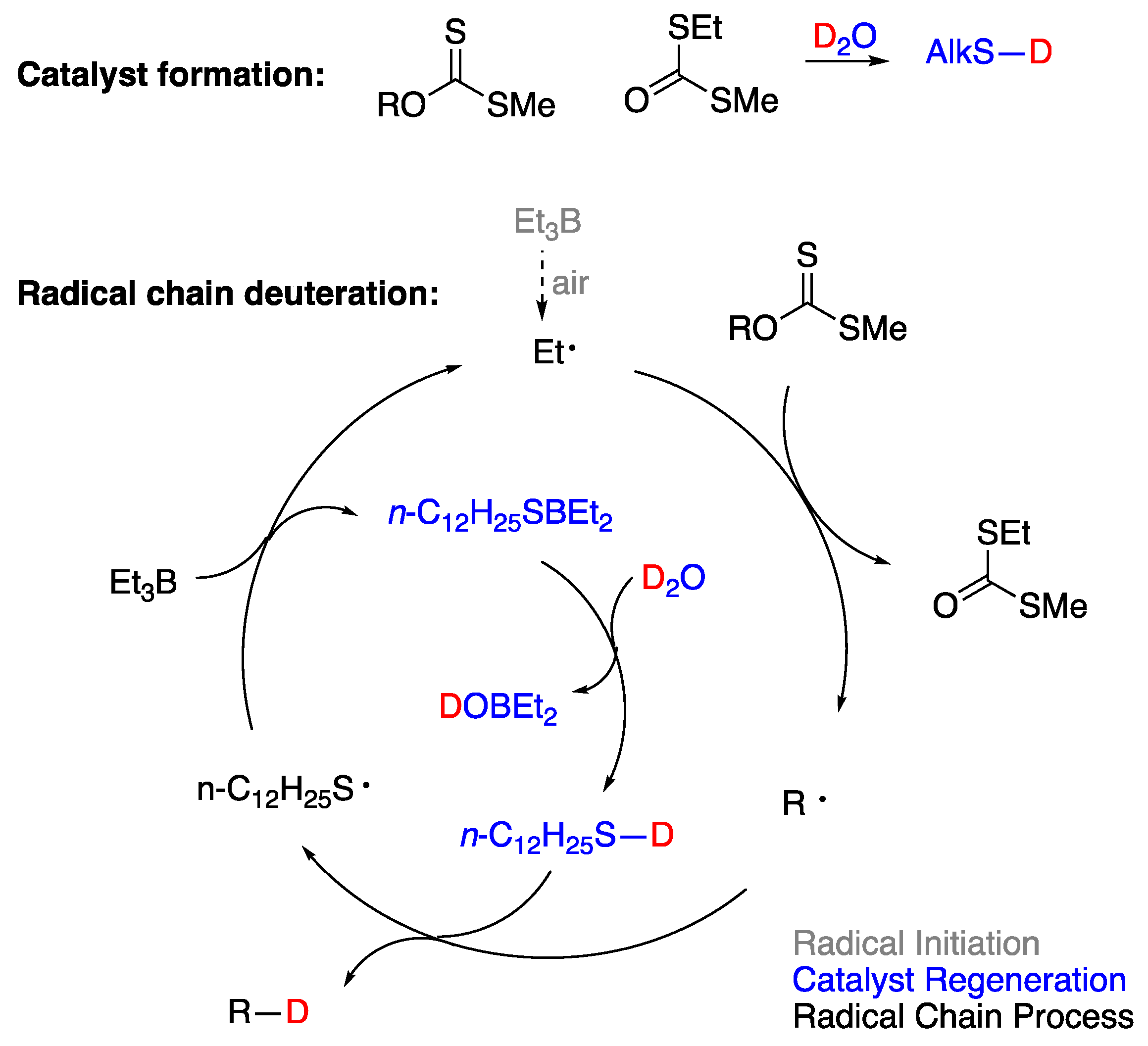
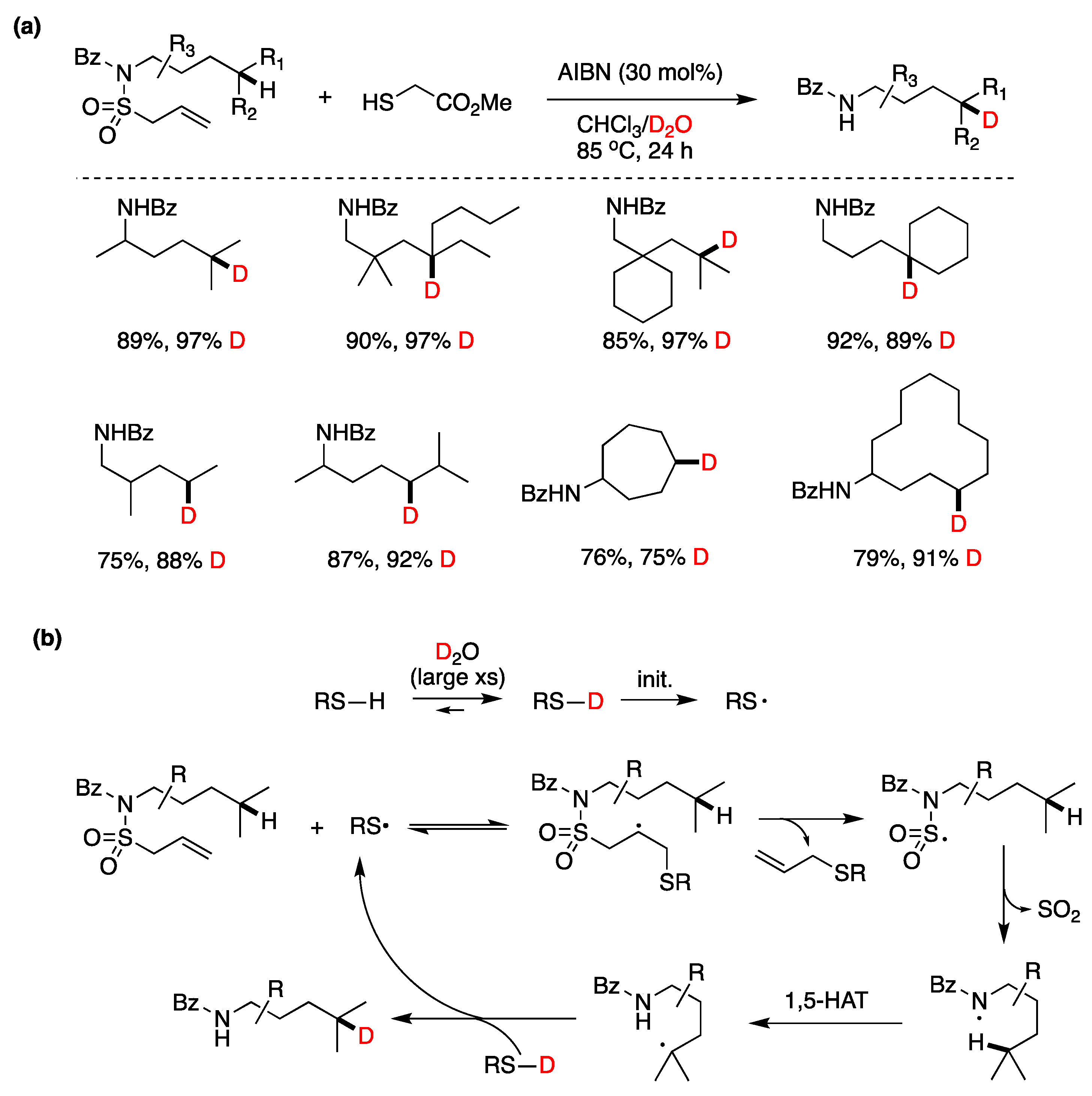
5.1. Boron Derivatives
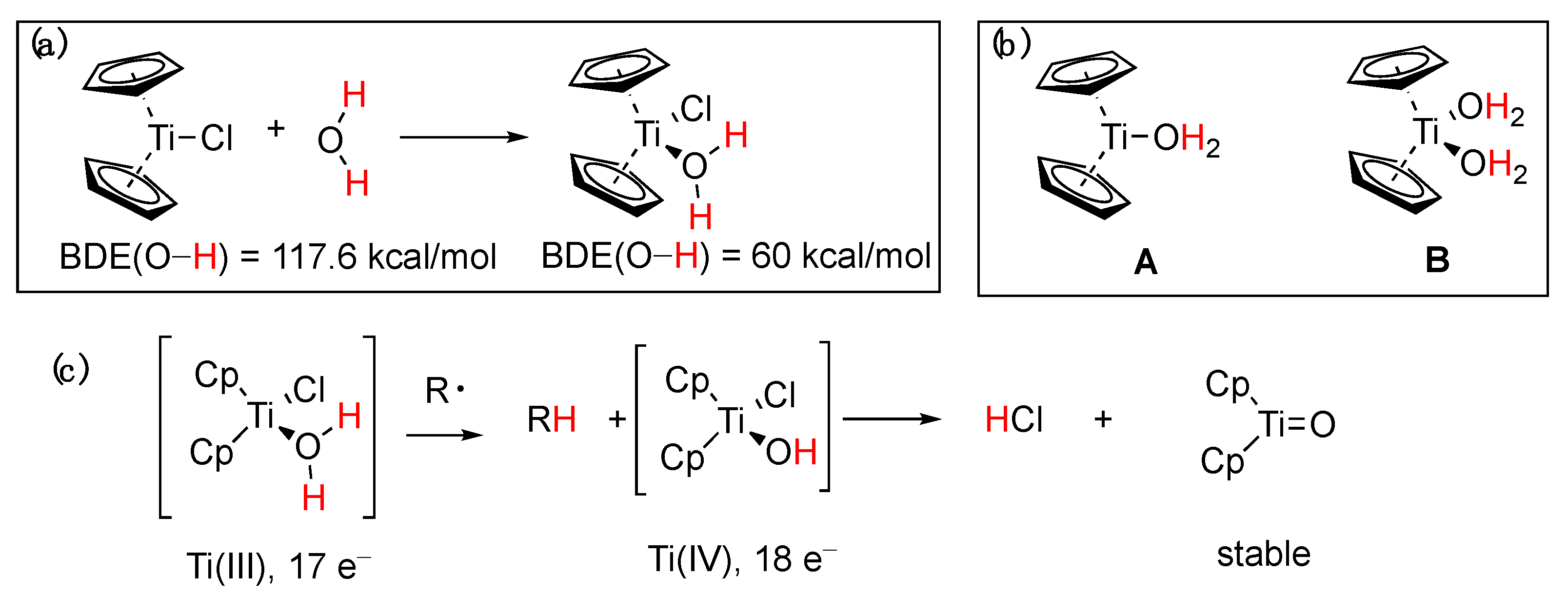
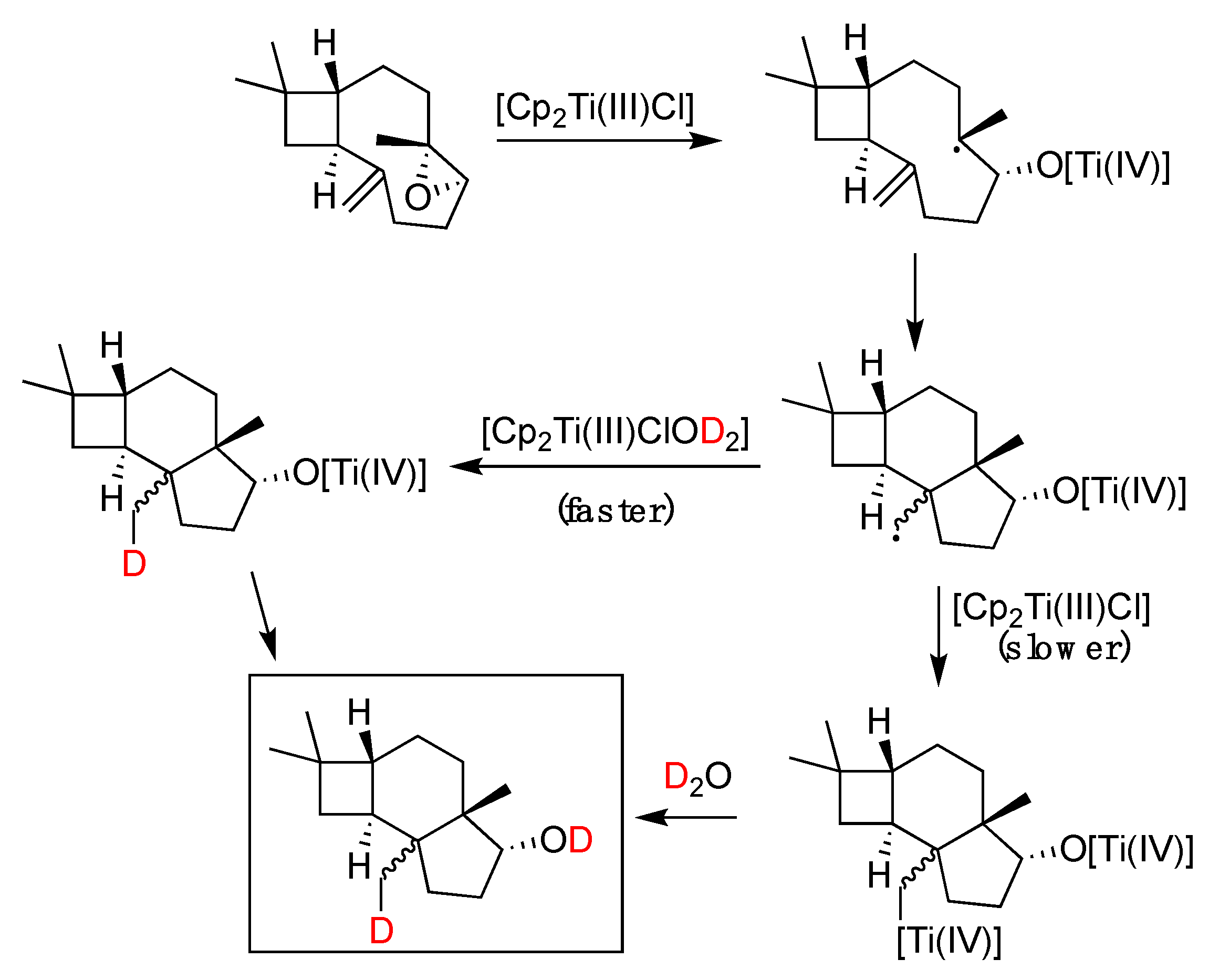
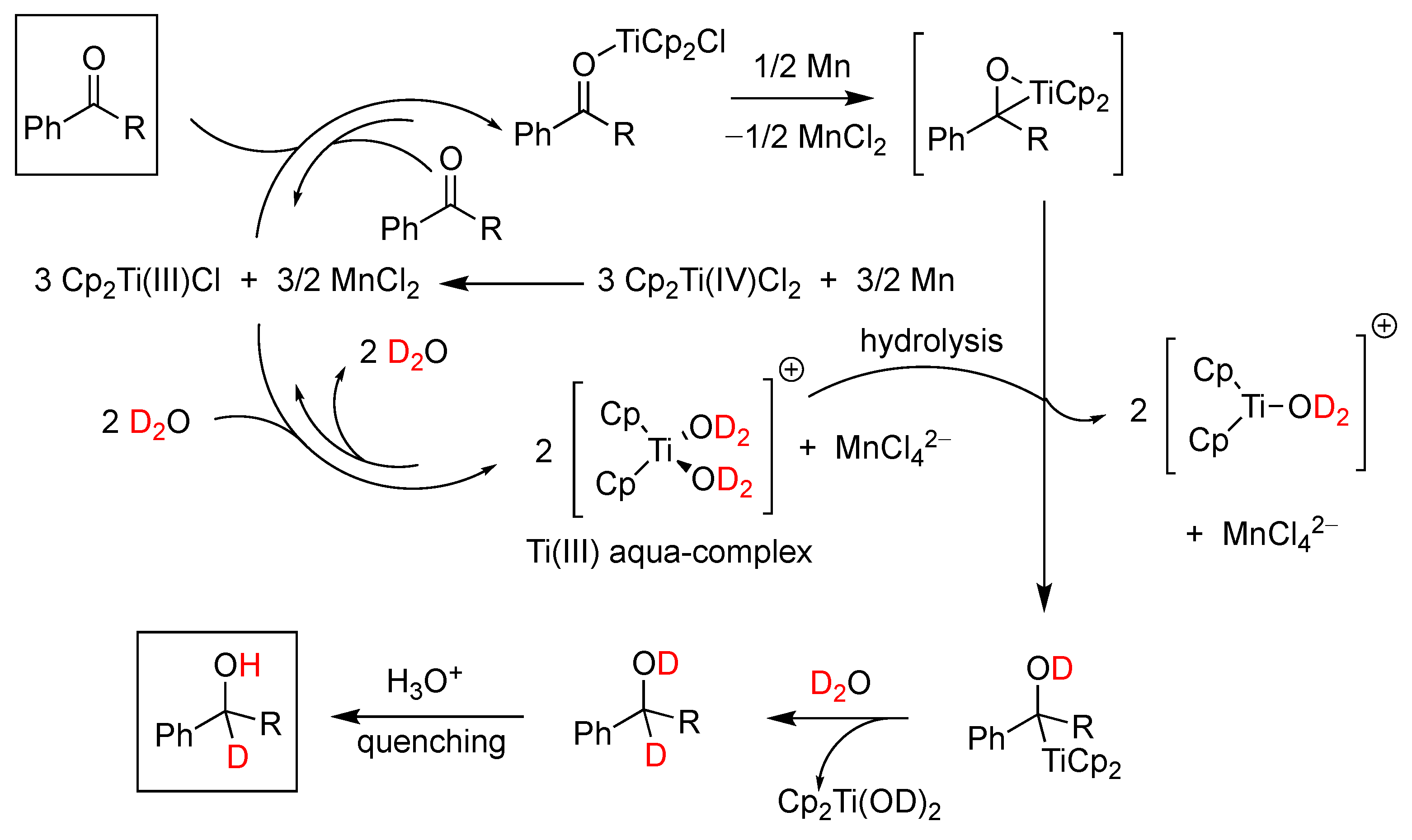
5.2. Titanium Complexes

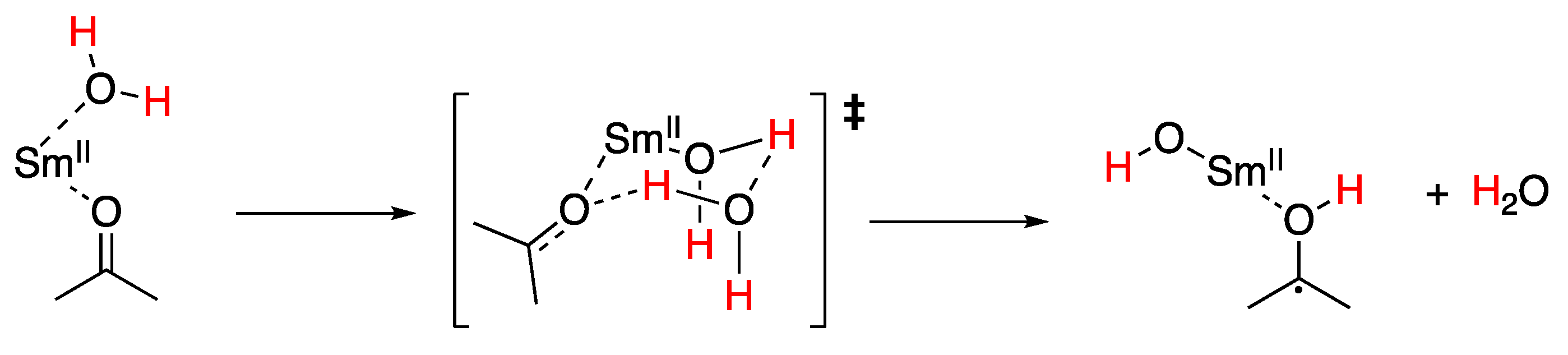
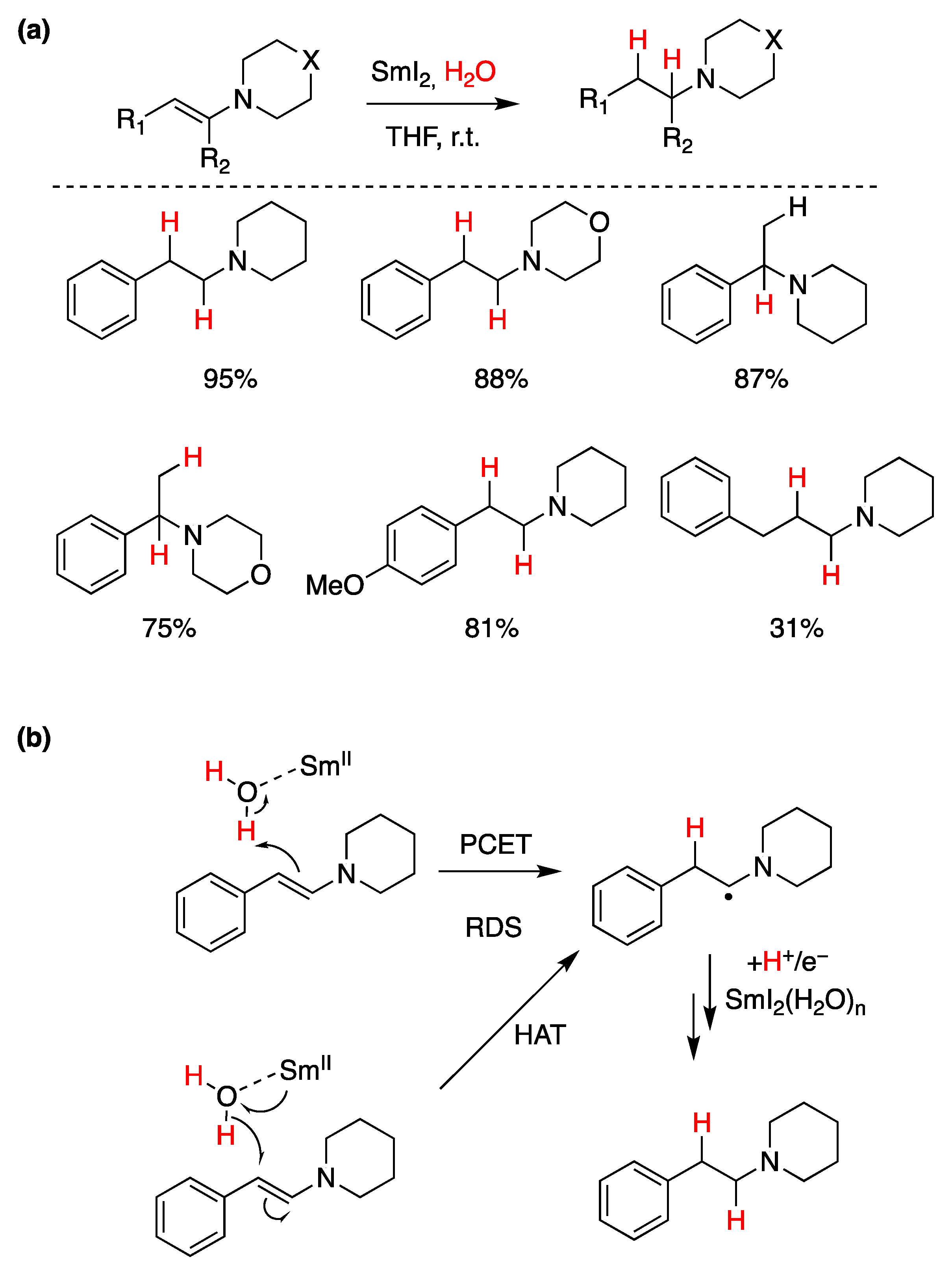
5.3. Samarium(II) Iodide (SmI2)
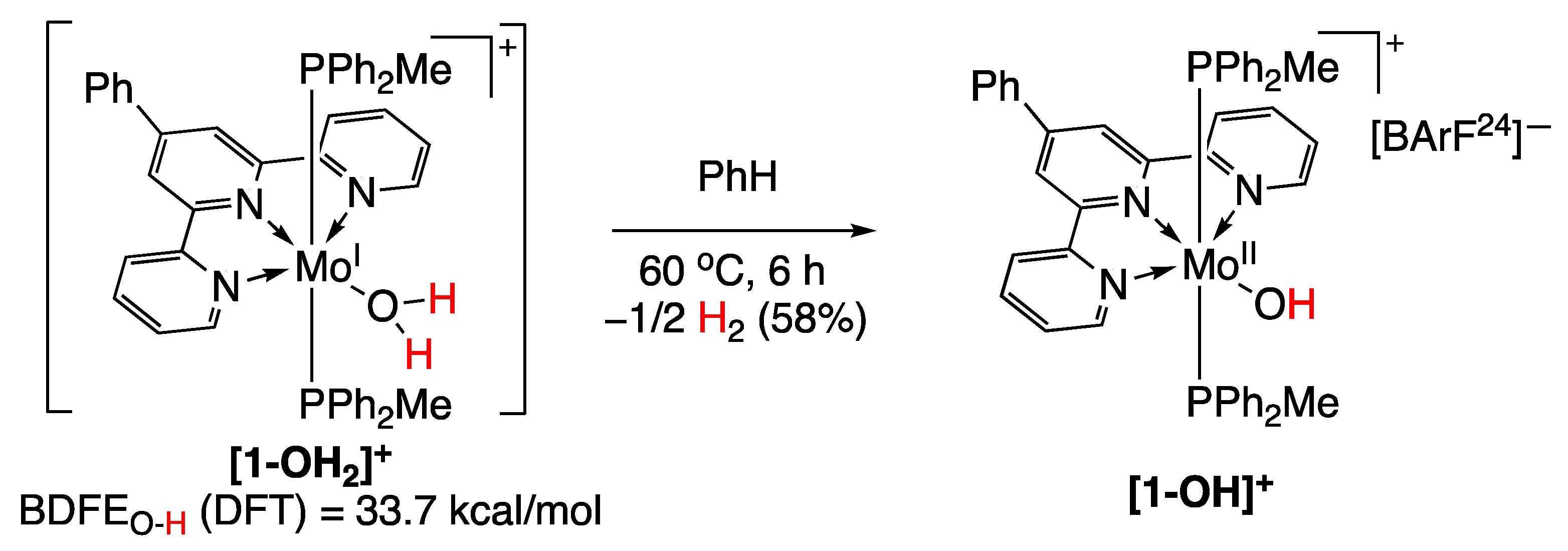
5.4. Molybdenum Complexes
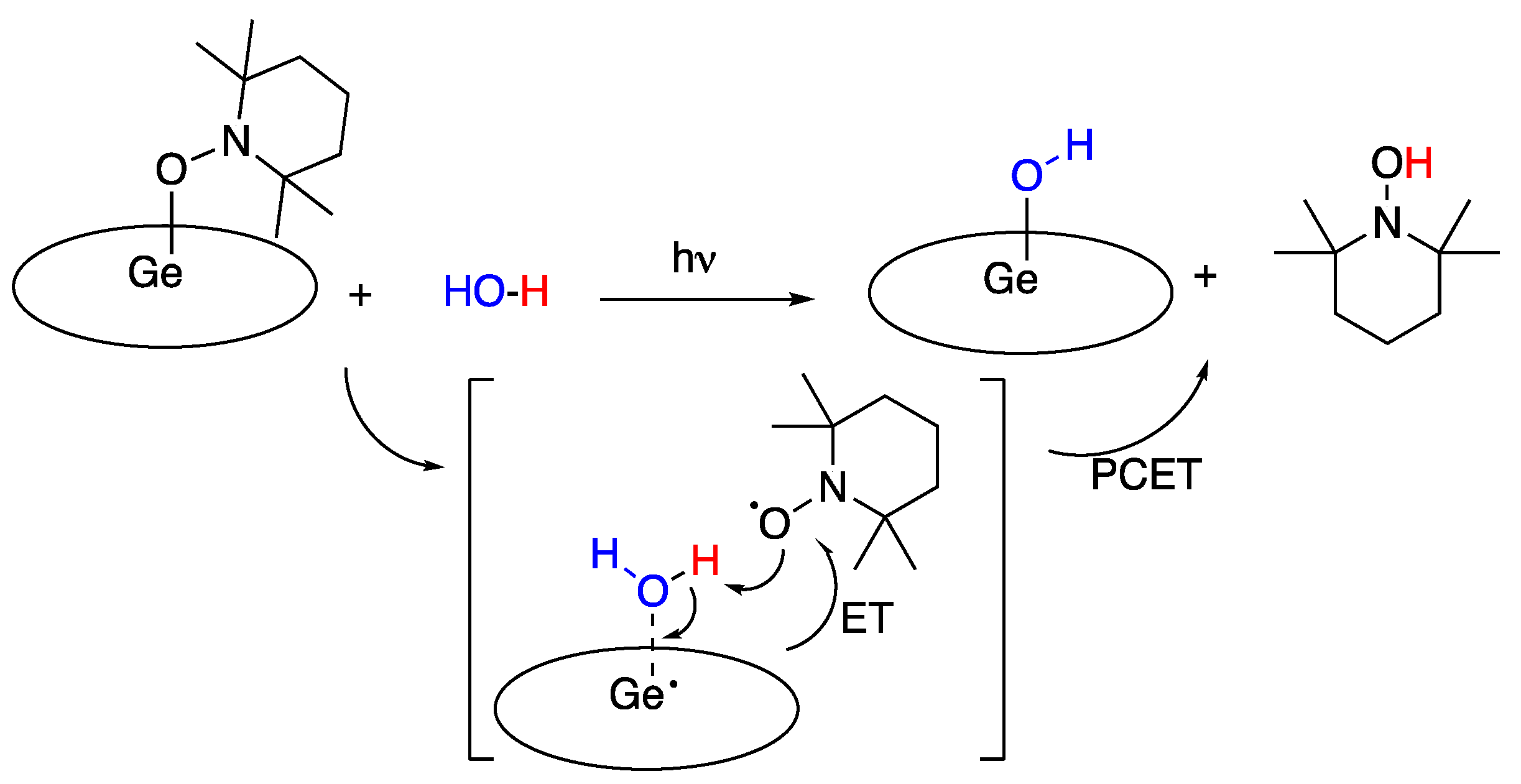
5.5. Germanium Corrole Complex
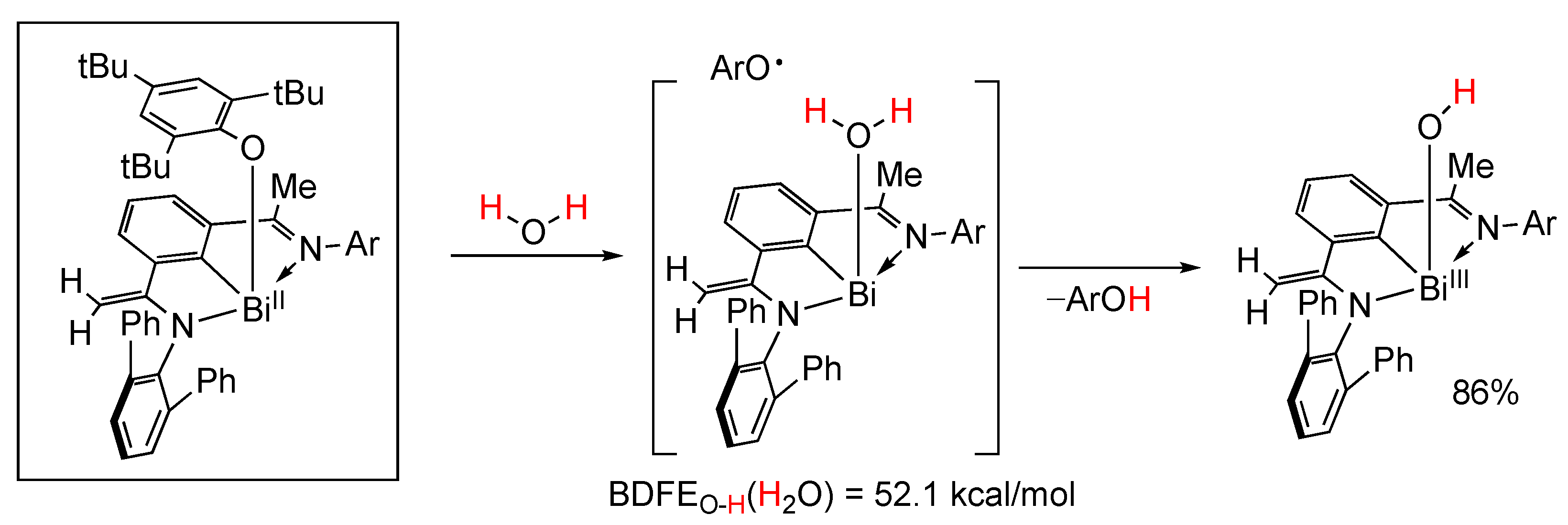
5.6. Organobismuth(II) Species
6. Photocatalysis in Water and Aqueous Media
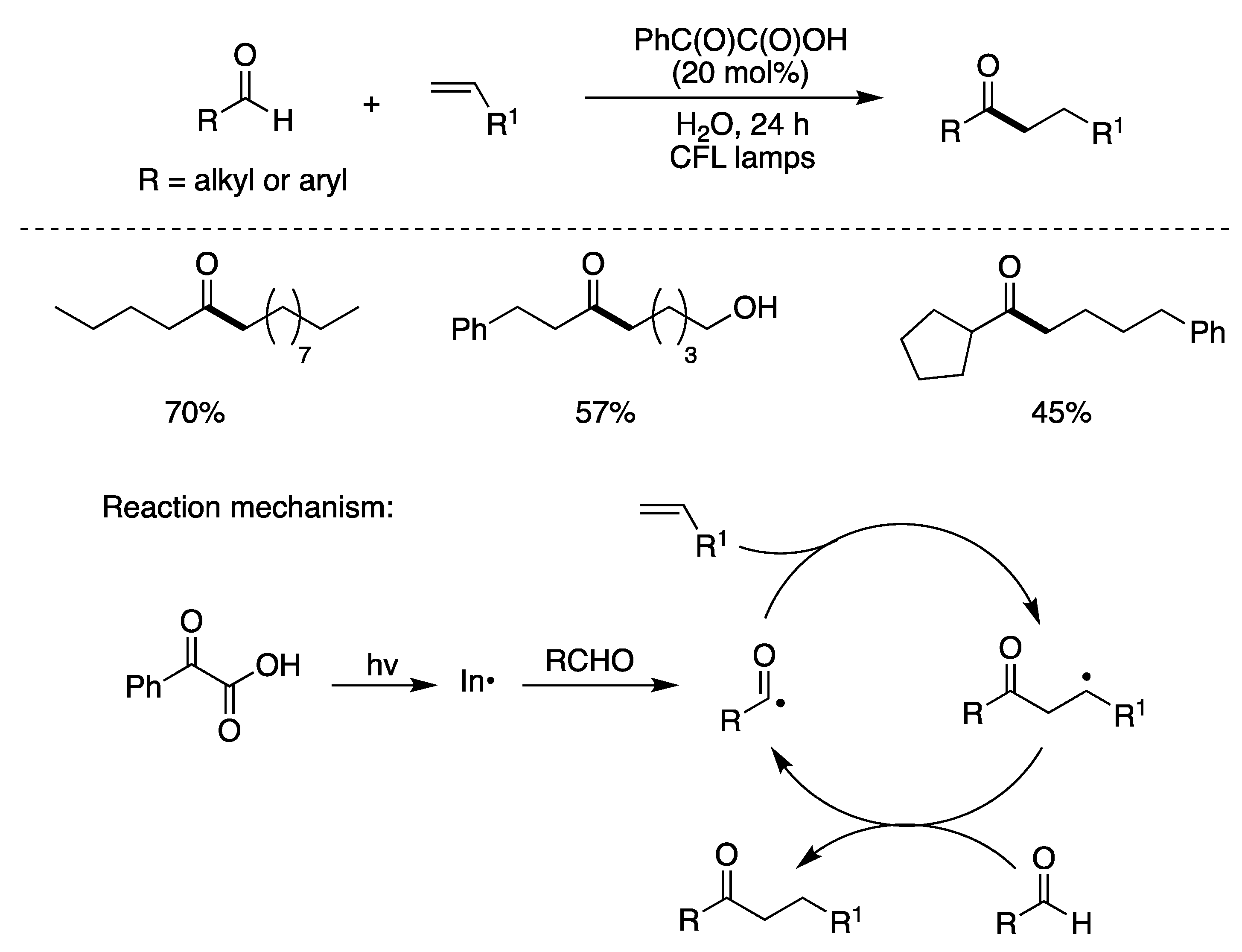
6.1. Photocatalyzed Radical Additions to Carbon–Carbon Multiple Bonds in Aqueous Media
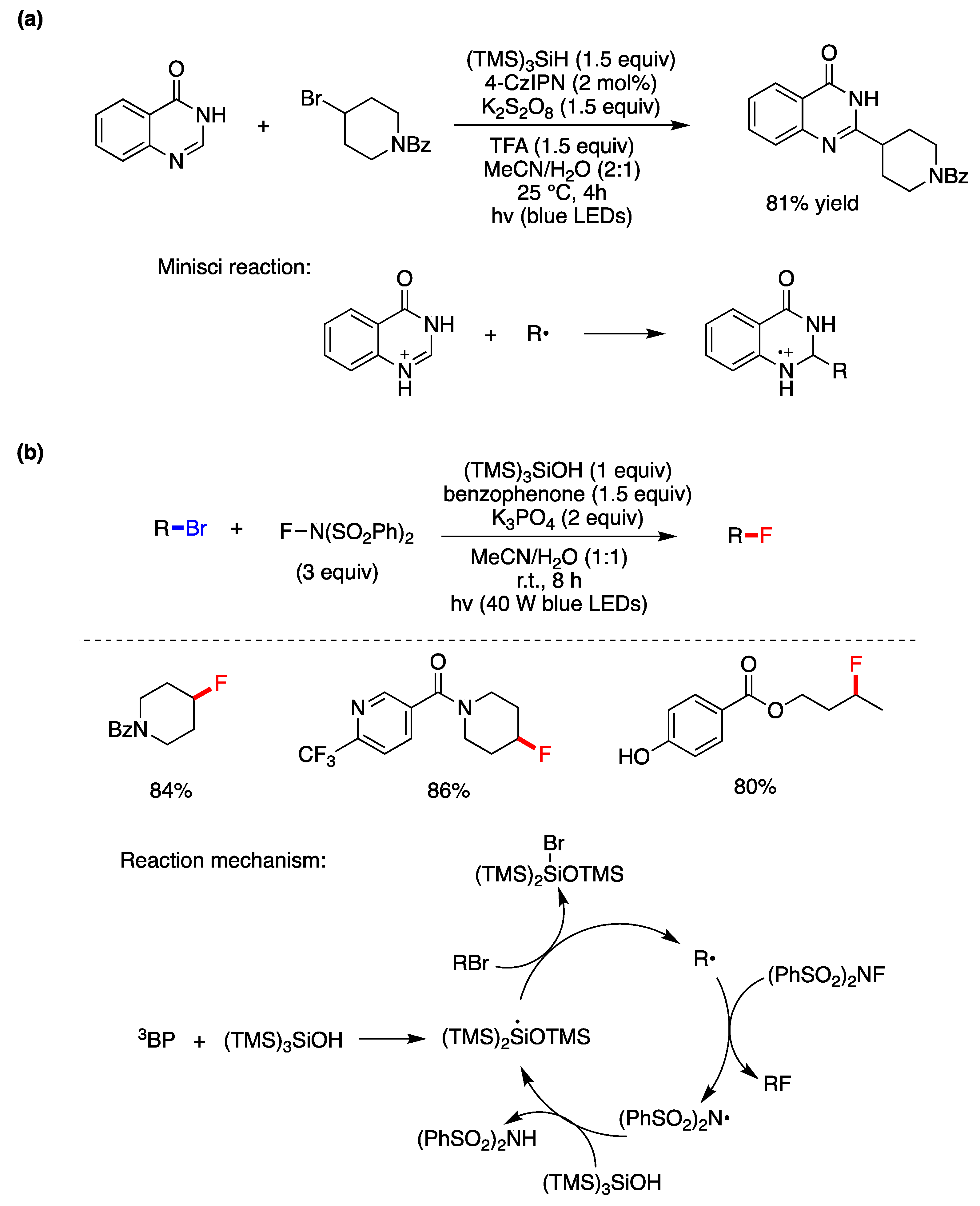
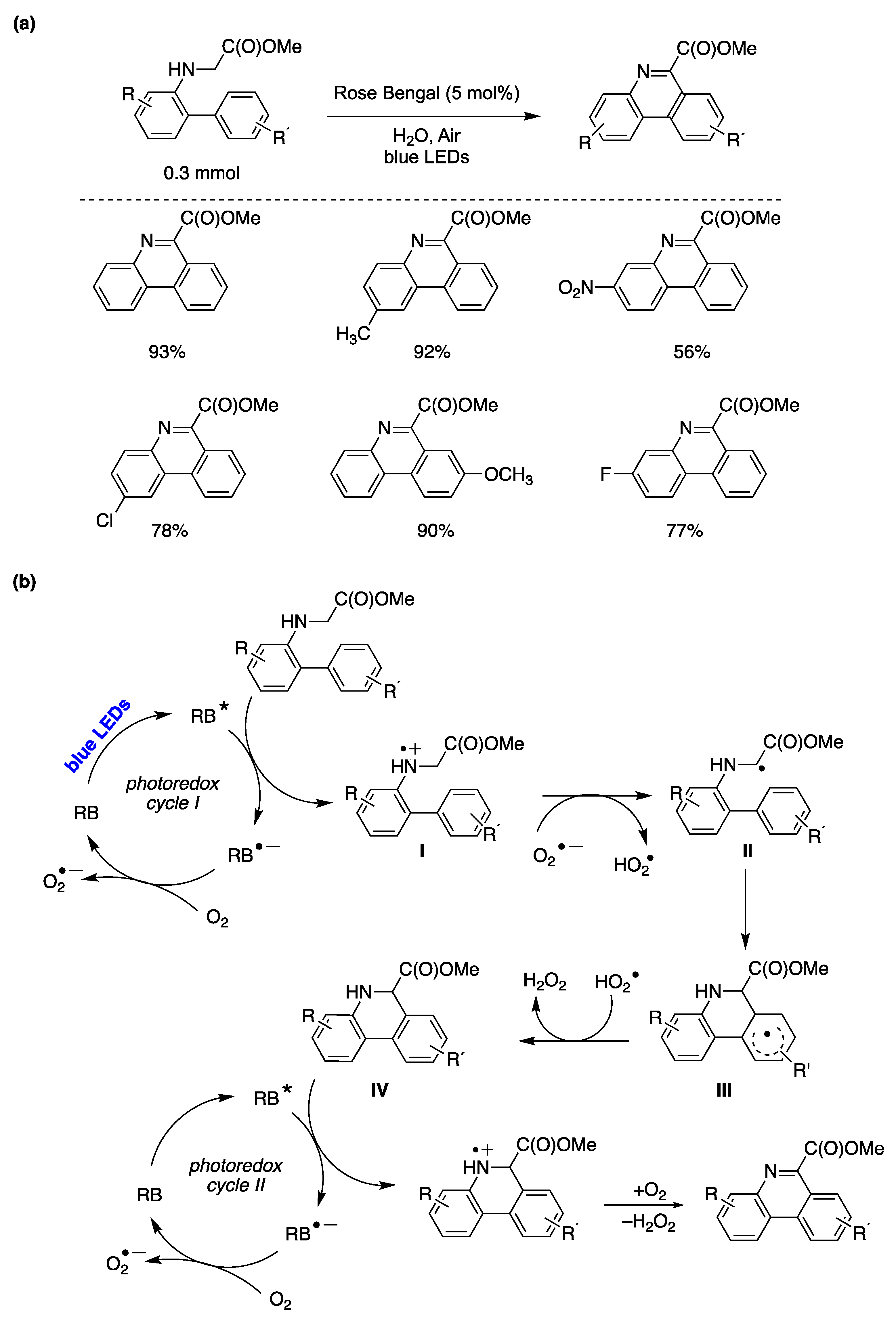
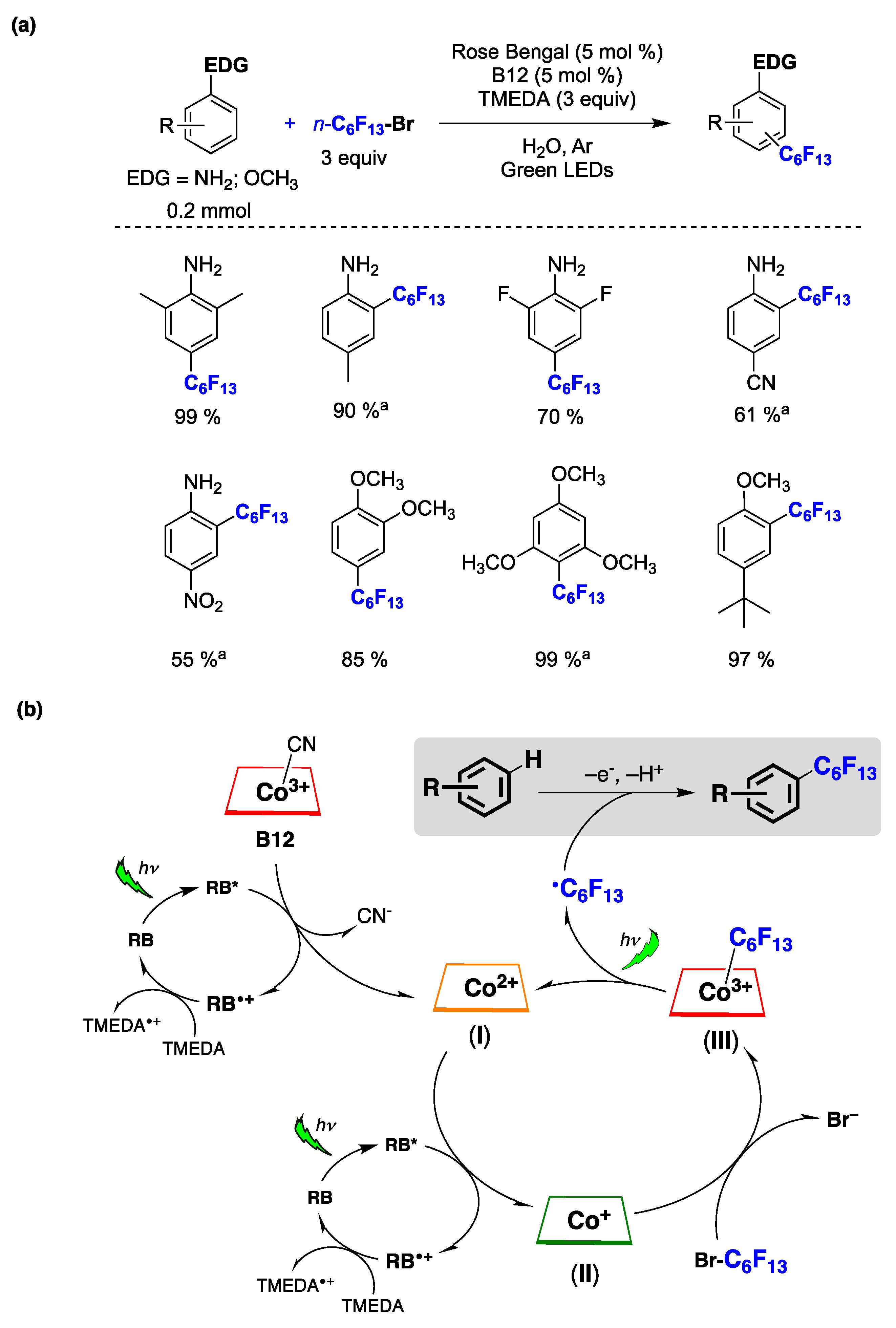
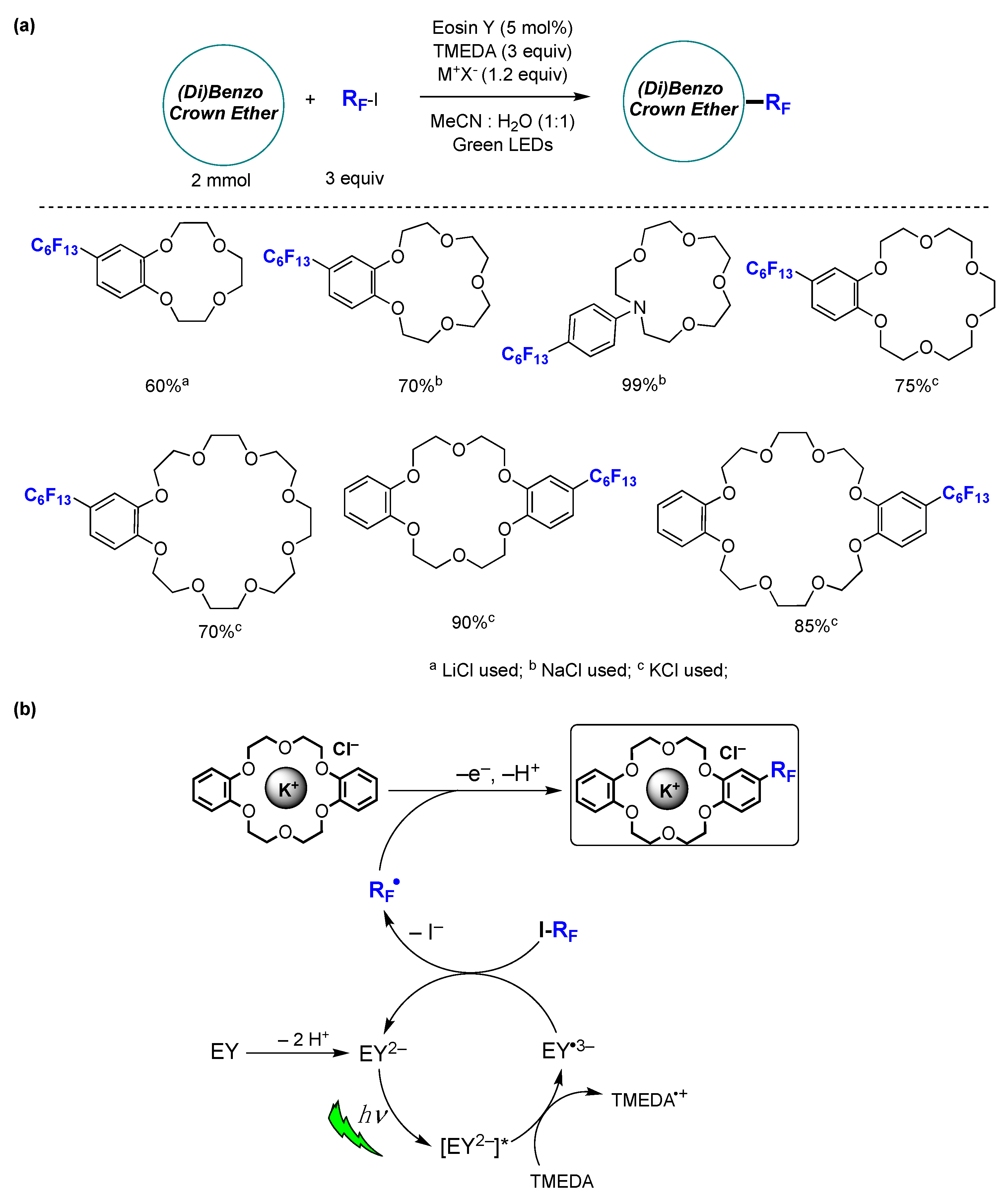
6.2. Photocatalyzed Homolytic Aromatic Substitutions in Aqueous Media
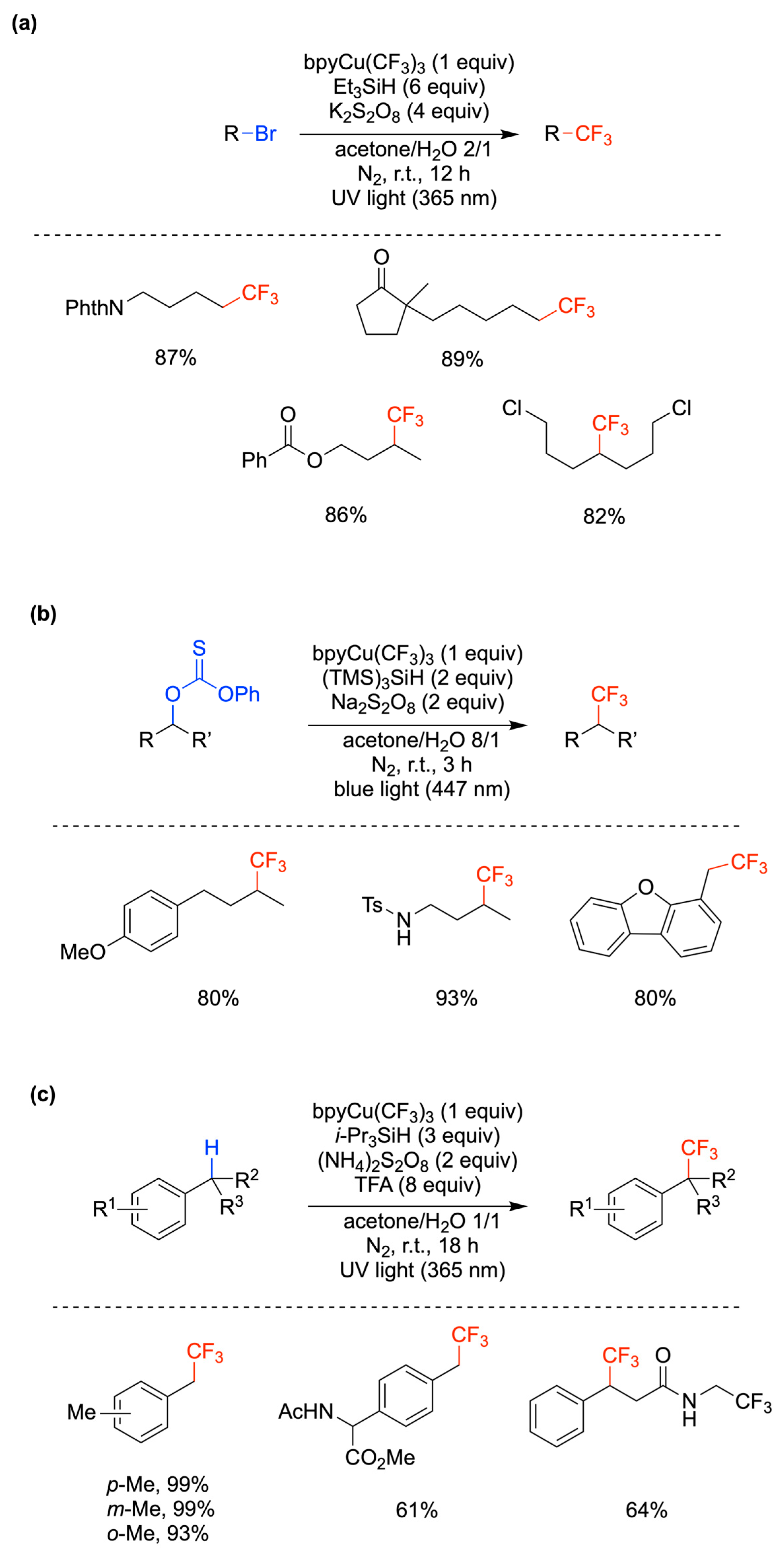
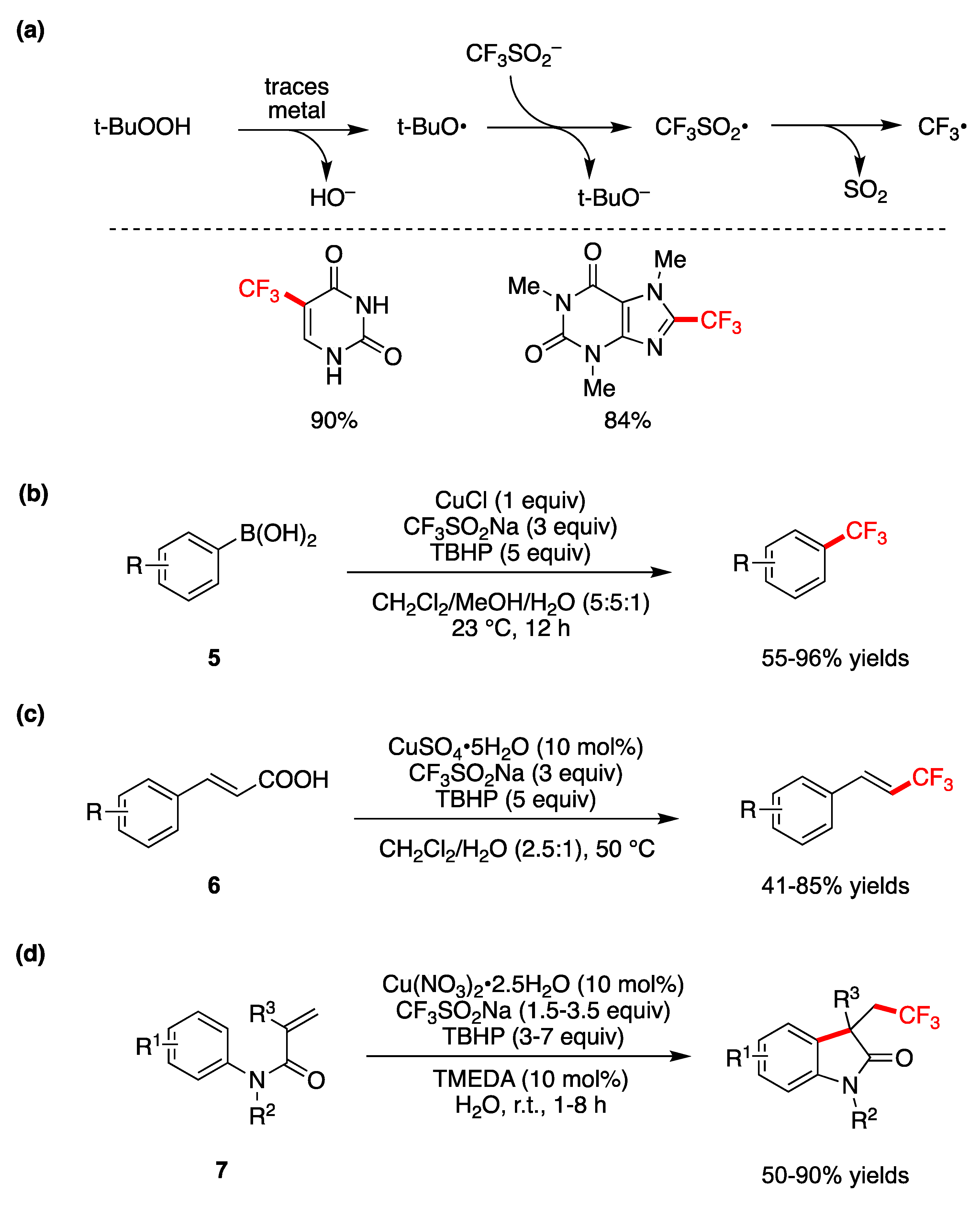
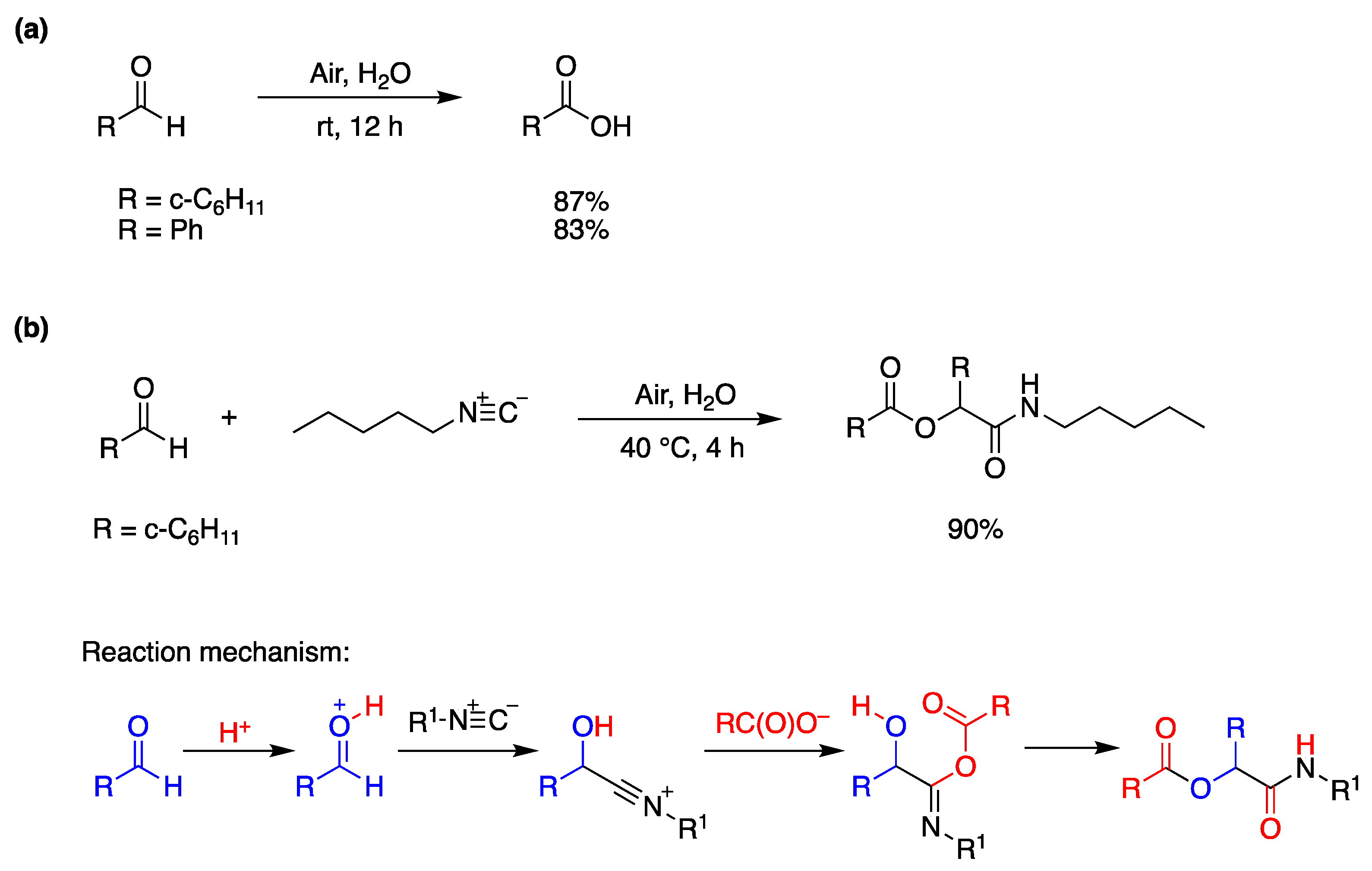
6.3. Miscellaneous
7. Bioinspiration from Biological Reactivity
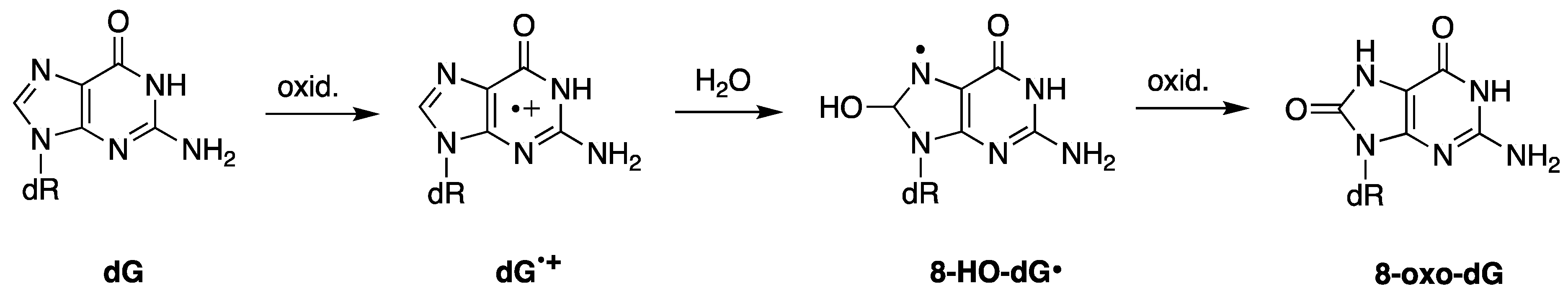
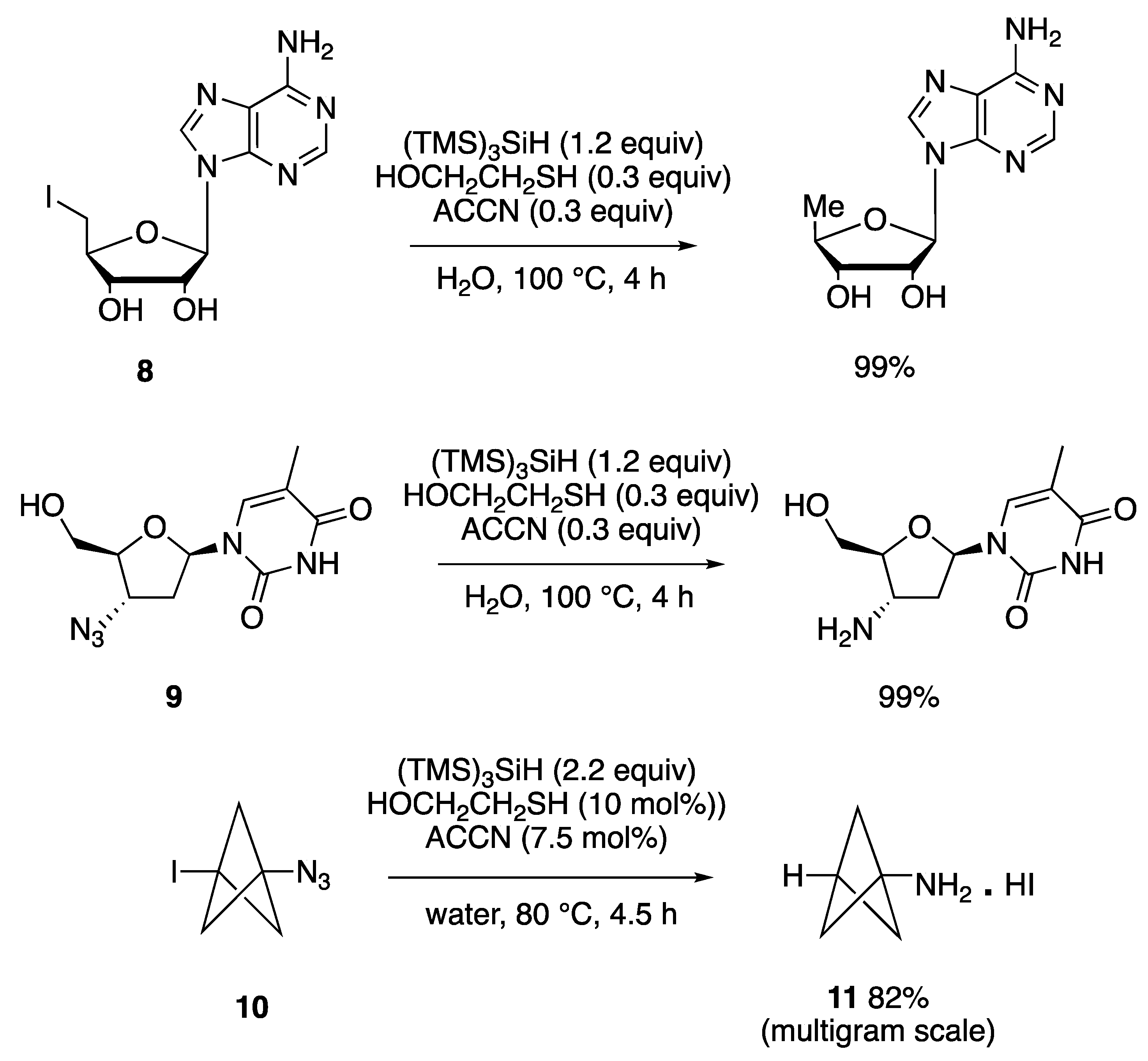
7.1. Purine 5′,8-Cyclo-2′-deoxynucleosides
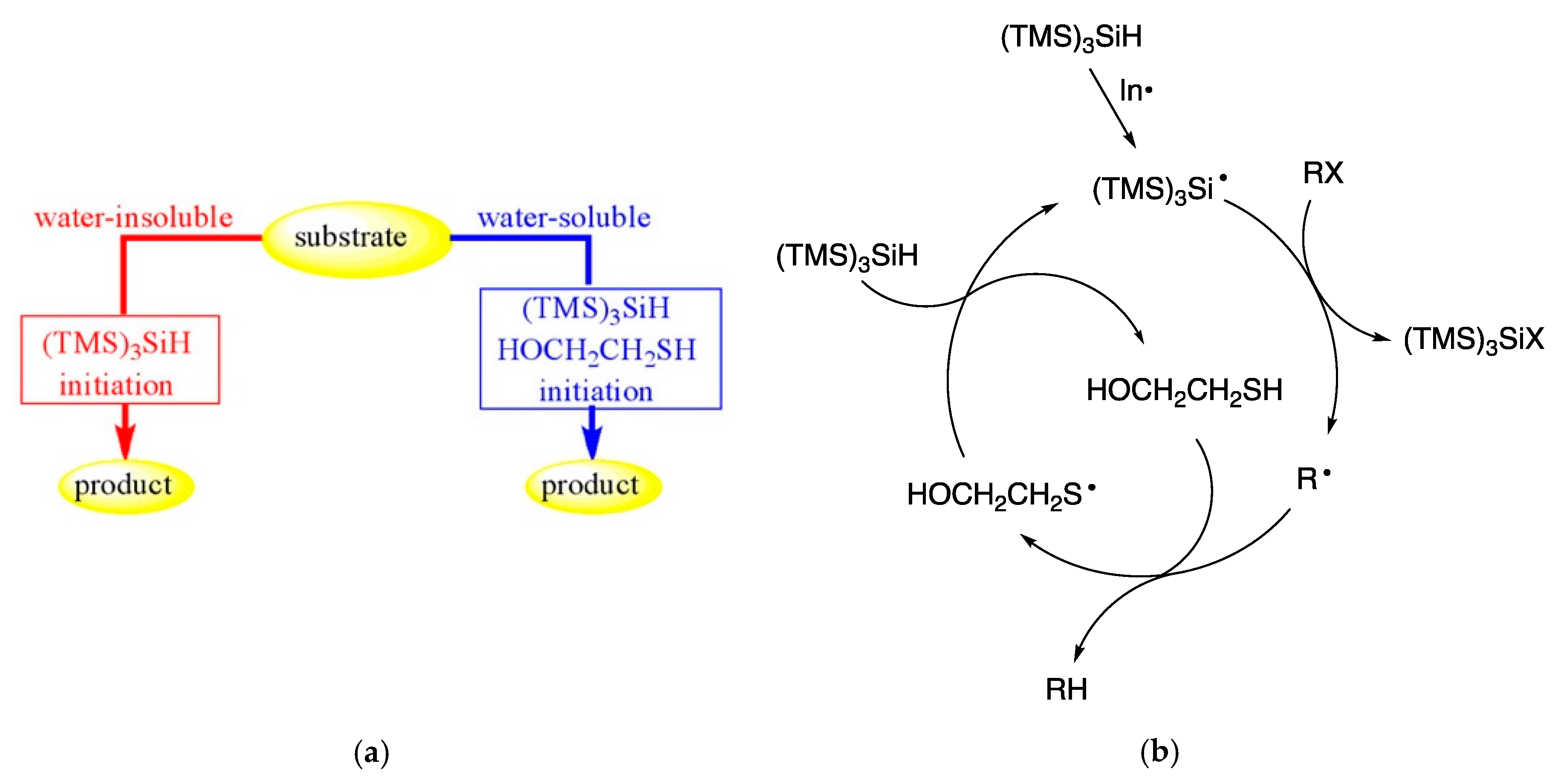
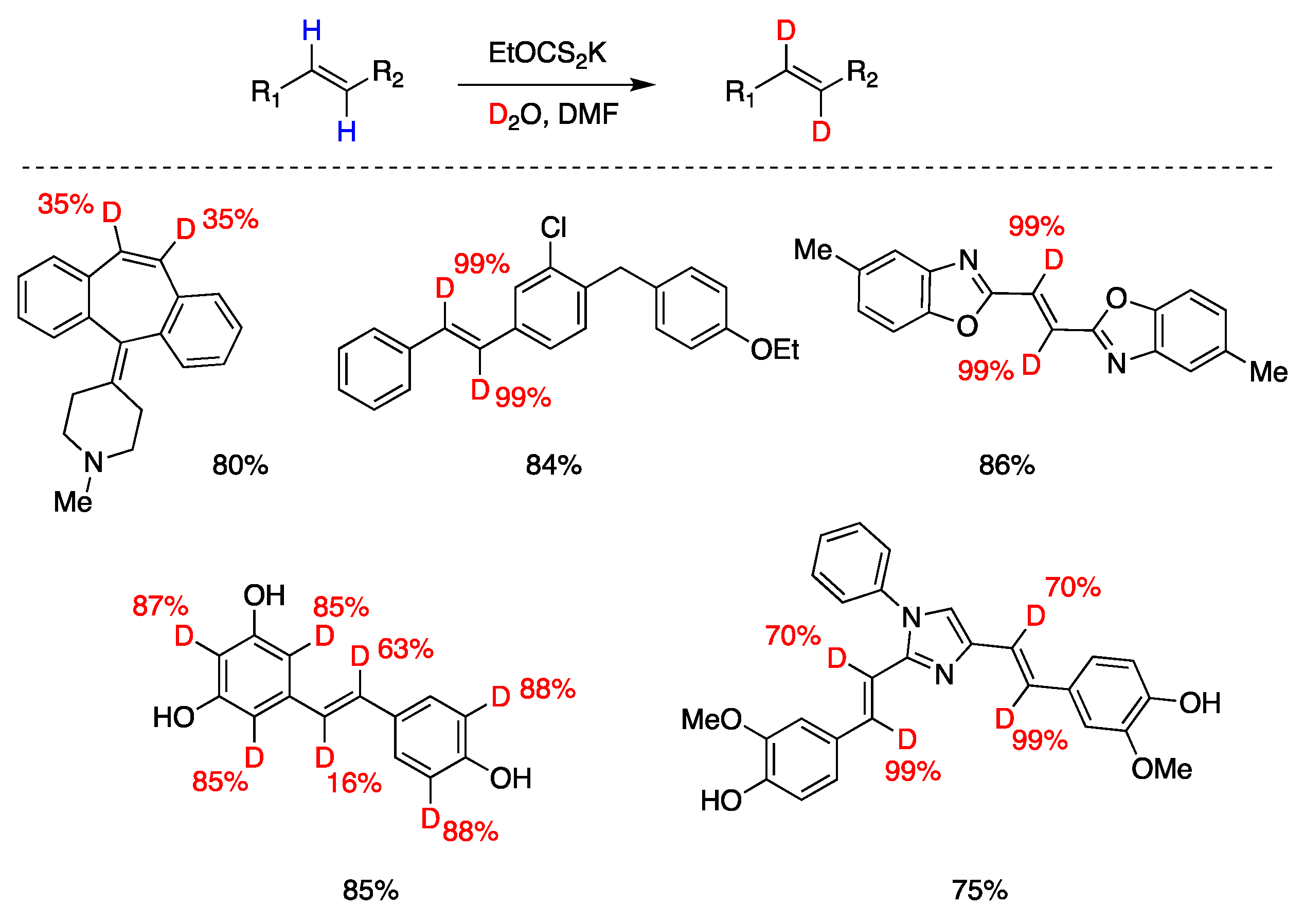
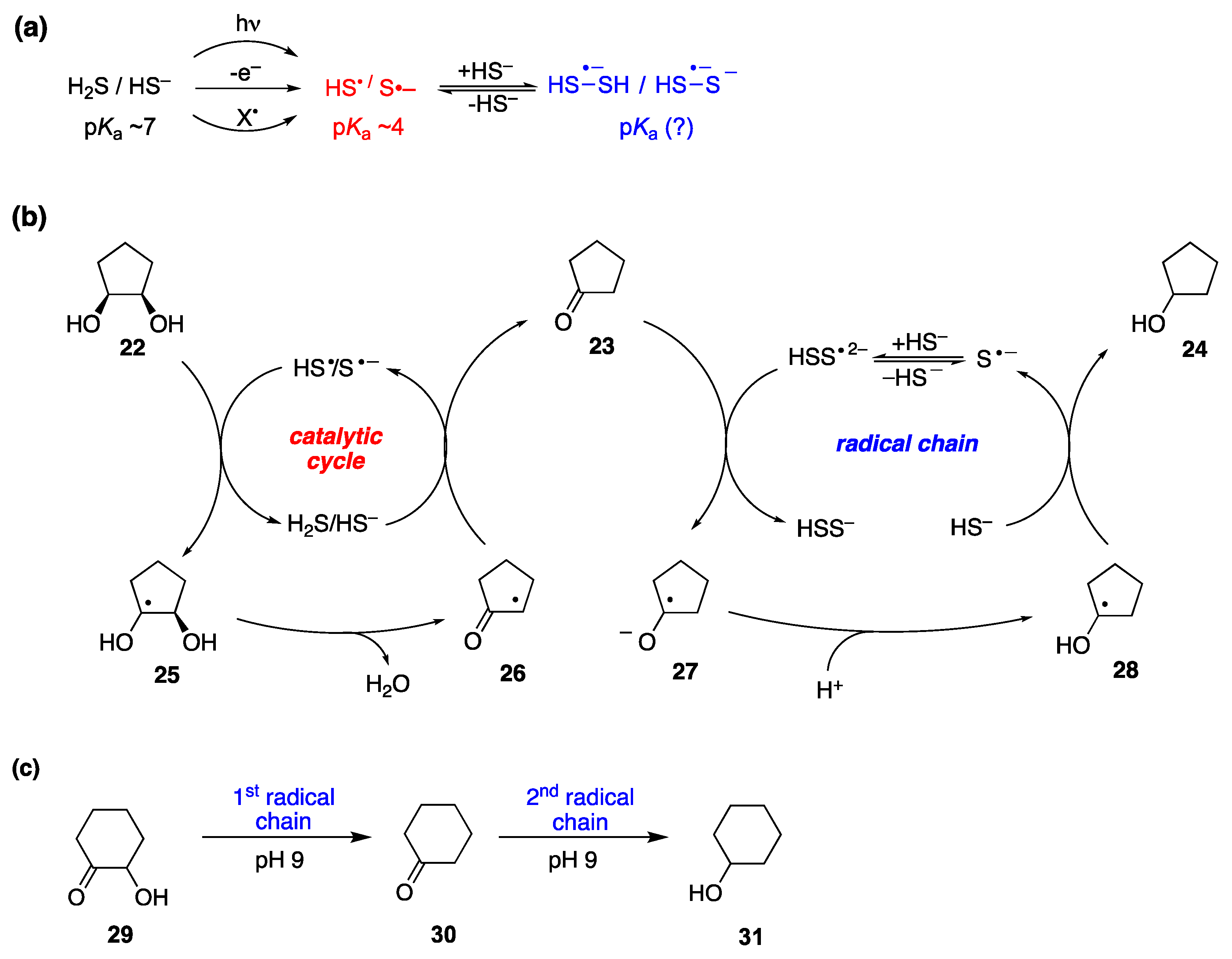
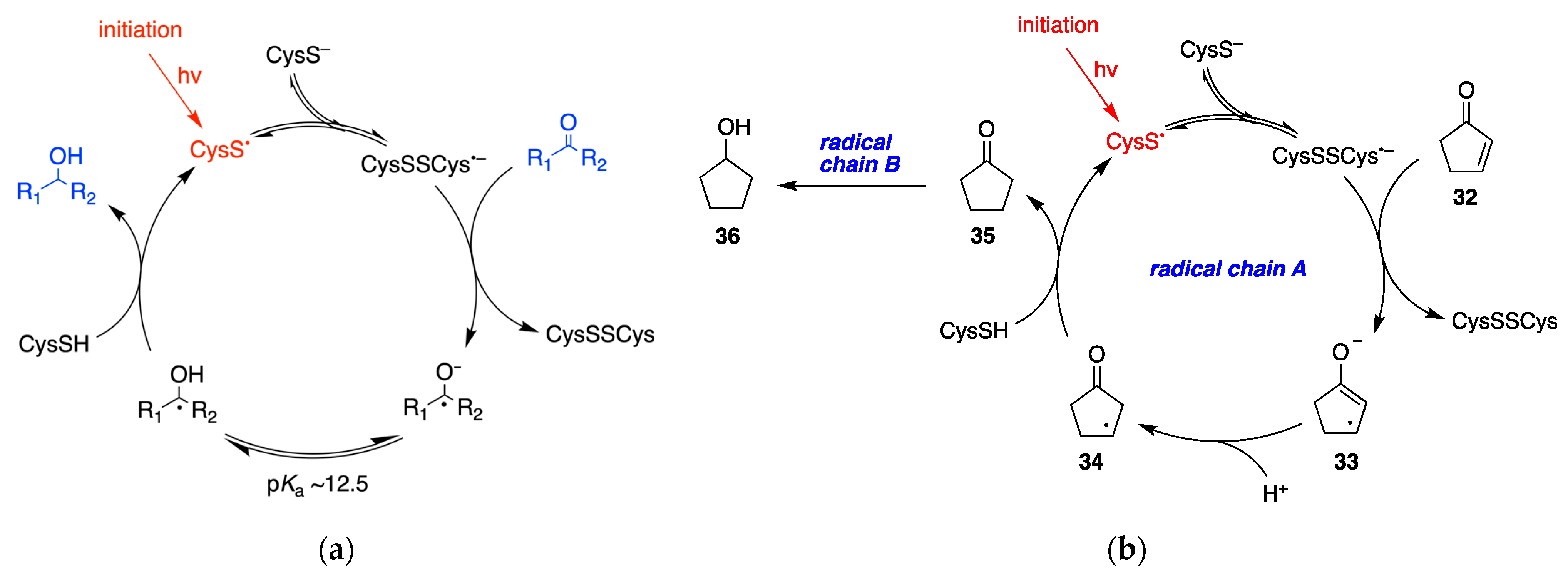
7.2. Disulfide Radical Anion as a Reductant


8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breslow, R. Hydrophobic Effects on Simple Organic Reactions in Water. Acc. Chem. Res. 1991, 24, 159–164. [Google Scholar] [CrossRef]
- Li, C.-J. Organic Reactions in Aqueous Media with a Focus on Carbon-Carbon Bond Formations. Chem. Rev. 1993, 93, 2023–2035. [Google Scholar] [CrossRef]
- Lindström, U.M. Stereoselective Organic Reactions in Water. Chem. Rev. 2002, 102, 2751–2772. [Google Scholar] [CrossRef] [PubMed]
- Breslow, R. Determining the Geometries of Transition States by Use of Antihydrophobic Additives in Water. Acc. Chem. Res. 2004, 37, 471–478. [Google Scholar] [CrossRef]
- Li, C.-J. Organic Reactions in Aqueous Media with a Focus on Carbon-Carbon Bond Formations: A Decade Update. Chem. Rev. 2005, 105, 3095–3165. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. (Ed.) Water in Organic Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Levin, E.; Ivry, E.; Diesendruck, C.E.; Lemcoff, N.G. Water in N-Heterocyclic Carbene-Assisted Catalysis. Chem. Rev. 2015, 115, 4607–4692. [Google Scholar] [CrossRef]
- Narayan, S.; Muldoon, J.; Finn, M.G.; Fokin, V.V.; Kolb, H.C.; Sharpless, K.B. “On Water”: Unique Reactivity of Organic Compounds in Aqueous Suspension. Angew. Chem. Int. Ed. 2005, 44, 3275–3279. [Google Scholar] [CrossRef]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef]
- Butler, R.N.; Coyne, A.G. Water: Nature’s Reaction Enforcer–Comparative Effects for Organic Synthesis “In-Water” and “On-Water”. Chem. Rev. 2010, 110, 6302–6337. [Google Scholar] [CrossRef]
- Simon, M.O.; Li, C.J. Green Chemistry Oriented Organic Synthesis in Water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef]
- Butler, R.N.; Coyne, A.G. Organic Synthesis Reactions on-Water at the Organic—Liquid Water Interface. Org. Biomol. Chem. 2016, 14, 9945–9960. [Google Scholar] [CrossRef] [PubMed]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Romney, D.K.; Arnold, F.H.; Lipshutz, B.H.; Li, C.-J. Chemistry Takes a Bath: Reactions in Aqueous Media. J. Org. Chem. 2018, 83, 7319–7322. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Shinokubo, H.; Oshima, K. Synthetic Radical Reactions in Aqueous Media. Synlett 2002, 674–686. [Google Scholar] [CrossRef]
- Postigo, A. Synthetically useful carbon–carbon and carbon–sulphur bond construction mediated by carbon- and sulphur-centred radicals in water and aqueous media. RSC Adv. 2011, 1, 14–32. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Oshima, K. Free-Radical Reactions. In Water in Organic Synthesis; Kobayashi, S., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2012; pp. 645–677. [Google Scholar]
- Nicewicz, D.A.; MacMillan, D.W.C. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [Google Scholar] [CrossRef]
- Dedon, P.C. The chemical toxicology of 2-deoxyribose oxidation in DNA. Chem. Res. Toxicol. 2008, 24, 206–219. [Google Scholar] [CrossRef]
- Emanuel, C.J.; Newcomb, M.; Ferreri, C.; Chatgilialoglu, C. Kinetics of 2′-deoxyuridin-1′-yl radical reactions. J. Am. Chem. Soc. 1999, 121, 2927–2928. [Google Scholar] [CrossRef]
- Gimisis, T.; Chatgilialoglu, C. Fate of the C-1′ peroxyl radical in the 2′-deoxyuridine system. Chem. Commun. 1998, 1249–1250. [Google Scholar] [CrossRef]
- Schmittel, M.; Burghart, A. Understanding Reactivity Patterns of Radical Cations. Angew. Chem. Int. Ed. Engl. 1997, 36, 2550–2589. [Google Scholar] [CrossRef]
- Chatgilialoglu, C. The Two Faces of the Guanyl Radical: Molecular Context and Behavior. Molecules 2021, 26, 3511. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Capobianco, A.; Bobrowski, K.; Peluso, A.; Chatgilialoglu, C. Hydroxyl Radical vs. One-Electron Oxidation Reactivities in an Alternating GC Double-Stranded Oligonucleotide: A New Type Electron Hole Stabilization. Biomolecules 2023, 13, 1493. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Lloyd, R.S. (Eds.) DNA Damage, DNA Repair and Disease; Royal Society of Chemistry: Croydon, UK, 2021. [Google Scholar]
- Voutyritsa, E.; Kokotos, C.G. Green Metal-Free Photochemical Hydroacylation of Unactivated Olefins. Angew. Chem. Int. Ed. 2020, 59, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. Thiyl Radicals in Biology: Their Role as a “Molecular Switch” Central to Cellular Oxidative Stress. In S-Centered Radicals; Alfassi, Z.B., Ed.; J. Wiley & Sons: Chichester, UK, 1999; p. 289. [Google Scholar]
- Chatgilialoglu, C. RSH/(TMS)3SiH Reducing System: A Tool for Measuring Rate Constants for Reactions of Radicals with Thiols. Helv. Chim. Acta 2006, 89, 2387–2398. [Google Scholar] [CrossRef]
- Ross, A.B.; Mallard, W.G.; Helman, W.P.; Buxton, G.V.; Huie, R.E.; Neta, P. 5NDRL-NIST Solution Kinetic Database—Vers. 36; Notre Dame Radiation Laboratory, Notre Dame, IN and NIST Standard Reference Data: Gaithersburg, MD, USA, 1998.
- Tronche, C.; Martinez, F.N.; Horner, J.H.; Newcomb, M.; Senn, M.; Giese, B. Polar Substituent and Solvent Effects on the Kinetics of Radical Reactions with Thiols. Tetrahedron Lett. 1996, 37, 5845–5848. [Google Scholar] [CrossRef]
- Von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair, A Chemical Perspective; Springer: Berlin/Heidelberg, Germany, 2006; p. 144. [Google Scholar]
- Reid, D.L.; Shustov, G.V.; Armstrong, D.A.; Rauk, A.; Schuchmann, M.N.; Akhlaq, M.S.; von Sonntag, C. H-atom abstraction from thiols by C-centered radicals. A theoretical and experimental study of reaction rates. Phys. Chem. Chem. Phys. 2002, 4, 2965–2974. [Google Scholar] [CrossRef]
- Buback, M.; Hutchinson, R.A.; Lacík, I. Radical polymerization kinetics of water-soluble monomers. Prog. Polym. Sci. 2023, 138, 101645. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Studer, A. (Eds.) Encyclopedia of Radicals in Chemistry, Biology and Materials; Wiley: Chichester, UK, 2012. [Google Scholar]
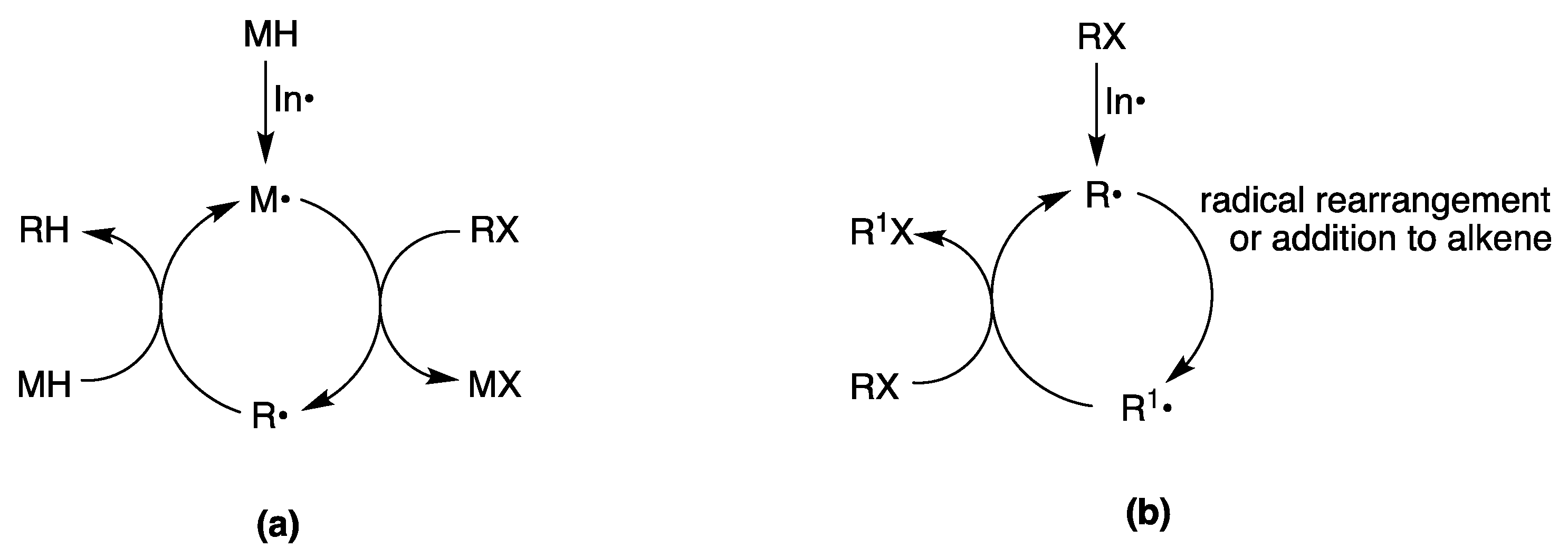
- Baguley, P.A.; Walton, J.C. Flight from the Tyranny of Tin: The Quest for Practical Radical Sources Free from Metal Encumbrances. Angew. Chem. Int. Ed. 1998, 37, 3072–3082. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Landais, Y.; Timokhin, V.I. Thirty Years of (TMS)3SiH: A Milestone in Radical-Based Synthetic Chemistry. Chem. Rev. 2018, 118, 6516–6572. [Google Scholar] [CrossRef]
- Light, J.; Breslow, R. A water soluble tin hydride reagent. Tetrahedron Lett. 1990, 31, 2957–2958. [Google Scholar] [CrossRef]
- Rai, R.; Collum, D.B. Reductions and radical cyclizations of aryl and alkyl bromides mediated by NaBH4 in aqueous base. Tetrahedron Lett. 1994, 35, 6221–6224. [Google Scholar] [CrossRef]
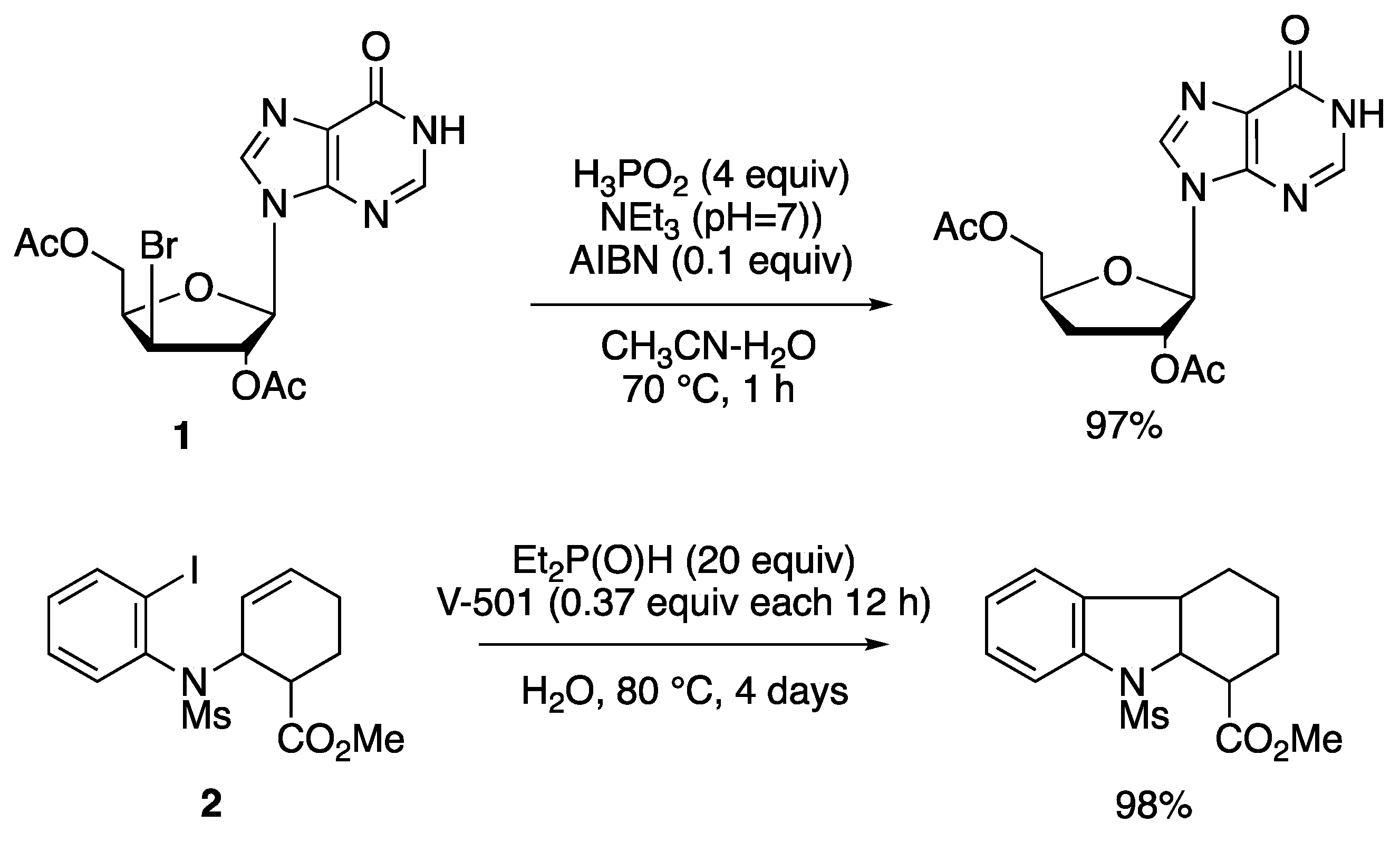
- Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Hypophosphorous acid and its salts: New reagents for radical chain deoxygenation, dehalogenation and deamination. Tetrahedron Lett. 1992, 33, 5709–5712. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Jang, D.O.; Jaszberenyi, J.C. The invention of radical reactions. Part 32. Radical deoxygenations, dehalogenations, and deaminations with dialkyl phosphites and hypophosphorous acid as hydrogen sources. J. Org. Chem. 1993, 58, 6838–6842. [Google Scholar] [CrossRef]
- Kita, Y.; Nambu, H.; Ramesh, N.G.; Anilkumar, G.; Matsugi, M. A Novel and Efficient Methodology for the C–C Bond Forming Radical Cyclization of Hydrophobic Substrates in Water. Org. Lett. 2001, 3, 1157–1160. [Google Scholar] [CrossRef]
- Guyon, C.; Metay, E.; Popowycz, F.; Lemaire, M. Synthetic applications of hypophosphite derivatives in reduction. Org. Biomol. Chem. 2015, 13, 7879–7906. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Katayama, S.; Hirose, N.; Naito, M.; Izawa, K. Radical deoxygenation and dehalogenation of nucleoside derivatives with hypophosphorous acid and dialkyl phosphites. Tetrahedron Lett. 2001, 42, 7605–7608. [Google Scholar] [CrossRef]
- Khan, T.A.; Tripoli, R.; Crawford, J.J.; Martin, C.G.; Murphy, J.A. Diethylphosphine Oxide (DEPO): High-Yielding and Facile Preparation of Indolones in Water. Org. Lett. 2003, 5, 2971–2974. [Google Scholar] [CrossRef]
- Perkins, J.J.; Schubert, J.W.; Streckfuss, E.C.; Balsells, J.; ElMarrouni, A. Photoredox catalysis for silyl-mediated C-H alkylation of heterocycles with non-activated alkyl bromides. Eur. J. Org. Chem. 2020, 2020, 1515–1522. [Google Scholar] [CrossRef]
- Lovett, G.H.; Chen, S.; Xue, X.S.; Houk, K.N.; MacMillan, D.W.C. Open-shell fluorination of alkyl bromides: Unexpected selectivity in a silyl radical-mediated chain process. J. Am. Chem. Soc. 2019, 141, 20031–20036. [Google Scholar] [CrossRef]
- Le, C.; Chen, T.Q.; Liang, T.; Zhang, P.; MacMillan, D.W.C. A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010–1014. [Google Scholar] [CrossRef]
- Jasperse, C.P.; Curran, D.P.; Fevig, T.L. Radical reactions in natural product synthesis. Chem. Rev. 1991, 91, 1237–1286. [Google Scholar] [CrossRef]
- Juliá, F.; Constantin, T.; Leonori, D. Applications of Halogen-Atom Transfer (XAT) for the Generation of Carbon Radicals in Synthetic Photochemistry and Photocatalysis. Chem. Rev. 2022, 122, 2292–2352. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; McFadden, T.R. Understanding Initiation with Triethylboron and Oxygen: The Differences between Low-Oxygen and High-Oxygen Regimes. J. Am. Chem. Soc. 2016, 138, 7741–7752. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, H.; Nakamura, T.; Shinokubo, H.; Oshima, K.; Omoto, K.; Fujimoto, H. Powerful Solvent Effect of Water in Radical Reaction: Triethylborane-Induced Atom-Transfer Radical Cyclization in Water. J. Am. Chem. Soc. 2000, 122, 11041–11047. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K. Triethylborane-Induced Bromine Atom-Transfer Radical Addition in Aqueous Media: Study of the Solvent Effect on Radical Addition Reactions. J. Org. Chem. 2001, 66, 7776–7785. [Google Scholar] [CrossRef]
- McNabb, S.B.; Ueda, M.; Naito, T. Addition of Electrophilic and Heterocyclic Carbon-Centered Radicals to Glyoxylic Oxime Ethers. Org. Lett. 2004, 6, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Shinokubo, H.; Oshima, K. From Alkenylsilanes to Ketones with Air as the Oxidant. Angew. Chem. Int. Ed. 2003, 42, 849–851. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Romine, A.M.; Nebra, N.; Konovalov, A.I.; Martin, E.; Benet-Buchholz, J.; Grushin, V.V. Easy Access to the Copper(III) Anion [Cu(CF3)4]−. Angew. Chem. Int. Ed. 2015, 54, 2745–2749. [Google Scholar] [CrossRef]
- Xiao, H.; Zhang, Z.; Fang, Y.; Zhu, L.; Li, C. Radical trifluoromethylation. Chem. Soc. Rev. 2021, 50, 6308–6319. [Google Scholar] [CrossRef]
- Shen, H.; Liu, Z.; Zhang, P.; Tan, X.; Zhang, Z.; Li, C. Trifluoromethylation of Alkyl Radicals in Aqueous Solution. J. Am. Chem. Soc. 2017, 139, 9843–9846. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Cook, S.P. Interrupting the Barton–McCombie Reaction: Aqueous Deoxygenative Trifluoromethylation of O-Alkyl Thiocarbonates. Org. Lett. 2021, 23, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; AbuSalim, D.I.; Cook, S.P. Aqueous Benzylic C−H Trifluoromethylation for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 12378–12382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Shen, H.; Zhu, L.; Cao, W.; Li, C. Radical C(sp2)–H Trifluoromethylation of Aldehydes in Aqueous Solution. Org. Lett. 2018, 20, 7062–7065. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, Z.; Shen, H.; Zhang, P.; Zhang, Z.; Li, C. Silver-Catalyzed Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc. 2017, 139, 12430–12433. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.N.; Brueckl, T.; Baxter, R.D.; Fujiwara, Y.; Seiple, I.B.; Su, S.; Blackmond, D.G.; Baran, P.S. Innate C-H trifluoromethylation of heterocycles. Proc. Natl. Acad. Sci. USA 2011, 108, 14411–14415. [Google Scholar] [CrossRef]
- Ye, Y.D.; Kuenzi, S.A.; Sanford, M.S. Practical Method for the Cu-Mediated Trifluoromethylation of Arylboronic Acids with CF3 Radicals Derived from NaSO2CF3 and tert-Butyl Hydroperoxide (TBHP). Org. Lett. 2012, 14, 4979–4981. [Google Scholar] [CrossRef]
- Li, Z.J.; Cui, Z.L.; Liu, Z.Q. Copper- and Iron-Catalyzed Decarboxylative Tri- and Difluoromethylation of α,β-Unsaturated Carboxylic Acids with CF3SO2Na and (CF2HSO2)2Zn via a Radical Process. Org. Lett. 2013, 15, 406–409. [Google Scholar] [CrossRef]
- Yang, F.; Klumphu, P.; Liang, Y.-M.; Lipshutz, B.H. Copper-catalyzed trifluoromethylation of N-arylacrylamides “on water” at room temperature. Chem. Commun. 2014, 50, 936–938. [Google Scholar] [CrossRef]
- Shapiro, N.; Vigalok, A. Highly Efficient Organic Reactions “on Water”, “in Water”, and Both. Angew. Chem. Int. Ed. 2008, 47, 2849–2852. [Google Scholar] [CrossRef]
- Papadopoulos, G.N.; Voutyritsa, E.; Kaplaneris, N.; Kokotos, C.G. Green Photo-Organocatalytic C—H Activation of Aldehydes: Selective Hydroacylation of Electron-Deficient Alkenes. Chem. Eur. J. 2018, 24, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Crich, D.; Komatsu, M.; Ryu, I. Chemistry of acyl radicals. Chem. Rev. 1999, 99, 1991–2070. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C. Organosilanes in Radical Chemistry; Wiley: Chichester, UK, 2004. [Google Scholar]
- Chatgilialoglu, C. (Me3Si)3SiH: Twenty years after its discovery as a radical-based reducing agent. Chem.—Eur. J. 2008, 14, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Lalevée, J. Recent Applications of the (TMS)3SiH Radical-Based Reagent. Molecules 2012, 17, 527–555. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.; Ferreri, C.; Navacchia, M.L.; Chatgilialoglu, C. The radical-based reduction with (TMS)3SiH “on water”. Synlett 2005, 2854–2856. [Google Scholar] [CrossRef]
- Postigo, A.; Kopsov, S.; Ferreri, C.; Chatgilialoglu, C. Radical reactions in aqueous medium using (Me3Si)3SiH. Org. Lett. 2007, 9, 5159–5162. [Google Scholar] [CrossRef]
- Postigo, A.; Kopsov, S.; Zlotsky, S.S.; Ferreri, C.; Chatgilialoglu, C. Hydrosilylation of C-C Multiple Bonds Using (Me3Si)3SiH in Water. Comparative Study of the Radical Initiation Step. Organometallics 2009, 28, 3282–3287. [Google Scholar] [CrossRef]
- Goh, Y.L.; Tam, E.K.W.; Bernardo, P.H.; Cheong, C.B.; Johannes, C.W.; William, A.D.; Adsool, V.A. A New Route to Bicyclo [1.1.1]pentan-1-amine from 1-Azido-3-iodobicyclo[1.1.1]pentane. Org. Lett. 2014, 16, 1884–1887. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Postigo, A. (Me3Si)3SiH-Mediated Intermolecular Radical Perfluoroalkylation. Reactions of Olefins in Water. J. Org. Chem. 2010, 75, 6141–6148. [Google Scholar] [CrossRef]
- Wu, L.-J.; Tan, F.-L.; Li, M.; Song, R.-J.; Li, J.-H. Fe-Catalyzed oxidative spirocyclization of N-arylpropiolamides with silanes and TBHP involving the formation of C–Si bonds. Org. Chem. Front. 2017, 4, 350–353. [Google Scholar] [CrossRef]
- Zhu, J.; Cui, W.-C.; Wang, S.; Yao, Z.-J. Radical Hydrosilylation of Alkynes Catalyzed by Eosin Y and Thiol under Visible Light Irradiation. Org. Lett. 2018, 20, 3174–3178. [Google Scholar] [CrossRef] [PubMed]
- Pickford, H.D.; Nugent, J.; Owen, B.; Mousseau, J.J.; Smith, R.C.; Anderson, E.A. Twofold Radical-Based Synthesis of N,C-Difunctionalized Bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2021, 143, 9729–9736. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.K.; Jang, D.O. Radical “On Water” Addition to the C═N Bond of Hydrazones: A Synthesis of Isoindolinone Derivatives. J. Org. Chem. 2018, 83, 7373–7379. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.K.; Jang, D.O. A synthetic route toward α,α-dialkyl α-amino ester derivatives via radical addition to hydrazone derivatives of α-ketoesters under ‘‘on-water” conditions. Tetrahedron Lett. 2020, 61, 151411. [Google Scholar] [CrossRef]
- Mori, K.; Shimizu, A.; Horibe, M.; Takei, M.; Awano, N.; Matsuoka, S.; Suzuki, M. Lewis Pair Radical Polymerization “On-Water”. Macromolecules 2021, 54, 3–10. [Google Scholar] [CrossRef]
- Ruscic, B.; Wagner, A.F.; Harding, L.B.; Asher, R.L.; Feller, D.; Dixon, D.A.; Peterson, K.A.; Song, Y.; Qian, X.; Ng, C.-Y.; et al. On the Enthalpy of Formation of Hydroxyl Radical and Gas-Phase Bond Dissociation Energies of Water and Hydroxyl. J. Phys. Chem. A 2002, 106, 2727–2747. [Google Scholar] [CrossRef]
- Mardyukov, A.; Sanchez-Garcia, E.; Crespo-Otero, R.; Sander, W. Interaction and Reaction of the Phenyl Radical with Water: A Source of OH Radicals. Angew. Chem. Int. Ed. 2009, 48, 4804–4807. [Google Scholar] [CrossRef]
- Boekell, N.G.; Flowers, R.A. Coordination-Induced Bond Weakening. Chem. Rev. 2022, 122, 13447–13477. [Google Scholar] [CrossRef]
- Agarwal, R.G.; Coste, S.C.; Groff, B.D.; Heuer, A.M.; Noh, H.; Parada, G.A.; Wise, C.F.; Nichols, E.M.; Warren, J.J.; Mayer, J.M. Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications. Chem. Rev. 2022, 122, 1–49. [Google Scholar] [CrossRef]
- Cuerva, J.M.; Campaña, A.G.; Justicia, J.; Rosales, A.; Oller-López, J.L.; Robles, R.; Cárdenas, D.J.; Buñuel, E.; Oltra, J.E. Water: The Ideal Hydrogen-Atom Source in Free-Radical Chemistry Mediated by TiIII and Other Single-Electron-Transfer Metals? Angew. Chem. Int. Ed. 2006, 45, 5522–5526. [Google Scholar] [CrossRef]
- Kolmar, S.S.; Mayer, J.M. SmI2 (H2O) n Reduction of Electron Rich Enamines by Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2017, 139, 10687–10692. [Google Scholar] [CrossRef] [PubMed]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. Proton-Coupled Electron Transfer in the Reduction of Carbonyls by Samarium Diiodide-Water Complexes. J. Am. Chem. Soc. 2016, 138, 8738–8741. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, D.A.; Wiberg, K.B.; Schacherer, L.N.; Medeiros, M.R.; Wood, J.L. Deoxygenation of Alcohols Employing Water as the Hydrogen Atom Source. J. Am. Chem. Soc. 2005, 127, 12513–12515. [Google Scholar] [CrossRef] [PubMed]
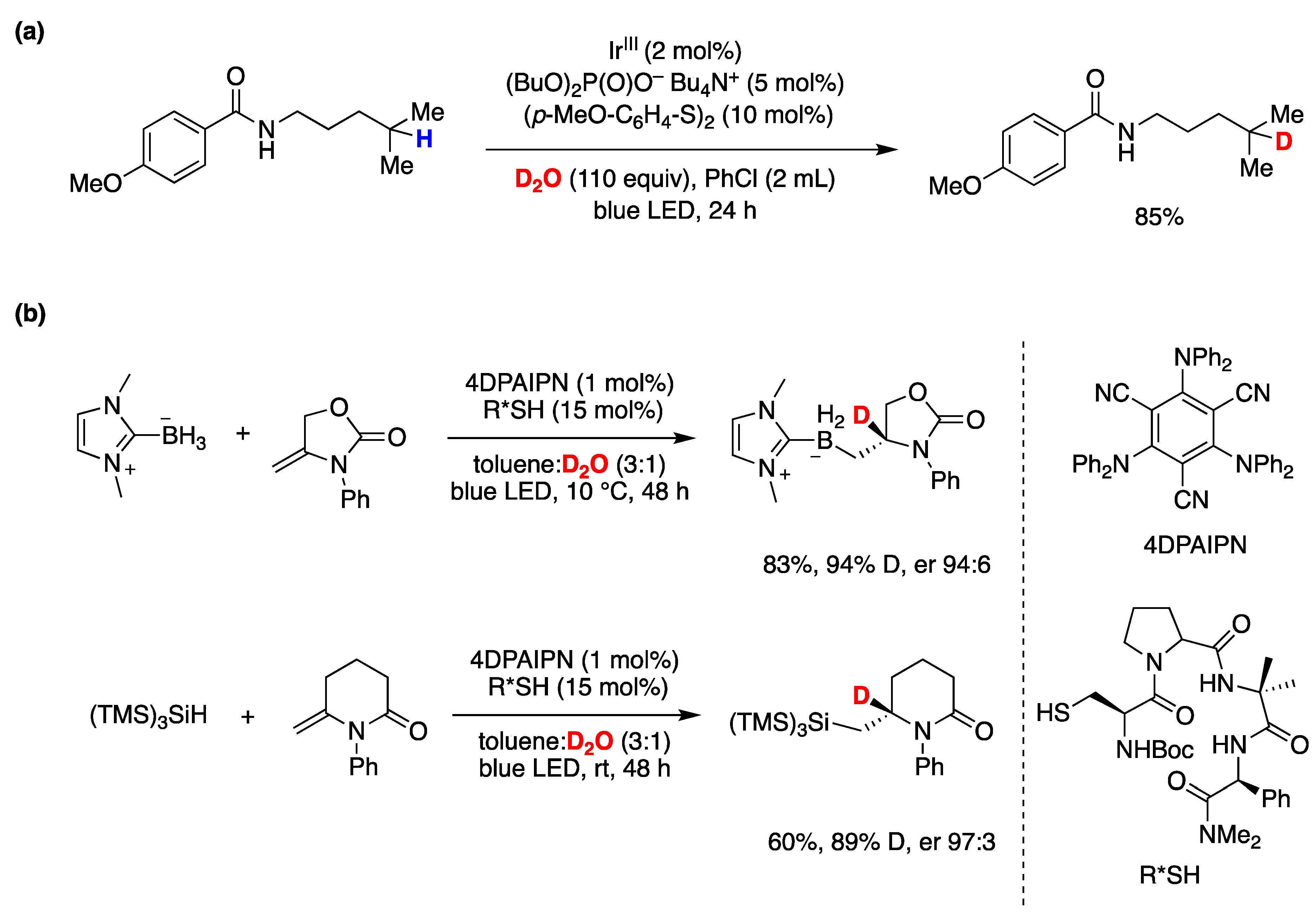
- Soulard, V.; Villa, G.; Vollmar, D.P.; Renaud, P. Radical Deuteration with D2O: Catalysis and Mechanistic Insights. J. Am. Chem. Soc. 2018, 140, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xia, Y.; Derdau, V.; Studer, A. Remote Site-Selective Radical C(sp3)–H Monodeuteration of Amides Using D2O. Angew. Chem. Int. Ed. 2021, 60, 18645–18650. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Jing, H.; Ge, H.; Brothers, P.J.; Fu, X.; Ye, S. The Mechanism of E–H (E = N, O) Bond Activation by a Germanium Corrole Complex: A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2015, 137, 7122–7127. [Google Scholar] [CrossRef]
- Bezdek, M.J.; Guo, S.; Chirik, P.J. Coordination-Induced Weakening of Ammonia, Water, and Hydrazine X–H Bonds in a Molybdenum Complex. Science 2016, 354, 730–733. [Google Scholar] [CrossRef]
- Yang, X.; Reijerse, E.J.; Bhattacharyya, K.; Leutzsch, M.; Kochius, M.; Nöthling, N.; Busch, J.; Schnegg, A.; Auer, A.A.; Cornella, J. Radical Activation of N–H and O–H Bonds at Bismuth(II). J. Am. Chem. Soc. 2022, 144, 16535–16544. [Google Scholar] [CrossRef]
- Medeiros, M.R.; Schacherer, L.N.; Spiegel, D.A.; Wood, J.L. Expanding the Scope of Trialkylborane/Water-Mediated Radical Reactions. Org. Lett. 2007, 9, 4427–4429. [Google Scholar] [CrossRef]
- Pozzi, D.; Scanlan, E.M.; Renaud, P. A Mild Radical Procedure for the Reduction of B -Alkylcatecholboranes to Alkanes. J. Am. Chem. Soc. 2005, 127, 14204–14205. [Google Scholar] [CrossRef] [PubMed]
- Boivin, J.; Nguyen, V.T. Part 1. Reduction of S -Alkyl-Thionocarbonates and Related Compounds in the Presence of Trialkylboranes/Air. Beilstein J. Org. Chem. 2007, 3, 45. [Google Scholar] [CrossRef] [PubMed]
- Allais, F.; Boivin, J.; Nguyen, V.T. Part 2. Mechanistic Aspects of the Reduction of S -Alkyl-Thionocarbonates in the Presence of Triethylborane and Air. Beilstein J. Org. Chem. 2007, 3, 46. [Google Scholar] [CrossRef] [PubMed]
- Boivin, J.; Nguyen, V.T. Part 3. Triethylborane-Air: A Suitable Initiator for Intermolecular Radical Additions of S-2-Oxoalkyl-Thionocarbonates (S-Xanthates) to Olefins. Beilstein J. Org. Chem. 2007, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Newcomb, M. Rate Constants for Reactions of Alkyl Radicals with Water and Methanol Complexes of Triethylborane. J. Org. Chem. 2007, 72, 5098–5103. [Google Scholar] [CrossRef] [PubMed]
- Povie, G.; Marzorati, M.; Bigler, P.; Renaud, P. Role of Equilibrium Associations on the Hydrogen Atom Transfer from the Triethylborane-Methanol Complex. J. Org. Chem. 2013, 78, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Deuterated Drugs Draw Heavier Backing. Nat. Rev. Drug Discov. 2016, 15, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Y.; Wu, X.; Zhu, C.; Xie, J. Radical Deuteration. Chem. Soc. Rev. 2022, 51, 6291–6306. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Ji, X.; He, R.; Liu, Y.; Chen, Z.; Huang, Y.; Liu, Q.; Li, Y. Synthesis of Deuterated (E)-Alkene through Xanthate-Mediated Hydrogen–Deuterium Exchange Reactions. Org. Lett. 2021, 23, 7412–7417. [Google Scholar] [CrossRef]
- Li, N.; Li, J.; Qin, M.; Li, J.; Han, J.; Zhu, C.; Li, W.; Xie, J. Highly Selective Single and Multiple Deuteration of Unactivated C(Sp3)-H Bonds. Nat. Commun. 2022, 13, 4224. [Google Scholar] [CrossRef]
- Shi, Q.; Xu, M.; Chang, R.; Ramanathan, D.; Peñin, B.; Funes-Ardoiz, I.; Ye, J. Visible-Light Mediated Catalytic Asymmetric Radical Deuteration at Non-Benzylic Positions. Nat. Commun. 2022, 13, 4453. [Google Scholar] [CrossRef]
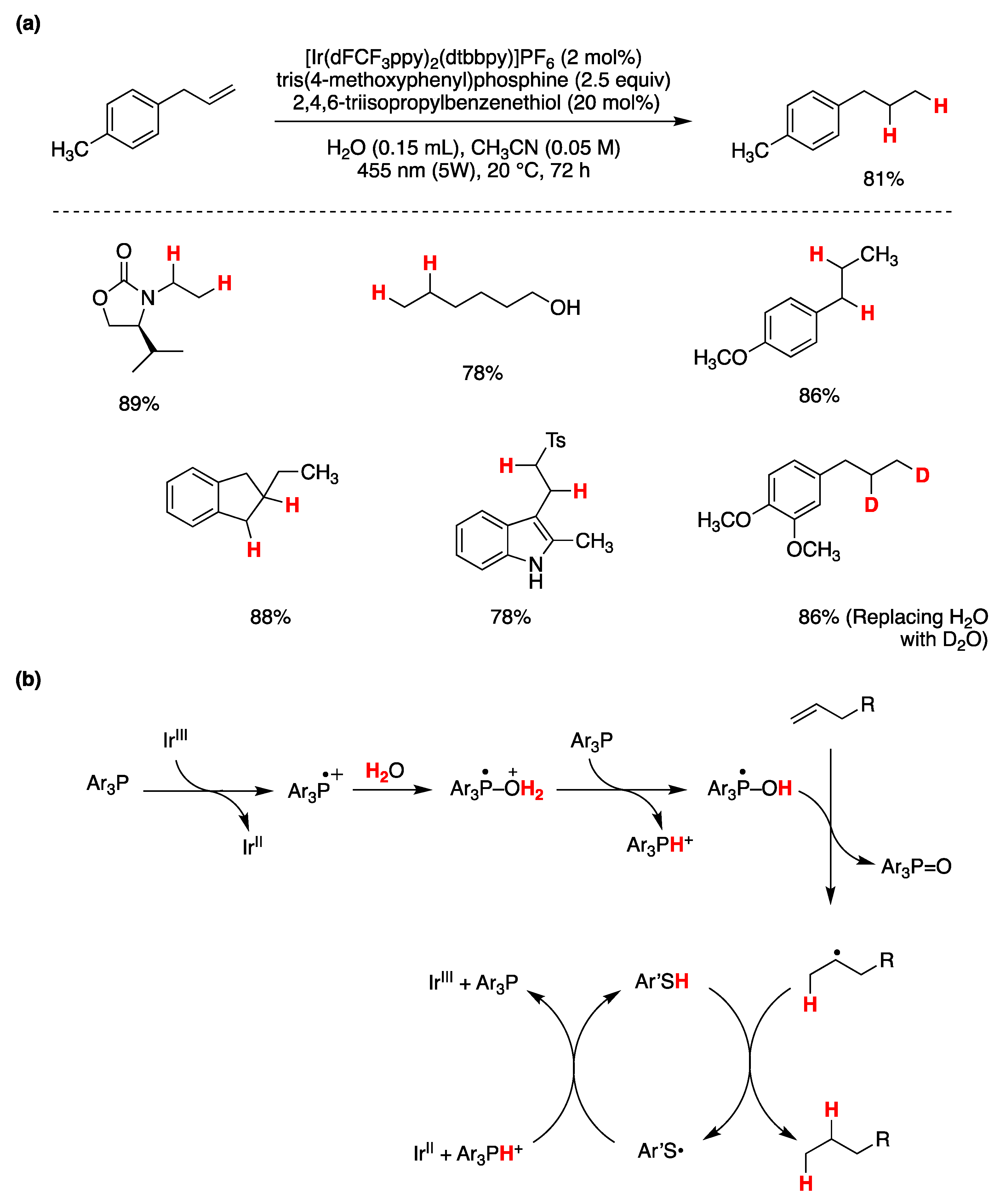
- Zhang, J.; Mück-Lichtenfeld, C.; Studer, A. Photocatalytic Phosphine-Mediated Water Activation for Radical Hydrogenation. Nature 2023, 619, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Newcomb, M. Rate Constants for Hydrogen Atom Transfer Reactions from Bis(Cyclopentadienyl)Titanium(III) Chloride-Complexed Water and Methanol to an Alkyl Radical. J. Org. Chem. 2008, 73, 7901–7905. [Google Scholar] [CrossRef] [PubMed]
- Paradas, M.; Campaña, A.G.; Jiménez, T.; Robles, R.; Oltra, J.E.; Buñuel, E.; Justicia, J.; Cárdenas, D.J.; Cuerva, J.M. Understanding the Exceptional Hydrogen-Atom Donor Characteristics of Water in Ti III -Mediated Free-Radical Chemistry. J. Am. Chem. Soc. 2010, 132, 12748–12756. [Google Scholar] [CrossRef] [PubMed]
- Gansäuer, A.; Behlendorf, M.; Cangönül, A.; Kube, C.; Cuerva, J.M.; Friedrich, J.; van Gastel, M. H2O Activation for Hydrogen-Atom Transfer: Correct Structures and Revised Mechanisms. Angew. Chem. Int. Ed. 2012, 51, 3266–3270. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bascón, J.; Hernández-Cervantes, C.; Padial, N.M.; Álvarez-Corral, M.; Rosales, A.; Rodríguez-García, I.; Oltra, J.E. Ti-Catalyzed Straightforward Synthesis of Exocyclic Allenes. Chem.-Eur. J. 2014, 20, 801–810. [Google Scholar] [CrossRef]
- Rosales, A.; Muñoz-Bascón, J.; Roldan-Molina, E.; Castañeda, M.A.; Padial, N.M.; Gansäuer, A.; Rodríguez-García, I.; Oltra, J.E. Selective Reduction of Aromatic Ketones in Aqueous Medium Mediated by Ti(III)/Mn: A Revised Mechanism. J. Org. Chem. 2014, 79, 7672–7676. [Google Scholar] [CrossRef] [PubMed]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. Reversibility of Ketone Reduction by SmI2—Water and Formation of Organosamarium Intermediates. Organometallics 2017, 36, 4579–4583. [Google Scholar] [CrossRef]
- Chciuk, T.V.; Anderson, W.R.; Flowers, R.A. High-Affinity Proton Donors Promote Proton-Coupled Electron Transfer by Samarium Diiodide. Angew. Chem. Int. Ed. 2016, 55, 6033–6036. [Google Scholar] [CrossRef]
- Boyd, E.A.; Peters, J.C. Sm(II)-Mediated Proton-Coupled Electron Transfer: Quantifying Very Weak N–H and O–H Homolytic Bond Strengths and Factors Controlling Them. J. Am. Chem. Soc. 2022, 144, 21337–21346. [Google Scholar] [CrossRef]
- Ciamician, G. The Photochemistry of the Future. Science 1912, 36, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. Efficient visible light photocatalysis of [2+2] enone cycloadditions. J. Am. Chem. Soc. 2008, 130, 12886–12887. [Google Scholar] [CrossRef] [PubMed]
- Narayanam, J.M.R.; Tucker, J.W.; Stephenson, C.R.J. Electron-transfer photoredox catalysis: Development of a tin-free reductive dehalogenation reaction. J. Am. Chem. Soc. 2009, 131, 8756–8757. [Google Scholar] [CrossRef]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates. Chem. Rev. 2016, 116, 9850–9913. [Google Scholar] [CrossRef]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Bell, J.D.; Murphy, J.A. Recent advances in visible light-activated radical coupling reactions triggered by (i) ruthenium, (ii) iridium and (iii) organic photoredox agents. Chem. Soc. Rev. 2021, 50, 9540–9685. [Google Scholar] [CrossRef]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef]
- Rueda-Marquez, J.J.; Levchuk, I.; Fernández Ibañez, P.; Sillanpää, M. A critical review on application of photocatalysis for toxicity reduction of real wastewaters. J. Clean. Prod. 2020, 258, 120694. [Google Scholar] [CrossRef]
- Arias-Rotondo, D.M.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Lv, Q.-Y.; Chen, X.-L.; Qu, L.-B.; Yu, B. Recent advances in visible-light-mediated organic transformations in water. Green Chem. 2021, 23, 232–248. [Google Scholar] [CrossRef]
- Russo, C.; Brunelli, F.; Tron, G.C.; Giustiniano, M. Visible-Light Photoredox Catalysis in Water. J. Org. Chem. 2023, 88, 6284–6293. [Google Scholar] [CrossRef] [PubMed]
- Barata-Vallejo, S.; Yerien, D.E.; Postigo, A. Advances in Photocatalytic Organic Synthetic Transformations in Water and Aqueous Media. ACS Sustain. Chem. Eng. 2021, 9, 10016–10047. [Google Scholar] [CrossRef]
- Dou, Q.; Zeng, H. Recent advances in photo-induced organic synthesis in water. Curr. Opin. Green Sustain. Chem. 2023, 40, 100766. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Yu, J.; Lu, C.; Wu, Z.; Wu, X.; Xue, X.; Zhu, C. Polarity Umpolung Strategy for the Radical Alkylation of Alkenes. Angew. Chem. Int. Ed. 2020, 59, 8195–8202. [Google Scholar] [CrossRef]
- Crespi, S.; Fagnoni, M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833. [Google Scholar] [CrossRef]
- Kvasovs, N.; Gevorgyan, V. Contemporary methods for generation of aryl radicals. Chem. Soc. Rev. 2021, 50, 2244–2259. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Lin, J.; Li, Z.; Huang, H. Radical acylation: Concepts, synthetic applications and directions. Org. Chem. Front. 2023, 10, 1056–1085. [Google Scholar] [CrossRef]
- Yerien, D.E.; Barata-Vallejo, S.; Postigo, A. Radical Perfluoroalkylation of Aliphatic Substrates. ACS Catal. 2023, 13, 7756–7794. [Google Scholar] [CrossRef]
- Wallentin, C.J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Hu, W.; Zhang, W. Difunctionalization of Alkenes and Alkynes via Intermolecular Radical and Nucleophilic Additions. Molecules 2020, 26, 105. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Huang, M.; Liu, C.; Xiao, J.C.; Chen, Q.Y.; Guo, Y.; Zhao, Z.G. 2,2,2-Trifluoroethylation of Styrenes with Concomitant Introduction of a Hydroxyl Group from Molecular Oxygen by Photoredox Catalysis Activated by Visible Light. Org. Lett. 2015, 17, 4714–4717. [Google Scholar] [CrossRef] [PubMed]
- Hari, D.P.; König, B. The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light. Angew. Chem. Int. Ed. 2013, 52, 4734–4743. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.R. Intermolecular Olefin Functionalisation Involving Aryl Radicals Generated from Arenediazonium Salts. Chem.—A Eur. J. 2009, 15, 820–833. [Google Scholar] [CrossRef]
- Prasad Hari, D.; Hering, T.; König, B. The photoredox-catalyzed meerwein addition reaction: Intermolecular amino-arylation of alkenes. Angew. Chem.—Int. Ed. 2014, 53, 725–728. [Google Scholar] [CrossRef]
- Kim, S. Free Radical-Mediated Acylation and Carboxylation Reactions. Adv. Synth. Catal. 2004, 346, 19–32. [Google Scholar] [CrossRef]
- Zhang, M.; Xie, J.; Zhu, C. A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes. Nat. Commun. 2018, 9, 3517. [Google Scholar] [CrossRef]
- Nguyen, J.D.; Tucker, J.W.; Konieczynska, M.D.; Stephenson, C.R.J. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef]
- Yerien, D.E.; Barata-Vallejo, S.; Mansilla, D.; Postigo, A. Rose Bengal-photocatalyzed perfluorohexylation reactions of organic substrates in water. Applications to late-stage syntheses. Photochem. Photobiol. Sci. 2022, 21, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Tong, C.L.; Zhang, Y.Y.; Xu, X.H.; Qing, F.L. Photoredox-Catalyzed and Silane-Mediated Hydrofluoromethylation of Unactivated Alkenes with Fluoroiodomethane in Water. Org. Lett. 2023, 25, 1035–1039. [Google Scholar] [CrossRef]
- Wang, T.; Zong, Y.Y.; Huang, T.; Jin, X.L.; Wu, L.Z.; Liu, Q. Photocatalytic redox-neutral selective single C(sp3)-F bond activation of perfluoroalkyl iminosulfides with alkenes and water. Chem. Sci. 2023, 14, 11566–11572. [Google Scholar] [CrossRef] [PubMed]
- Gurry, M.; Aldabbagh, F. A new era for homolytic aromatic substitution: Replacing Bu 3 SnH with efficient light-induced chain reactions. Org. Biomol. Chem. 2016, 14, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Jia, Z.H.; Zhao, C.J.; Zhang, Y.Y.; Wang, C.; Xiao, J. Direct arylation of n-heteroarenes with aryldiazonium salts by photoredox catalysis in water. Chem.—A Eur. J. 2014, 20, 2960–2965. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, P.; Chuskit, D. Priya Metal-free, visible-light-promoted oxidative radical cyclization of: N-biarylglycine esters: One-pot construction of phenanthridine-6-carboxylates in water. Green Chem. 2019, 21, 4406–4411. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Cooke, M.V.; Postigo, A. Radical Fluoroalkylation Reactions. ACS Catal. 2018, 8, 7287–7307. [Google Scholar] [CrossRef]
- Yerien, D.E.; Postigo, A.; Baroncini, M.; Barata-Vallejo, S. Bioinspired photocatalysed C-H fluoroalkylation of arenes in water promoted by native vitamin B12and Rose Bengal. Green Chem. 2021, 23, 8147–8153. [Google Scholar] [CrossRef]
- Nicoli, F.; Baroncini, M.; Silvi, S.; Groppi, J.; Credi, A. Direct synthetic routes to functionalised crown ethers. Org. Chem. Front. 2021, 8, 5531–5549. [Google Scholar] [CrossRef]
- Yerien, D.E.; Groppi, J.; Postigo, A.; Credi, A.; Conde, R.S.; Baroncini, M.; Barata-Vallejo, S. Late-Stage Photocatalytic Fluoroalkylation of Aromatic Crown Ethers in Aqueous Media. Eur. J. Org. Chem. 2023, 26, e202300478. [Google Scholar] [CrossRef]
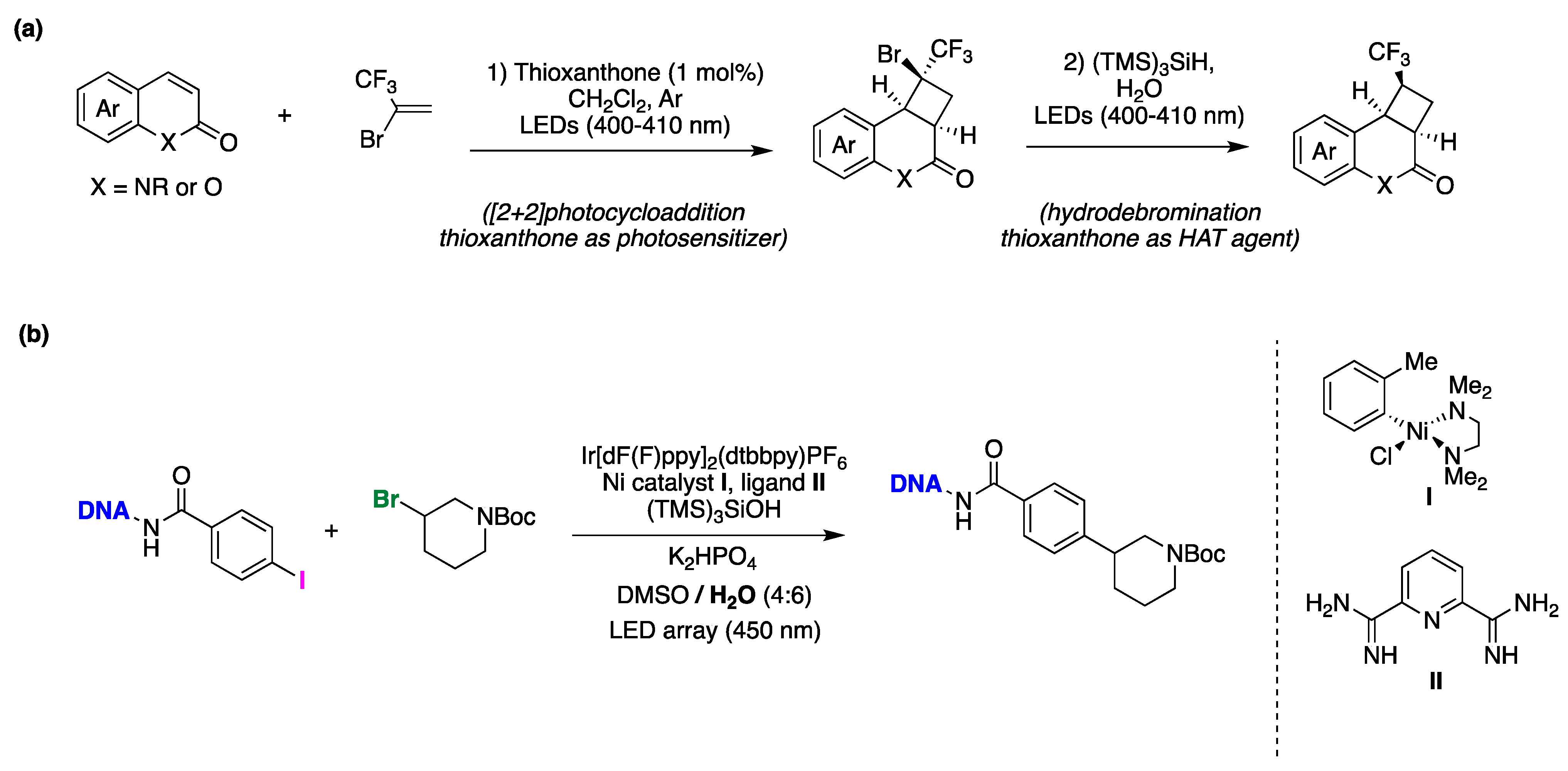
- Hu, X.; Xu, W.; Liu, Y.; Guo, H. Visible Light-Induced Diastereoselective Construction of Trifluoromethylated Cyclobutane Scaffolds through [2+2]-Photocycloaddition and Water-Assisted Hydrodebromination. J. Org. Chem. 2023, 88, 2521–2534. [Google Scholar] [CrossRef]
- Kölmel, D.K.; Ratnayake, A.S.; Flanagan, M.E. Photoredox cross-electrophile coupling in DNA-encoded chemistry. Biochem. Biophys. Res. Commun. 2020, 533, 201–208. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.T.; Benny, A.; Nolan, M.D.; Swinand, G.; Scanlan, W.M. Cysteinyl radicals in chemical synthesis and in nature. Chem. Soc. Rev. 2021, 50, 10857–10894. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef] [PubMed]
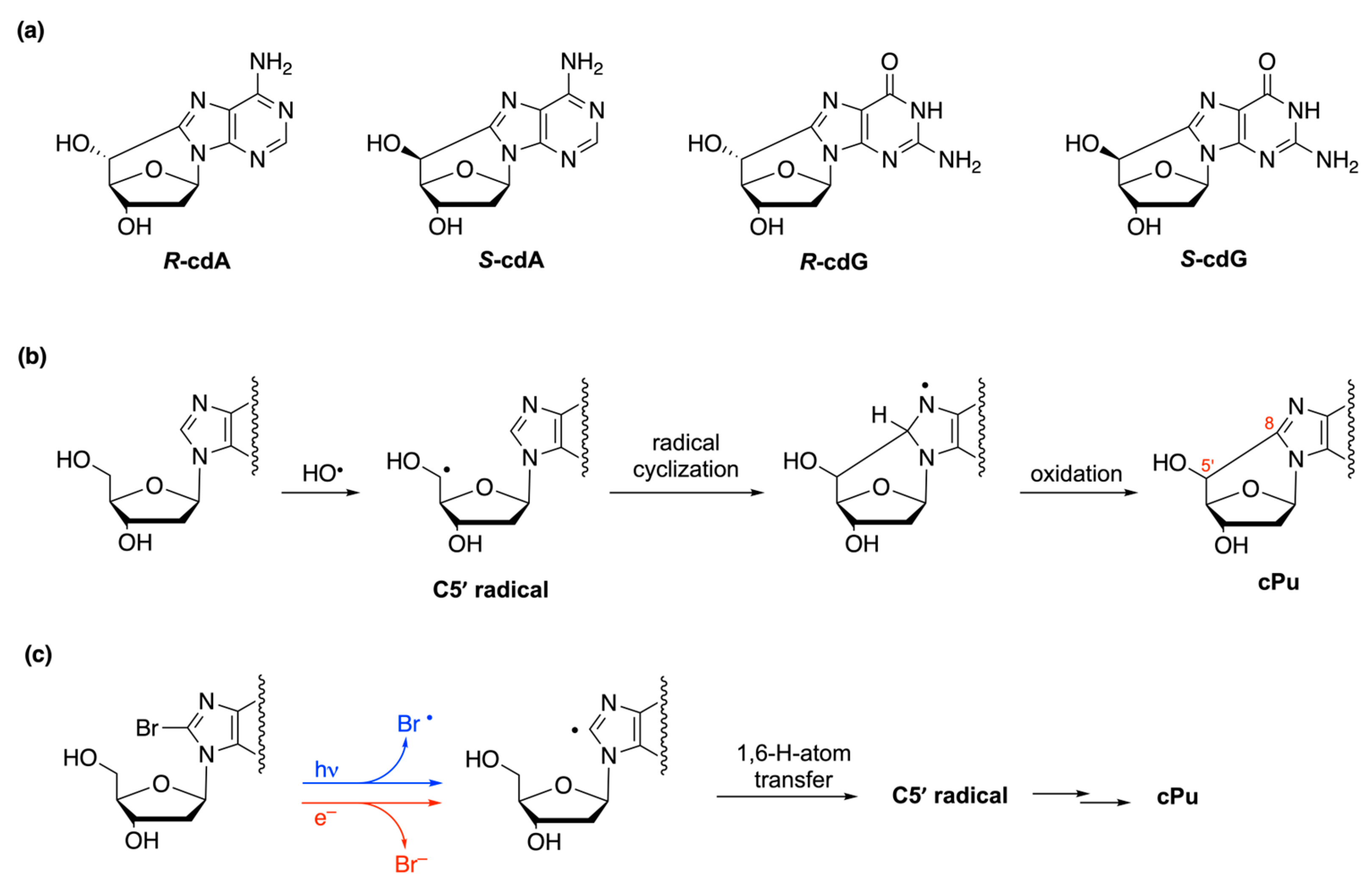
- Chatgilialoglu, C.; Ferreri, C.; Terzidis, M.A. Purine 5′,8-cyclonucleoside lesions: Chemistry and biology. Chem. Soc. Rev. 2011, 40, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Geacintov, N.E.; Krokidis, M.G.; Liu, Y.; Masi, A.; Shafirovich, N.; Terzidis, M.A.; Tsegay, P.S. 5′,8-Cyclopurine lesions in DNA damage: Chemical, analytical, biological and diagnostic significance. Cells 2019, 8, 513. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Krokidis, M.G.; Masi, A.; Terzidis, M.A. On the relevance of hydroxyl radical to purine DNA damage. Free Radic. Res. 2021, 55, 384–404. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Bazzanini, R.; Jimenez, L.B.; Miranda, M.A. (5′S)- and (5′R)-5′,8-Cyclo-2′-deoxyguanosine: Mechanistic insights on the 2′-deoxyguanosin-5′-yl radical cyclization. Chem. Res. Toxicol. 2007, 20, 1820–1824. [Google Scholar] [CrossRef]
- Flyunt, R.; Bazzanini, R.; Chatgilialoglu, C.; Mulazzani, Q.G. Fate of the 2′-deoxyadenosin-5′-yl radical under anaerobic conditions. J. Am. Chem. Soc. 2000, 122, 4225–4226. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Guerra, M.; Mulazzani, Q.G. Model studies of DNA C5′ radicals. Selective generation and reactivity of 2′-deoxyadenosin-5′-yl radical. J. Am. Chem. Soc. 2003, 125, 3839–3848. [Google Scholar] [CrossRef]
- Boussicault, F.; Kaloudis, P.; Caminal, C.; Mulazzani, Q.G.; Chatgilialoglu, C. The fate of C5′ radicals of purine nucleosides under oxidative conditions. J. Am. Chem. Soc. 2008, 130, 8377–8385. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; D’Angelantonio, M.; Kciuk, G.; Bobrowski, K. New insights into the reaction paths of hydroxyl radicals with 2′-deoxyguanosine. Chem. Res. Toxicol. 2011, 24, 2200–2206. [Google Scholar] [CrossRef] [PubMed]
- Navacchia, M.L.; Chatgilialoglu, C.; Montevecchi, P.C. C5′-adenosinyl radical cyclization. A stereochemical investigation. J. Org. Chem. 2006, 71, 4445–4452. [Google Scholar] [CrossRef] [PubMed]
- Terzidis, M.A.; Chatgilialoglu, C. Radical cascade protocol for the synthesis of (5′S)- and (5′R)-5′,8-cyclo-2′-deoxyguanosine derivatives. Aust. J. Chem. 2013, 66, 330–335. [Google Scholar] [CrossRef]
- Terzidis, M.A.; Chatgilialoglu, C. An ameliorative protocol for the quantification of purine 5′,8-cyclo-2′-deoxynucleosides in oxidized DNA. Front. Chem. 2015, 3, 47. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Terzidis, M.A.; Szreder, T.; Bobrowski, K. New Insights into the Reaction Paths of Hydroxyl Radicals with Purine Moieties in DNA and Double-Stranded Oligodeoxynucleotides. Molecules 2019, 24, 3860. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Eriksson, L.A.; Krokidis, M.G.; Masi, A.; Wang, S.; Zhang, R. Oxygen Dependent Purine Lesions in Double-Stranded Oligodeoxynucleotides: Kinetic and Computational Studies Highlight the Mechanism for 5′,8-Cyclopurine Formation. J. Am. Chem. Soc. 2020, 142, 5825–5833. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Terzidis, M.A.; Efthimiadou, E.; Zervou, S.; Kordas, G.; Papadopoulos, K.; Hiskia, A.; Kletsas, D.; Chatgilialoglu, C. Purine 5′,8-cyclo-2′-deoxynucleoside lesions: Formation by radical stress and repair in human breast epithelial cancer cells. Free Radic. Res. 2017, 51, 470–482. [Google Scholar] [CrossRef]
- Krokidis, M.G.; Parlanti, E.; D’Errico, M.; Pascucci, B.; Pino, A.; Alimonti, A.; Pietraforte, D.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Purine DNA Lesions at Different Oxygen Concentration in DNA Repair-Impaired Human Cells (EUE-siXPA). Cells 2019, 8, 1377. [Google Scholar] [CrossRef]
- Krokidis, M.G.; D’Errico, M.; Pascucci, B.; Parlanti, E.; Masi, A.; Ferreri, C.; Chatgilialoglu, C. Oxygen-Dependent Accumulation of Purine DNA Lesions in Cockayne Syndrome Cells. Cells 2020, 9, 1671. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Krokidis, M.G.; Masi, A.; Barata-Vallejo, S.; Ferreri, C.; Pascucci, B.; D’Errico, M. Assessing the Formation of Purine Lesions in Mitochondrial DNA of Cockayne Syndrome Cells. Biomolecules 2022, 12, 1630. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Louka, M.; Efthimiadou, E.K.; Zervou, S.K.; Papadopoulos, K.; Hiskia, A.; Ferreri, C.; Chatgilialoglu, C. Membrane Lipidome Reorganization and Accumulation of Tissue DNA Lesions in Tumor-Bearing Mice: An Exploratory Study. Cancers 2019, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Krokidis, M.G.; Prasinou, P.; Efthimiadou, E.K.; Boari, A.; Ferreri, C.; Chatgilialoglu, C. Effects of Aging and Disease Conditions in Brain of Tumor-Bearing Mice: Evaluation of Purine DNA Damages and Fatty Acid Pool Changes. Biomolecules 2022, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Fortini, P.; Krokidis, M.G.; Romeo, E.F.; Bascietto, C.; De Angelis, P.; Guglielmi, V.; Chatgilialoglu, C. Increased Levels of 5′,8-Cyclopurine DNA Lesions in Inflammatory Bowel Diseases. Redox Biol. 2020, 34, 101562. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Masi, A.; Sansone, A.; Terzidis, M.A.; Tsakos, M. A problem solving approach for the diastereoselective synthesis of (5′S)- and (5′R)-5′,8-cyclopurine lesions. Org. Chem. Front. 2014, 1, 698–702. [Google Scholar] [CrossRef]
- Kropachev, K.; Ding, S.; Terzidis, M.A.; Masi, A.; Liu, Z.; Cai, Y.; Kolbanovskiy, M.; Chatgilialoglu, C.; Broyde, S.; Geacintov, N.E.; et al. Structural basis for the recognition of diastereomeric 5′,8-cyclo-2′-deoxypurine lesions by the human nucleotide excision repair system. Nucl. Acids Res. 2014, 42, 5020–5032. [Google Scholar] [CrossRef]
- Xu, M.; Lai, Y.; Jiang, Z.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. A 5′, 8-cyclo-2′-deoxypurine lesion induces trinucleotide repeat deletion via a unique lesion bypass by DNA polymerase b. Nucl. Acids Res. 2014, 42, 13749–13763. [Google Scholar] [CrossRef]
- Jiang, Z.; Xu, M.; Lai, Y.; Laverde, E.E.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Liu, Y. Bypass of a 5′,8-cyclopurine-2′-deoxynucleoside by DNA polymerase b during DNA replication and base excision repair leads to nucleotide misinsertions and DNA strand breaks. DNA Repair 2015, 33, 24–34. [Google Scholar] [CrossRef]
- Shafirovich, V.; Kolbanovskiy, M.; Kropachev, K.; Liu, Z.; Cai, Y.; Terzidis, M.A.; Masi, A.; Chatgilialoglu, C.; Amin, S.; Dadali, A.; et al. Nucleotide Excision Repair and Impact of Site-Specific 5′,8-Cyclopurine and Bulky DNA Lesions on the Physical Properties of Nucleosomes. Biochemistry 2019, 58, 561–574. [Google Scholar] [CrossRef]
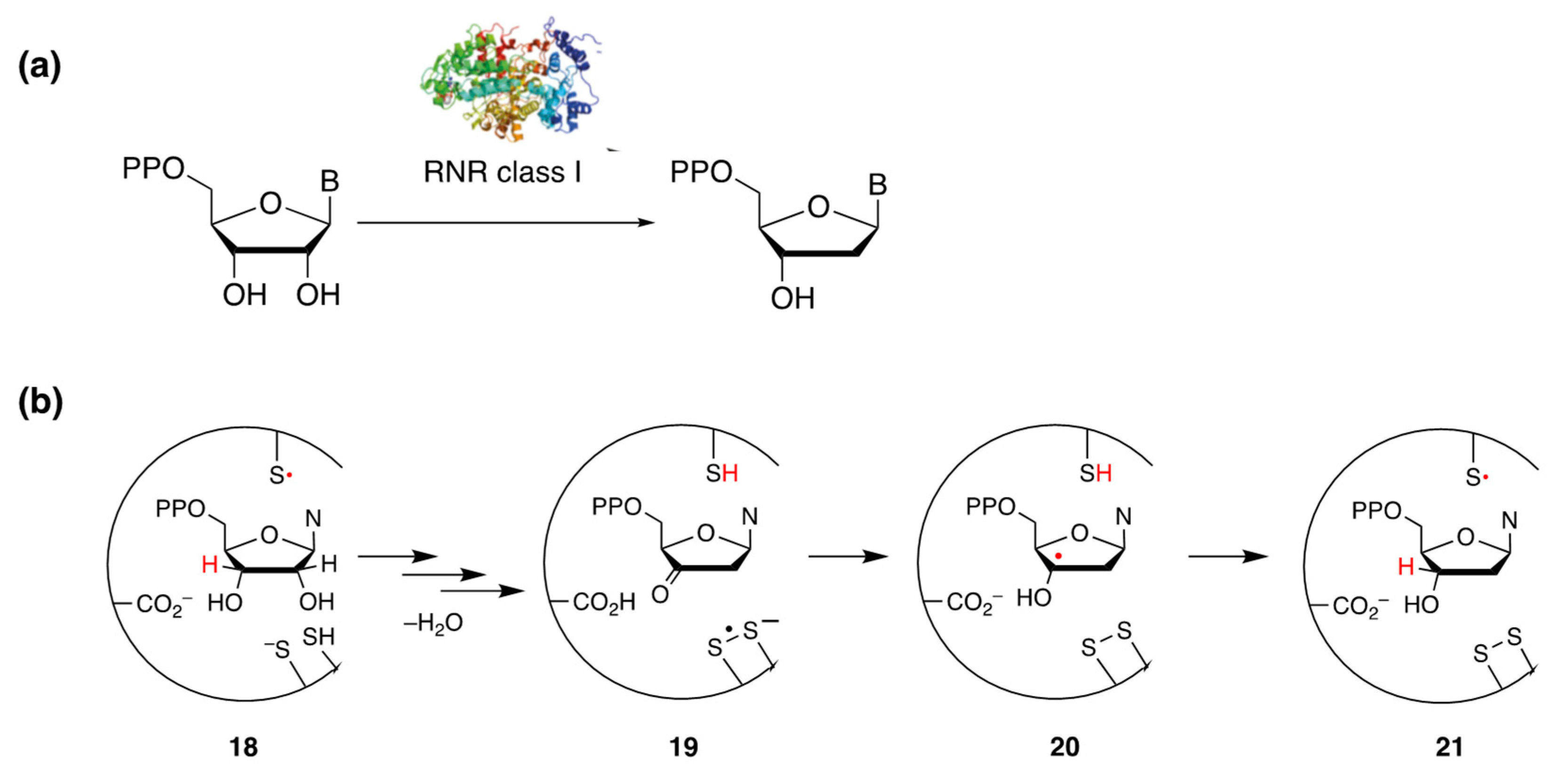
- Minnihan, E.C.; Nocera, D.G.; Stubbe, J. Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res. 2013, 46, 2524–2535. [Google Scholar] [CrossRef]
- Greene, B.L.; Taguchi, A.T.; Stubbe, J.; Nocera, D.G. Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc. 2017, 139, 16657–16665. [Google Scholar] [CrossRef] [PubMed]
- McCombie, S.W.; Motherwell, W.B.; Tozer, M.J. The Barton-McCombie Reaction. Org. React. 2012, 77, 161–438. [Google Scholar]
- Barata-Vallejo, S.; Ferreri, C.; Golding, B.T.; Chatgilialoglu, C. Hydrogen Sulfide: A Reagent for pH-Driven Bioinspired 1,2-Diol Mono-deoxygenation and Carbonyl Reduction in Water. Org. Lett. 2018, 20, 4290–4294. [Google Scholar] [CrossRef]
- Das, T.N.; Huie, R.E.; Neta, P.; Padmaja, S. Reduction Potential of the Sulfhydryl Radical: Pulse Radiolysis and Laser Flash Photolysis Studies of the Formation and Reactions of ‚SH and HSSH·- in Aqueous Solutions. J. Phys. Chem. A 1999, 103, 5221–5226. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L. Signaling by sulfur-containing molecules. Quantitative aspects. Arch. Biochem. Biophys. 2017, 617, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R.; Straub, K.D. The Role of Hydrogen Sulfide in Evolution and the Evolution of Hydrogen Sulfide in Metabolism and Signaling. Physiology 2016, 31, 60–72. [Google Scholar] [CrossRef]
- Green, N.J.; Xu, J.; Sutherland, J.D. Illuminating Life’s Origins: UV Photochemistry in Abiotic Synthesis of Biomolecules. J. Am. Chem. Soc. 2021, 143, 7219–7236. [Google Scholar] [CrossRef]
- Barata-Vallejo, S.; Skotnicki, K.; Ferreri, C.; Marciniak, B.; Bobrowski, K.; Chatgilialoglu, C. Biomimetic Ketone Reduction by Disulfide Radical Anion. Molecules 2021, 26, 5429. [Google Scholar] [CrossRef]
- Mezyk, S.P.; Armstrong, D.A. Disulfide anion radical equilibria: Effects of -NH3+, -CO2−, -NHC(O)- and -CH3 groups. J. Chem. Soc. Perkin Trans. 1999, 2, 1411–1419. [Google Scholar] [CrossRef]
- Zhu, Q.; Nocera, D.G. Catalytic C(β)–O Bond Cleavage of Lignin in a One-Step Reaction Enabled by a Spin-Center Shift. ACS Catal. 2021, 11, 14181–14187. [Google Scholar] [CrossRef]
- Zhu, Q.; Costentin, C.; Stubbe, J.; Nocera, D.G. Disulfide radical anion as a super-reductant in biology and photoredox chemistry. Chem. Sci. 2023, 14, 6876–6881. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.Z.; Hayon, E. One-Electron Reduction of the Disulfide Linkage in Aqueous Solution. Formation, Protonation, and Decay Kinetics of the RSSR- Radical. J. Am. Chem. Soc. 1972, 94, 795–7957. [Google Scholar] [CrossRef]












































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
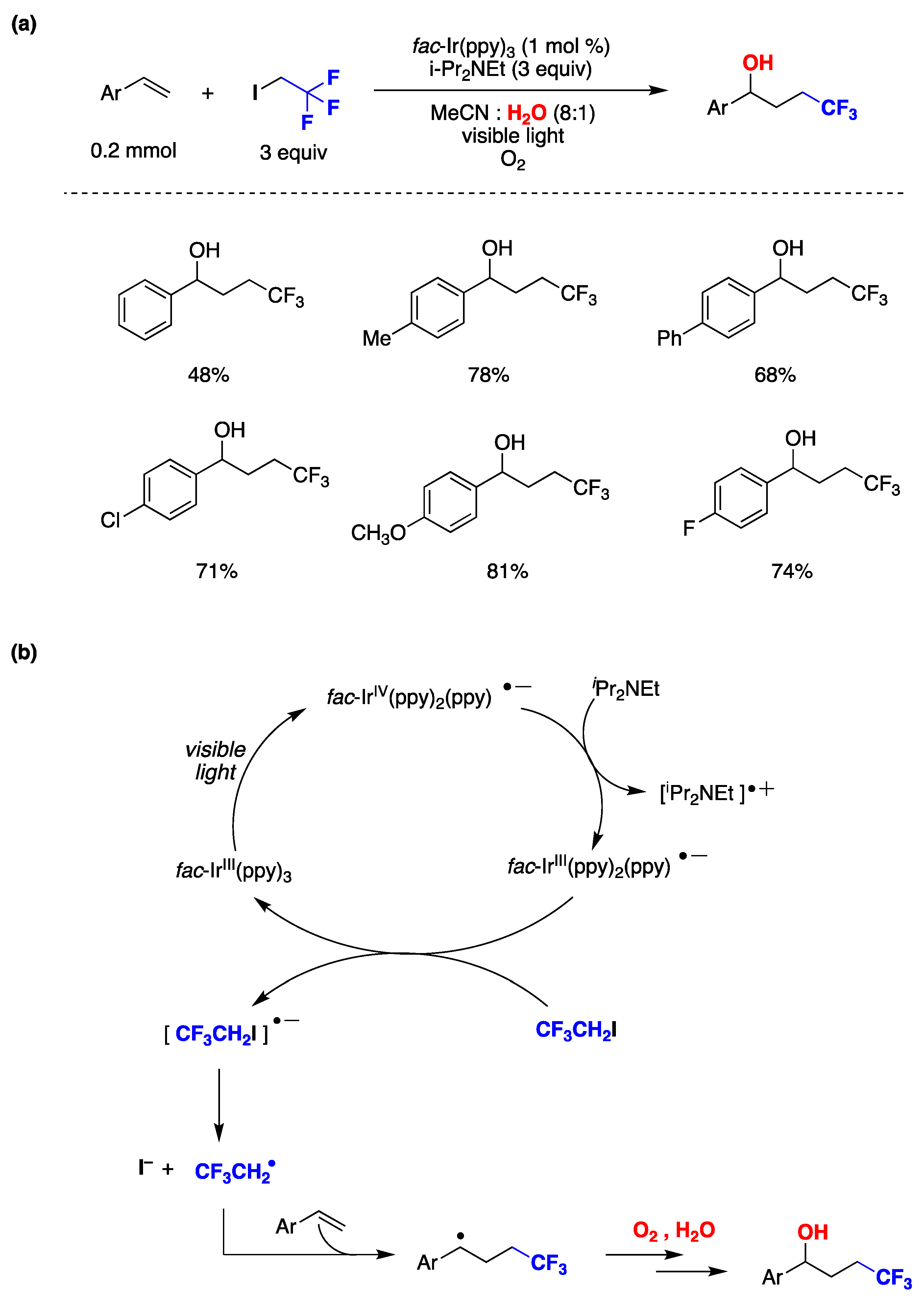{kind=link}
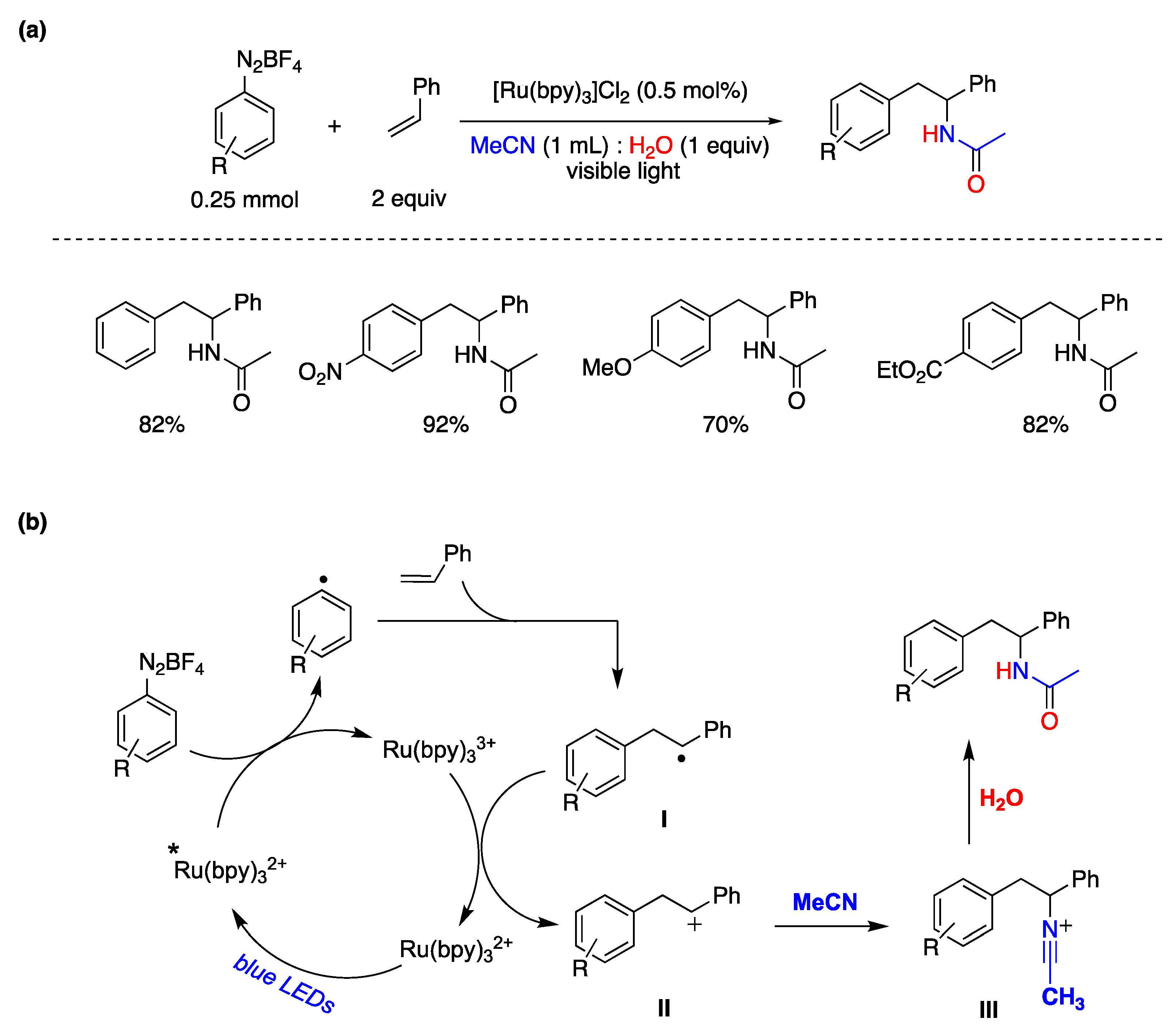{kind=link}
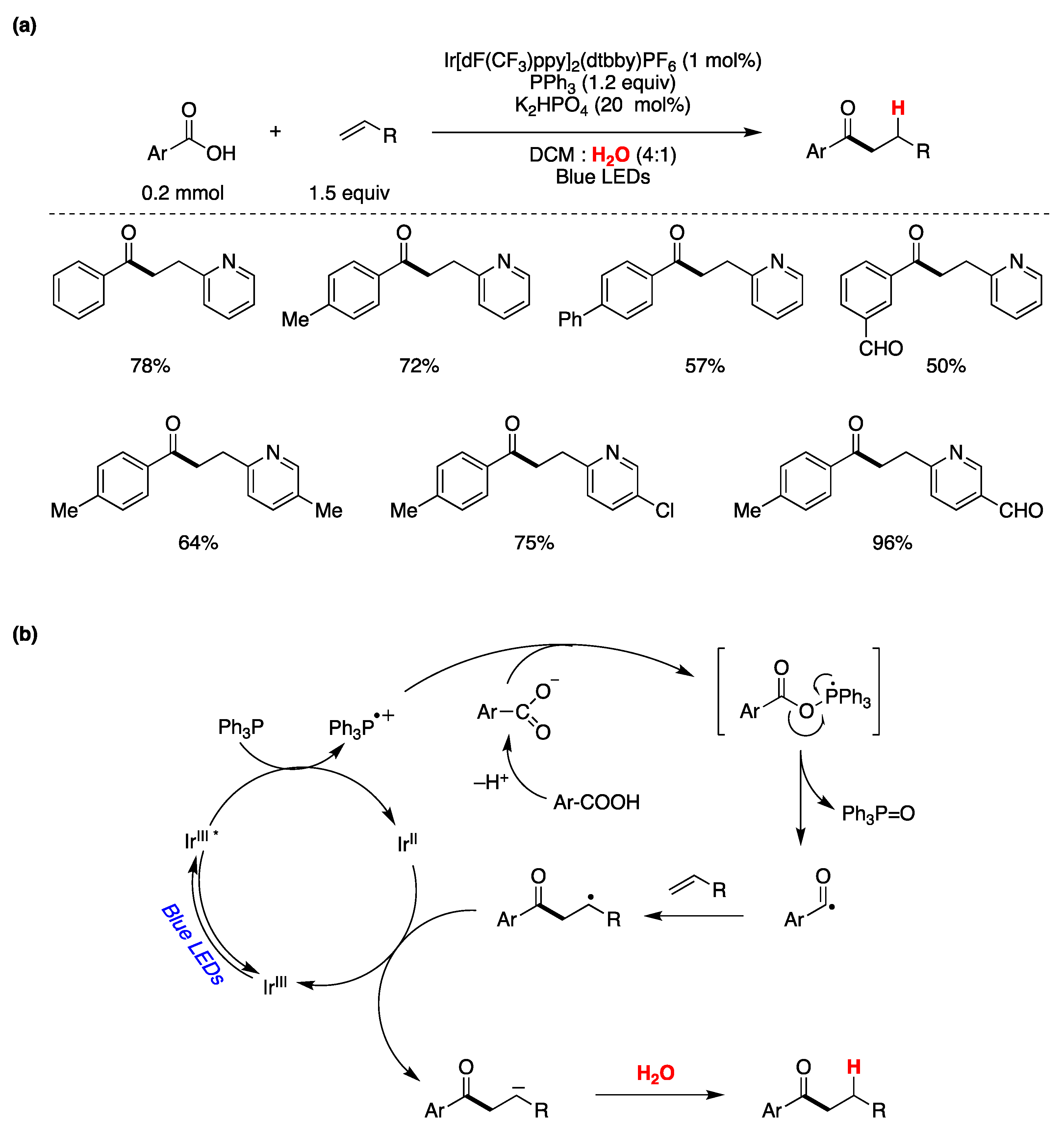{kind=link}
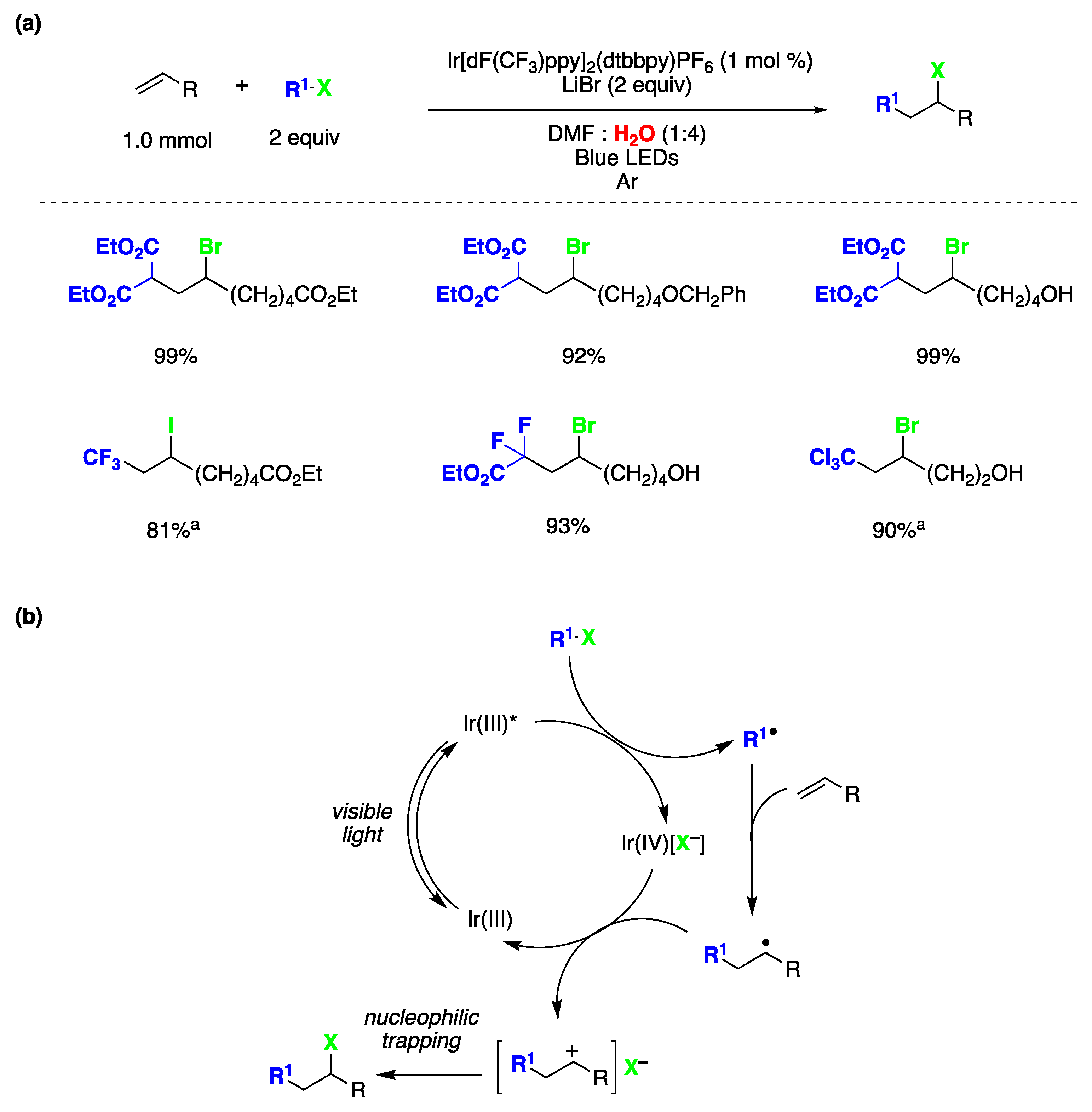{kind=link}
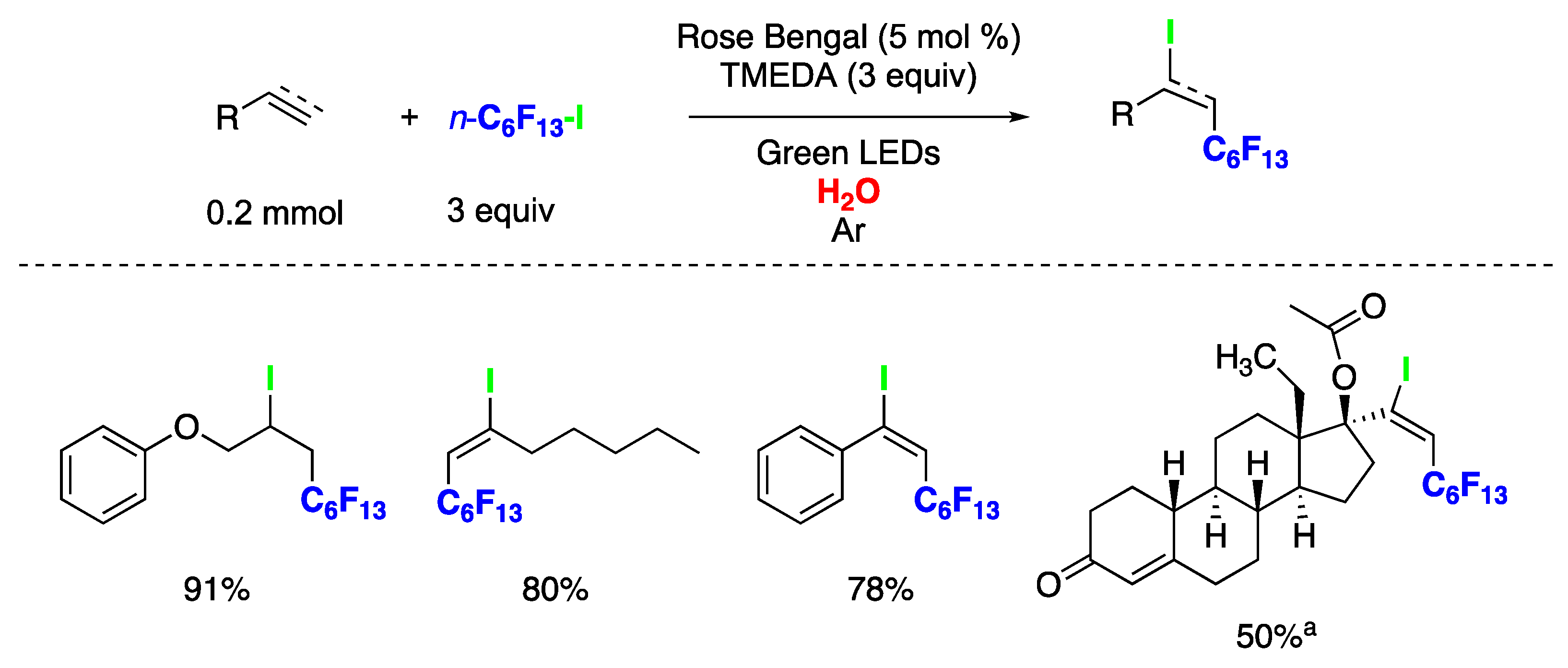{kind=link}
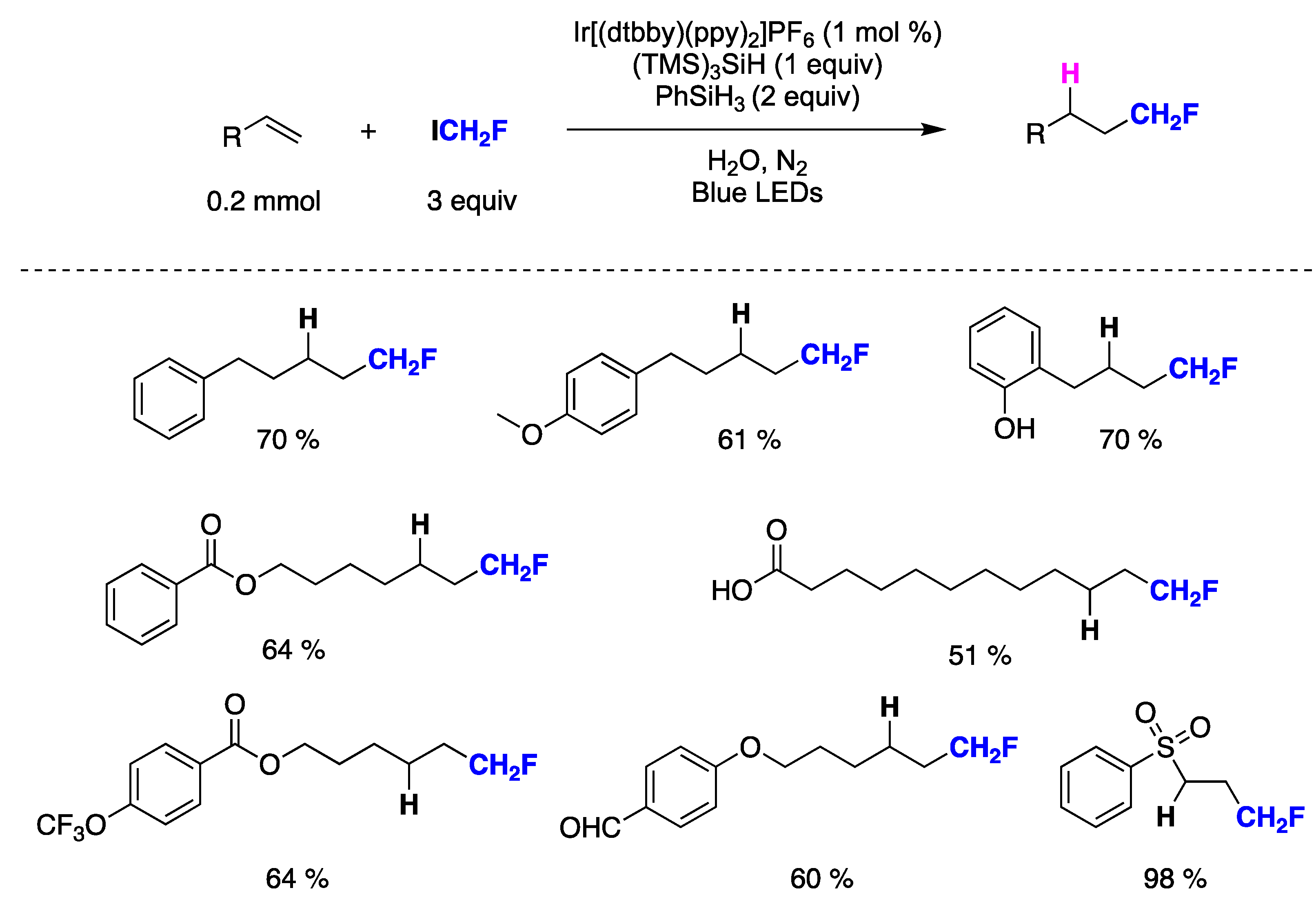{kind=link}
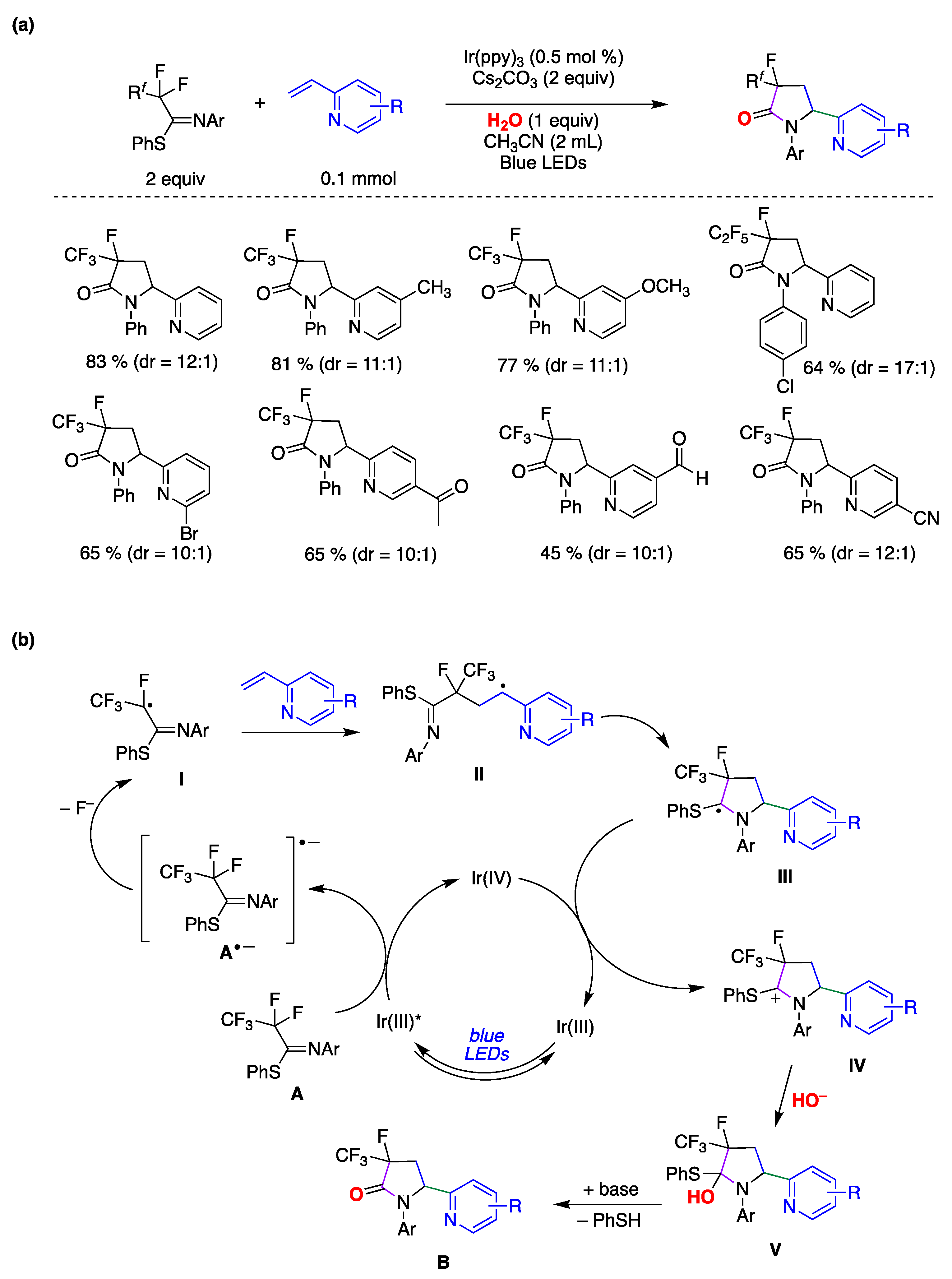{kind=link}
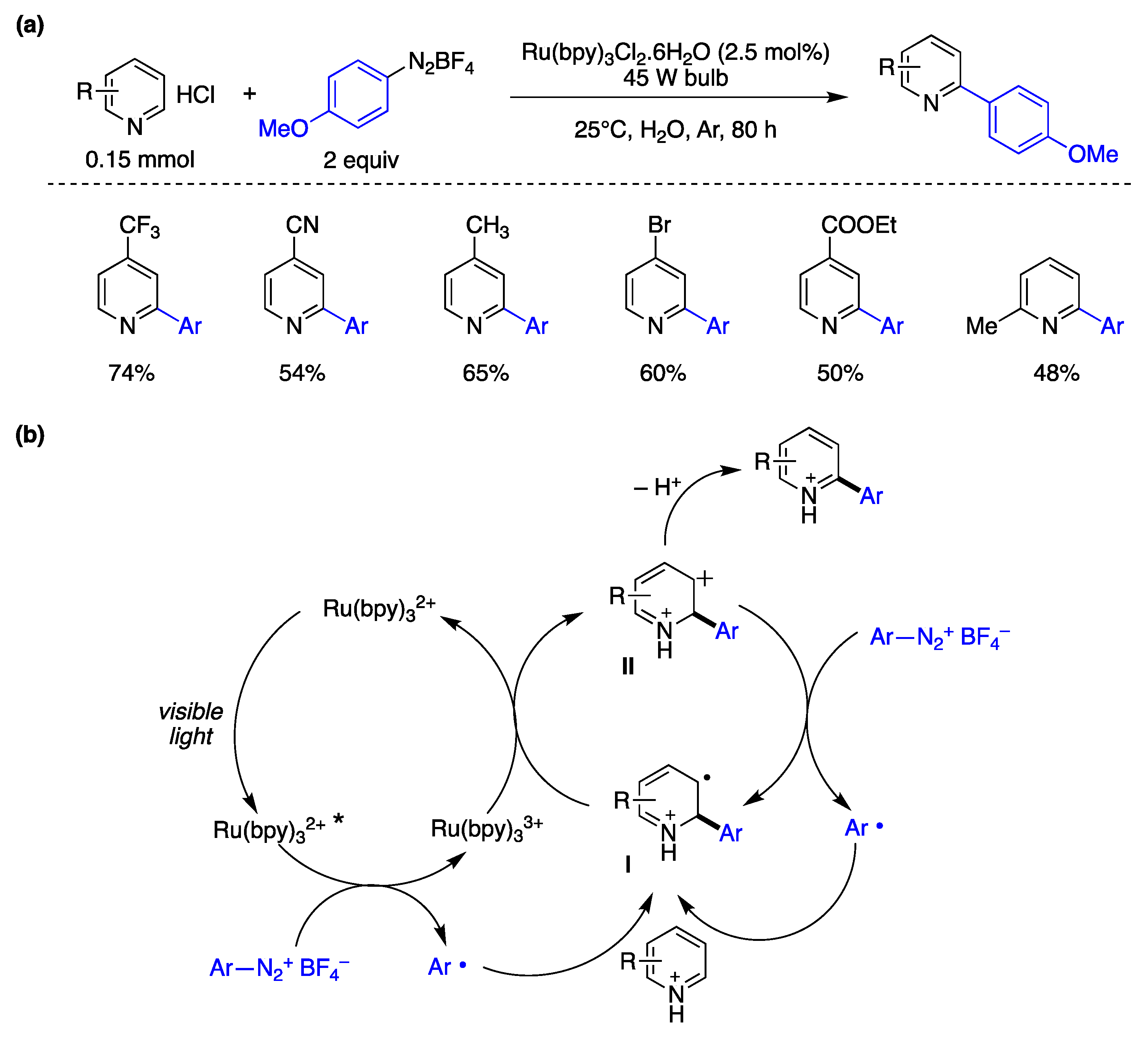{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkyl Radical | kH, M−1 s−1 |
|---|---|
| HOCH2• | 1.3 × 108 |
| HOC(•)Me | 2.3 × 108 |
| HOC(•)Me2 | 5.1 × 108 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatgilialoglu, C.; Barata-Vallejo, S.; Gimisis, T. Radical Reactions in Organic Synthesis: Exploring in-, on-, and with-Water Methods. Molecules 2024, 29, 569. https://doi.org/10.3390/molecules29030569
Chatgilialoglu C, Barata-Vallejo S, Gimisis T. Radical Reactions in Organic Synthesis: Exploring in-, on-, and with-Water Methods. Molecules. 2024; 29(3):569. https://doi.org/10.3390/molecules29030569
Chicago/Turabian StyleChatgilialoglu, Chryssostomos, Sebastian Barata-Vallejo, and Thanasis Gimisis. 2024. "Radical Reactions in Organic Synthesis: Exploring in-, on-, and with-Water Methods" Molecules 29, no. 3: 569. https://doi.org/10.3390/molecules29030569
APA StyleChatgilialoglu, C., Barata-Vallejo, S., & Gimisis, T. (2024). Radical Reactions in Organic Synthesis: Exploring in-, on-, and with-Water Methods. Molecules, 29(3), 569. https://doi.org/10.3390/molecules29030569