
Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane


Abstract
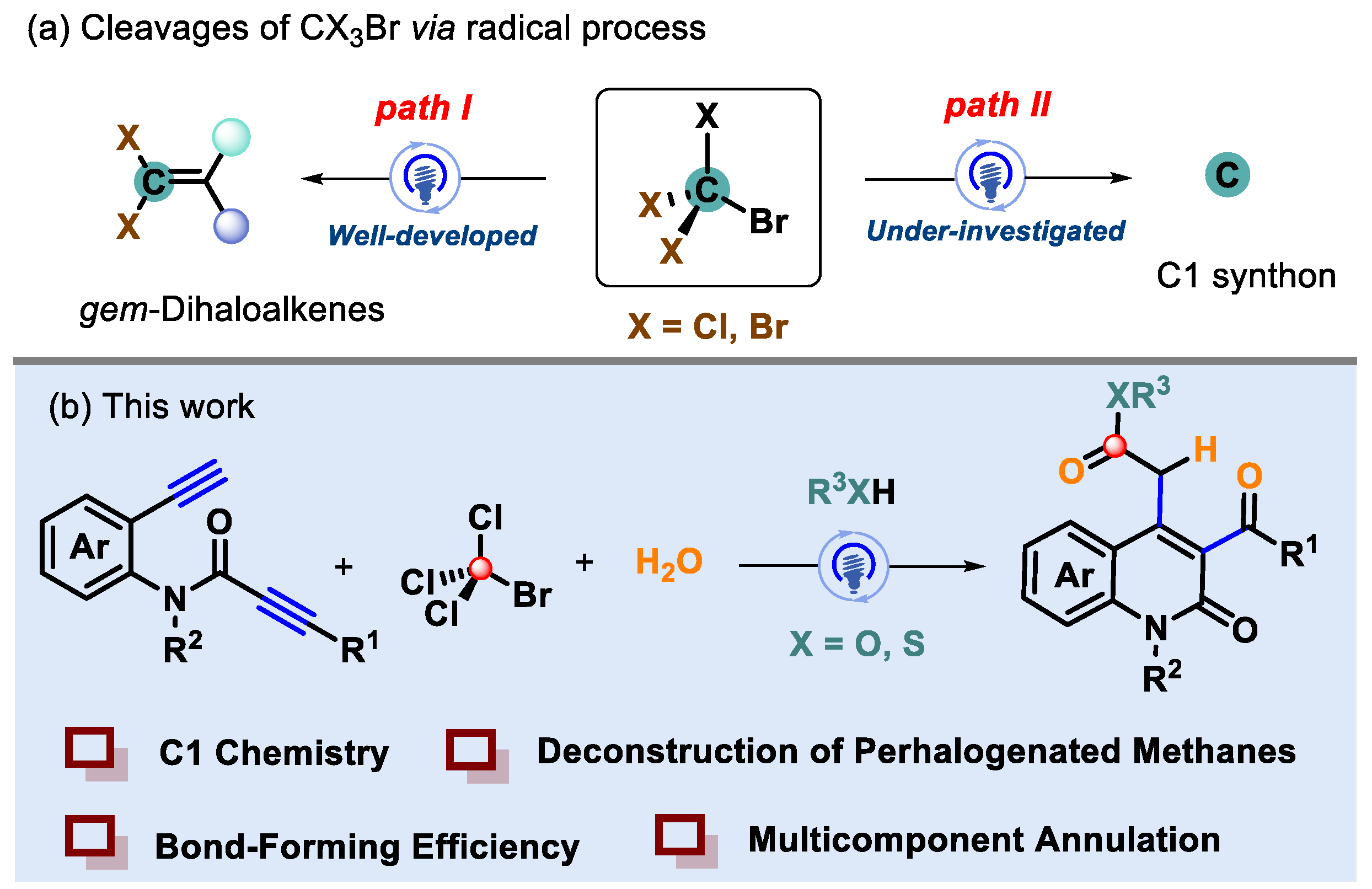
:1. Introduction
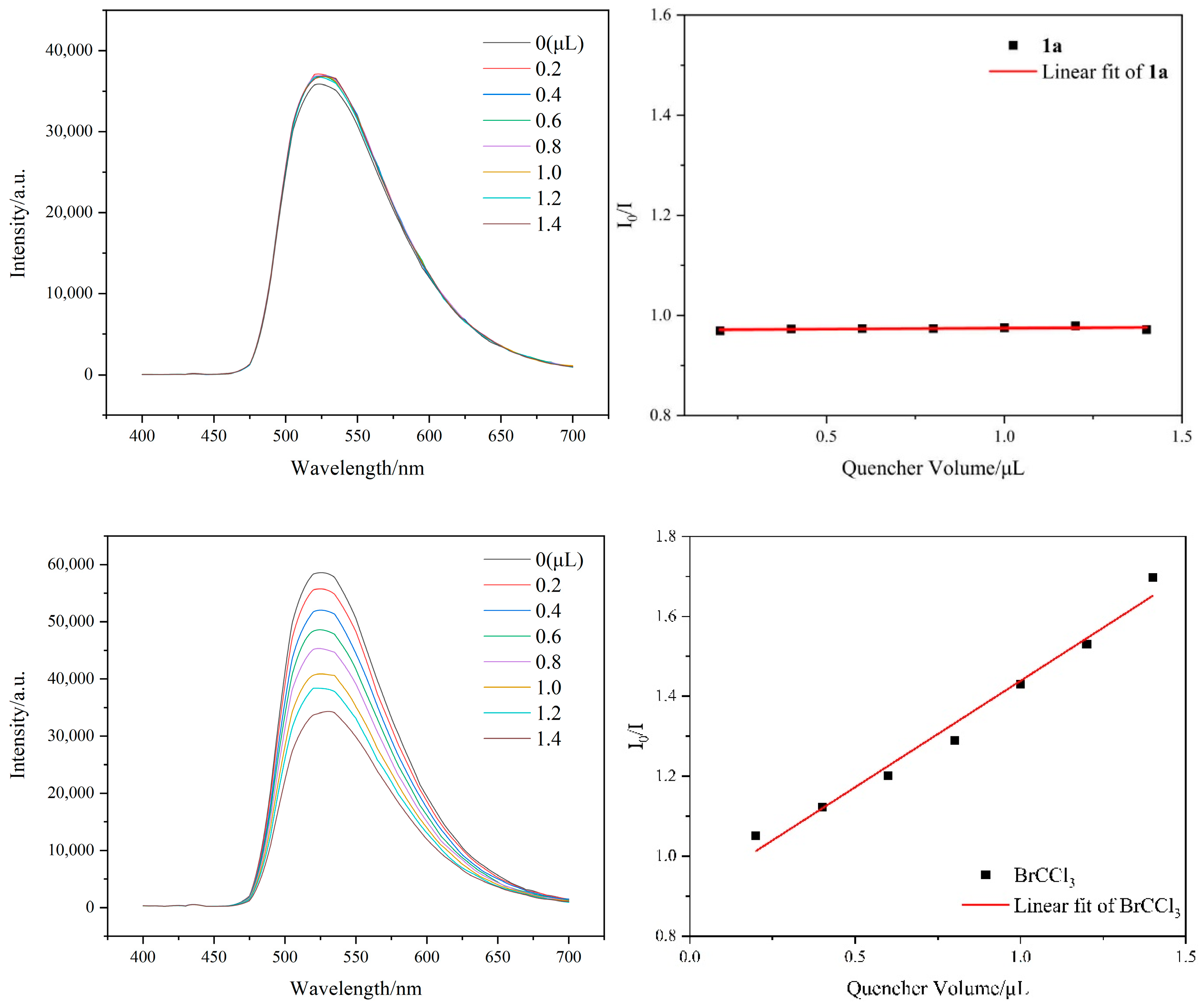
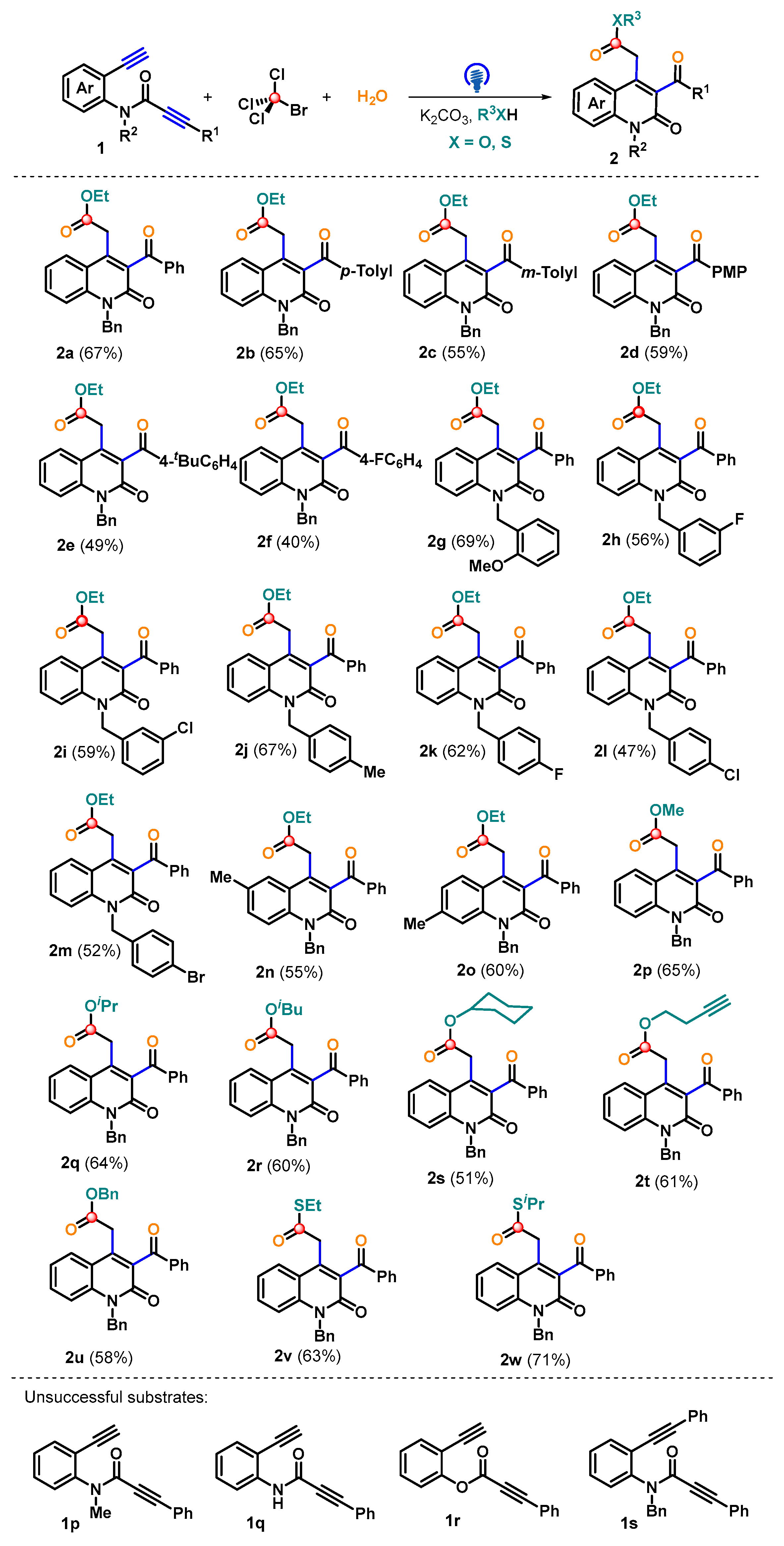
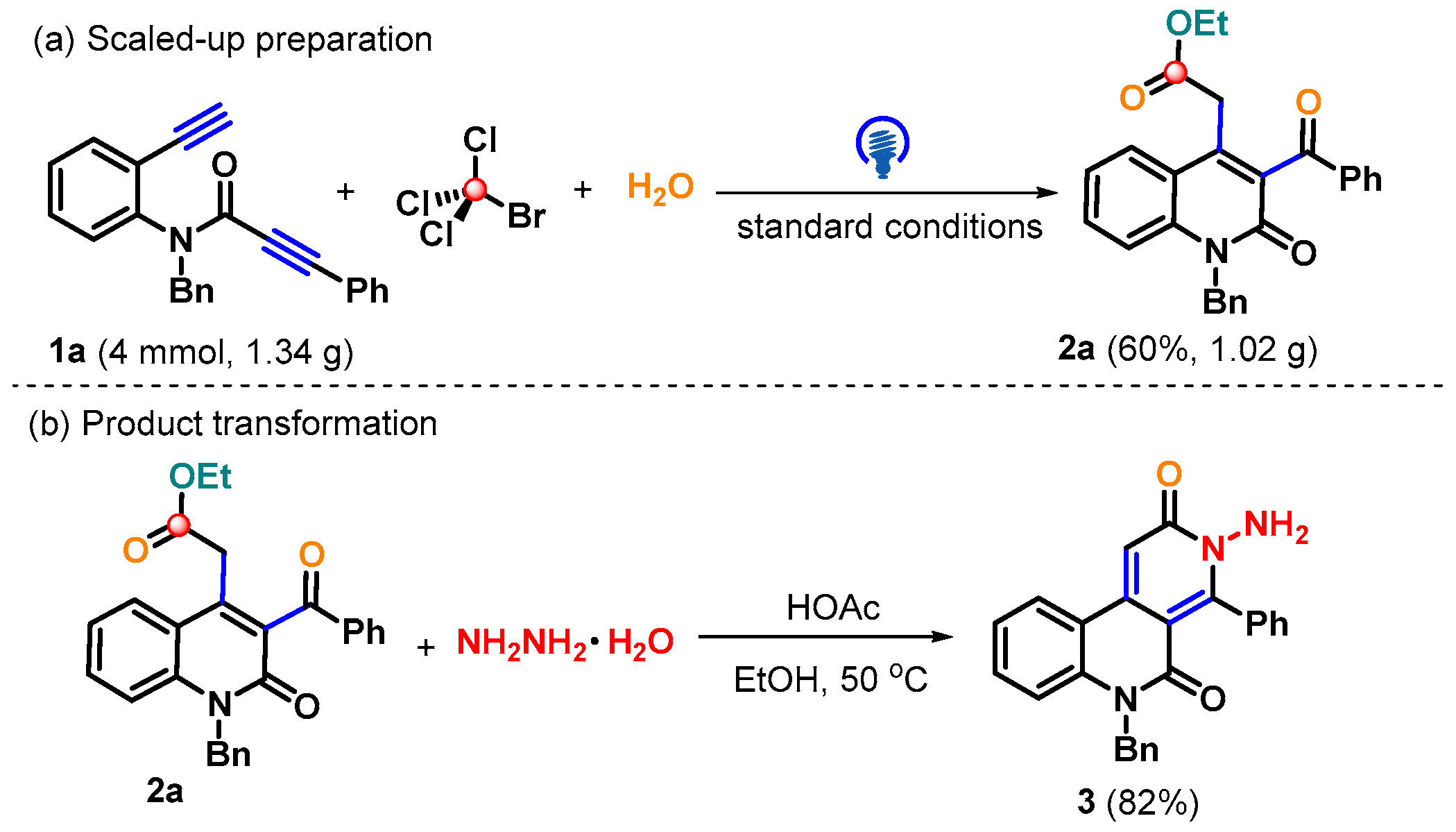
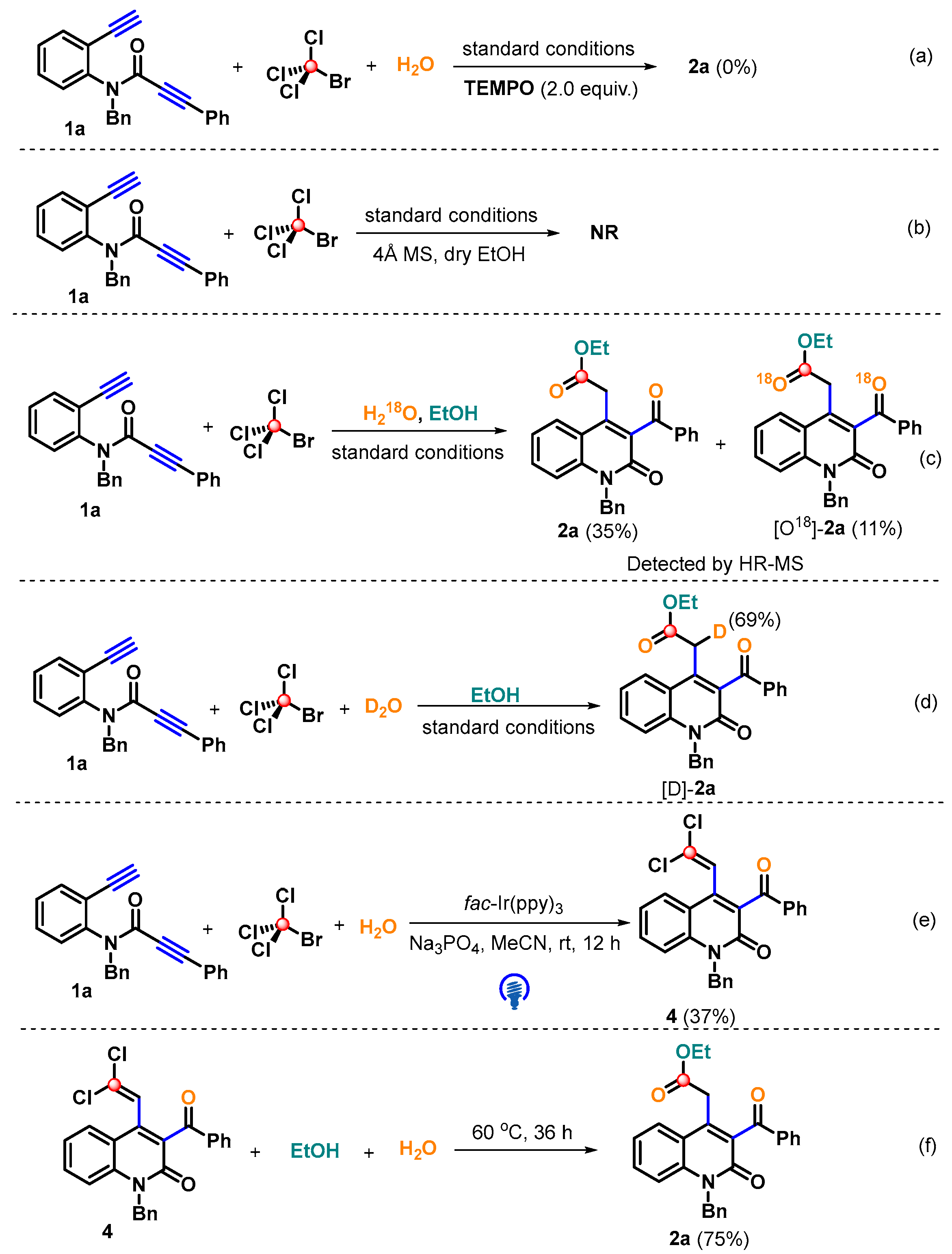
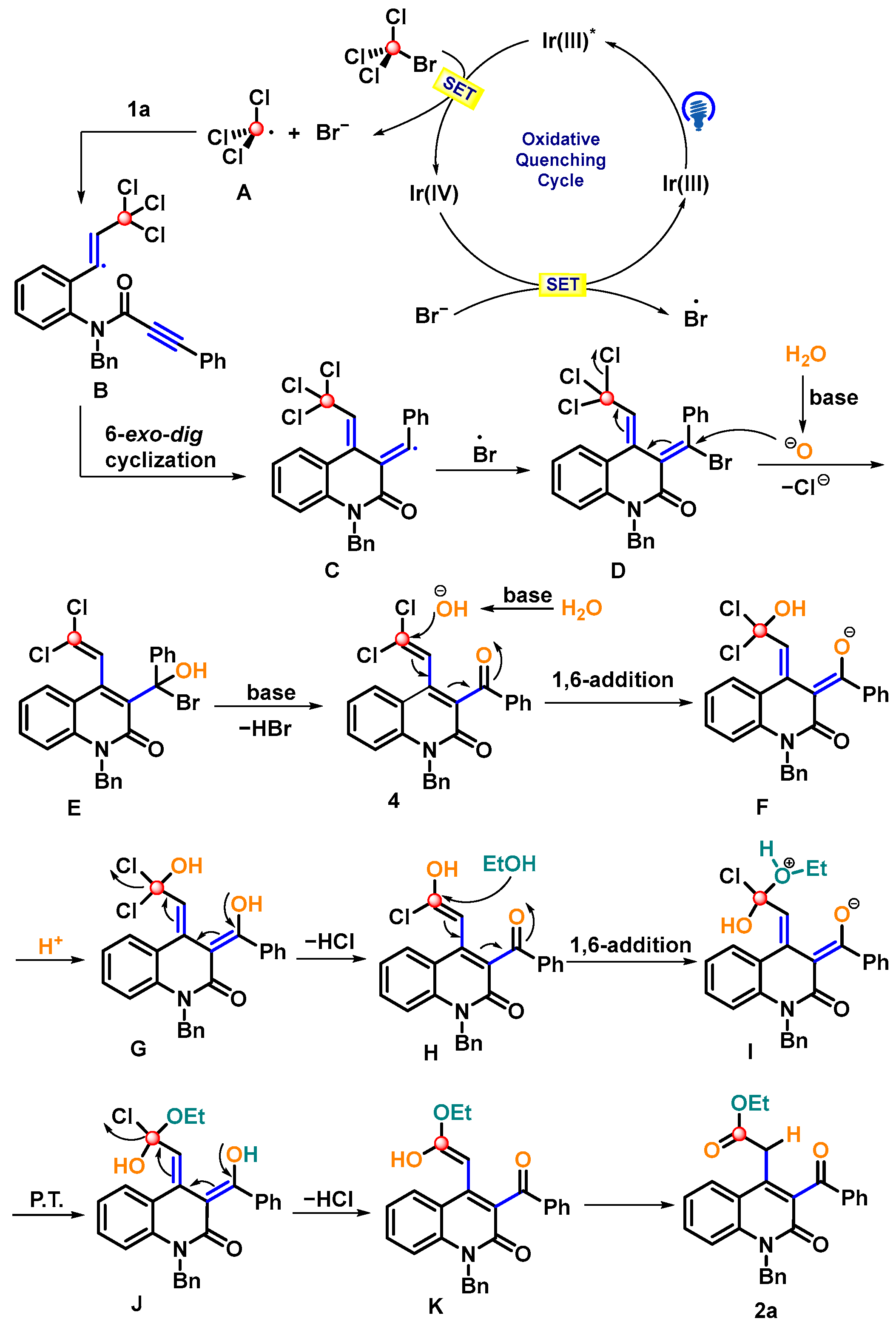
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of Carbon Dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-metal-catalyzed C–C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef]
- Yu, C.; Ma, X.; Song, Q. Palladium-catalyzed cyanation of aryl halides with in situ generated CN− from ClCF2H and NaNH2. Org. Chem. Front. 2020, 7, 2950–2954. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W.A.; Kühn, F.E. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Hu, X. Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction. Chem. Soc. Rev. 2013, 42, 2253–2261. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933–5947. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, Y.; Song, Q. Synthesis of β-Aminoenones via Cross-Coupling of In-Situ-Generated Isocyanides with 1,3-Dicarbonyl Compounds. Org. Lett. 2018, 20, 4777–4781. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Mai, S.; Zhou, Y.; Cheng, G.; Song, Q. Dual role of ethyl bromodifluoroacetate in the formation of fluorine-containing heteroaromatic compounds. Chem. Commun. 2018, 54, 8960–8963. [Google Scholar] [CrossRef]
- Ma, X.; Deng, S.; Song, Q. Halodifluoroacetates as formylation reagents for various amines via unprecedented quadruple cleavage. Org. Chem. Front. 2018, 5, 3505–3509. [Google Scholar] [CrossRef]
- Deng, S.; Chen, H.; Ma, X.; Zhou, Y.; Yang, K.; Lan, Y.; Song, Q. S8-Catalyzed triple cleavage of bromodifluoro compounds for the assembly of N-containing heterocycles. Chem. Sci. 2019, 10, 6828–6833. [Google Scholar] [CrossRef]
- Ma, X.; Su, J.; Zhang, X.; Song, Q. Chlorodifluoromethane as a C1 Synthon in the Assembly of N-Containing Compounds. iScience 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Ma, X.; Song, Q. Recent progress on selective deconstructive modes of halodifluoromethyl and trifluoromethylcontaining reagents. Chem. Soc. Rev. 2020, 49, 9197–9219. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, H.I.; Kim, D.Y. Visible light-mediated photocatalytic bromination of 2-arylimidazo[1,2-a]pyridines using CBr4 as bromine source. Synth. Commun. 2020, 50, 197–206. [Google Scholar] [CrossRef]
- Kumar, S.; Shah, T.A.; Punniyamurthy, T. Recent advances in the application of tetrabromomethane in organic synthesis. Org. Chem. Front. 2021, 8, 4288–4314. [Google Scholar] [CrossRef]
- Dinda, T.K.; Mal, P. Activation of C-Br Bond of CBr4 and CBrCl3 Using 9-Mesityl-10-methylacridinium Perchlorate Photocatalyst. J. Org. Chem. 2023, 88, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-B.; Chen, F.; Li, M.; Bu, Q.; Du, Z.; Liu, J.; Dai, B.; Liu, N. Visible-light-promoted synthesis of gem-dihaloenones. Green Chem. 2023, 25, 1191–1200. [Google Scholar] [CrossRef]
- Wu, D.; Hao, W.-J.; Rao, Q.; Lu, Y.; Tu, S.-J.; Jiang, B. Engaging 1,7-diynes in a photocatalytic Kharasch-type addition/1,5-(SN″)-substitution cascade toward β-gem-dihalovinyl carbonyls. Chem. Commun. 2021, 57, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Liu, Y.-P.; Liu, X.; Fu, R.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photocatalytic Chemodivergent Synthesis of α-gem-Dihalovinyl Ketones and Chromen-2-Ones from Monoalkynes. Adv. Synth. Catal. 2022, 364, 2666–2672. [Google Scholar] [CrossRef]
- Rossi-Ashton, J.-A.; Clarke, A.-K.; Unsworth, W.-P.; Taylor, R.-J. Phosphoranyl Radical Fragmentation Reactions Driven by Photoredox Catalysis. ACS Catal. 2020, 10, 7250–7261. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Pagire, S.-K.; Foell, T.; Reiser, O. Shining visible light on vinyl halides: Expanding the horizons of photocatalysis. Acc. Chem. Res. 2020, 53, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Qin, Y. Indole Alkaloid Synthesis Facilitated by Photoredox Catalytic Radical Cascade Reactions. Acc. Chem. Res. 2019, 52, 1877–1891. [Google Scholar] [CrossRef]
- Wen, J.; Zhao, W.; Gao, X.; Ren, X.; Dong, C.; Wang, C.; Liu, L.; Li, J. Synthesis of [1,2,3]Triazolo-[1,5-a]quinoxalin-4(5H)-ones through Photoredox-Catalyzed [3 + 2] Cyclization Reactions with Hypervalent Iodine(III) Reagents. J. Org. Chem. 2022, 87, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.; Zhao, W.; Wen, J.; Dong, C.; Hu, C.; Li, J. Photoredox-Catalyzed Cascade sp2 C−H Bond Functionalization to Construct Substituted Acridine with Diarylamine and Hypervalent Iodine(III) Reagents. Org. Lett. 2023, 25, 592–596. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Awad, J.M.; Xie, G.; Qiu, W.; Zhang, W. One-pot synthesis of tetrahydro-pyrrolobenzodiazepinones through sequential 1,3-dipolar cycloaddition/N-alkylation(N-acylation)Staudinger/aza-Witting reactions. Green Chem. 2019, 21, 4489–4494. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Q.; Zhang, W. Remote Radical 1,3-,1,4-,1,5-,1,6- and 1,7-Difunctionalization Reaction. Molecules 2023, 28, 3027. [Google Scholar] [CrossRef]
- Wang, C.-S.; Dixneuf, P.H.; Soule, J.-F. Photoredox Catalysis for Building C—C Bonds from C(sp2)—H Bonds. Chem. Rev. 2018, 118, 7532–7585. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Lang, X.; Zhao, J.; Chen, X. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef]
- Narayanama, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef]
- Rotondo, D.M.A.; McCusker, J.K. The photophysics of photoredox catalysis: A roadmap for catalyst design. Chem. Soc. Rev. 2016, 45, 5803–5820. [Google Scholar] [CrossRef]
- Zhu, S.-S.; Zhou, J.-N.; Wu, Q.-L.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photoinduced double [2 + 2] cycloaddition relay of yne-allenones for highly diastereoselective synthesis of hexacyclic 1-naphthols. Org. Chem. Front. 2020, 7, 2975–2980. [Google Scholar] [CrossRef]
- Zheng, J.-L.; Wu, D.; Lin, N.; Liu, Y.-P.; Wang, L.; Zhu, X.-T.; Hao, W.-J.; Wang, S.-L.; Jiang, B. Kharasch-type photocyclization of 1,7-diynes for the stereospecific synthesis of tetrahydronaphthalen-1-ols. Tetrahedron Lett. 2021, 85, 153485. [Google Scholar] [CrossRef]
- Wang, L.; Shen, Y.-T.; Wang, Y.-X.; Wang, H.-Y.; Hao, W.-J.; Jiang, B. Multicomponent Annulative SO2 Insertion of Heteroatom-Linked 1,7-Diynes for Accessing Tricyclic Sulfones. Adv. Synth. Catal. 2023, 365, 1693–1698. [Google Scholar] [CrossRef]
- Wang, L.; Xu, T.; Rao, Q.; Zhang, T.-S.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Photocatalytic Biheterocyclization of 1,7-Diynes for Accessing Skeletally Diverse Tricyclic 2-Pyranones. Org. Lett. 2021, 23, 7845–7850. [Google Scholar] [CrossRef] [PubMed]
- Nikitas, N.-F.; Voutyritsa, E.; Gkizis, P.-L.; Kokotos, C.-G. Metal-free Photochemical Atom Transfer Radical Addition (ATRA) of BrCCl3 to Alkenes. Eur. J. Org. Chem. 2021, 2021, 96–101. [Google Scholar] [CrossRef]
- Nguyen, D.; Tucker, J.-W.; Konieczynska, M.-D.; Stephenson, C.-R. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef] [PubMed]
- Voutyritsa, E.; Triandafillidi, L.; Tzouras, N.-V.; Nikitas, N.-F.; Pefkianakis, E.-K.; Vougioukalakis, G.-C.; Kokotos, C.-G. Photocatalytic Atom Transfer Radical Addition to Olefins Utilizing Novel Photocatalysts. Molecules 2019, 24, 1644. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.-Z.; Wang, S.-C.; Song, K.-X.; Hao, W.-J.; Jiang, B. Visible-Light-Driven Photocatalytic Kharasch-Type Addition of 1,6-Enynes. Chin. J. Org. Chem. 2021, 41, 4815–4824. [Google Scholar] [CrossRef]
- Ji, X.-S.; Fu, R.; Wang, S.-L.; Hao, W.-J.; Jiang, B. Visible Light Driven Phot ocatalytic Kharasch Reaction of Phenol/Arylamine Linked 1,6 Enynes with Perhalogenated Methane. Chin. J. Org. Chem. 2022, 42, 4282–4291. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, S.; Tang, Y.; Yan, S.; Li, G. Copper-Catalyzed Annulation-Trifluoromethyl Functionalization of Enynones. Org. Lett. 2023, 25, 2509–2514. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Li, G.; Hao, W.-J.; Jiang, B. Catalytic Benzannulation Reactions of Enynones for Accessing Heterocycle-Incorporating Diarylmethanes. Synlett 2023, 34, 243–248. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, S.; Yuan, Q.; Li, G.; Yan, S. Catalytic Radical-Triggered Annulation/Iododifluoromethylation of Enynones for the Stereospecific Synthesis of 1-Indenones. J. Org. Chem. 2023, 88, 8532–8541. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, D.; Wang, J.-Y.; Yan, S.; Li, G. Four-layer folding framework: Design, GAP synthesis, and aggregation-induced emission. Front. Chem. 2023, 11, 1259609. [Google Scholar] [CrossRef] [PubMed]
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition Metal Photoredox Catalysis of Radical Thiol-Ene Reactions. J. Org. Chem. 2013, 78, 2046–2050. [Google Scholar] [CrossRef]
- Keylor, M.H.; Park, J.E.; Wallentin, C.-J.; Stephenson, C.R.J. Photocatalytic initiation of thioleene reactions: Synthesis of thiomorpholin-3-ones. Tetrahedron 2014, 70, 4264–4269. [Google Scholar] [CrossRef]
- Bacauanu, V.; Cardinal, S.; Yamauchi, M.; Kondo, M.; Fernandez, D.F.; Remy, R.; MacMillan, D.W.C. Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem. Int. Ed. 2018, 57, 12543–12548. [Google Scholar] [CrossRef]
- Wang, S.-W.; Yu, J.; Zhou, Q.-Y.; Chen, S.-Y.; Xu, Z.-H.; Tang, S. Visible-Light-Induced Atom Transfer Radical Addition and Cyclization of Perfluoroalkyl Halides with 1,n-Enynes. ACS Sustain. Chem. Eng. 2019, 7, 10154–10162. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| Entry | [PC] | Base | Yield (%) b |
| 1 | fac-Ir(ppy)3 | - | 48 |
| 2 | fac-Ir(ppy)3 | K2CO3 | 67 |
| 3 c | fac-Ir(ppy)3 | K2CO3 | 35 |
| 4 d | fac-Ir(ppy)3 | K2CO3 | 52 |
| 5 | - | K2CO3 | NR |
| 6 | [Ir(dFCF3ppy)2dtbbpy]PF6 | K2CO3 | 47 |
| 7 | Eosin Y | K2CO3 | 28 |
| 8 | Mes-Acr+ClO4− | K2CO3 | 16 |
| 9 | fac-Ir(ppy)3 | Na2CO3 | 28 |
| 10 | fac-Ir(ppy)3 | Cs2CO3 | 27 |
| 11 | fac-Ir(ppy)3 | MeONa | 45 |
| 12 | fac-Ir(ppy)3 | Et3N | 55 |
| 13 | fac-Ir(ppy)3 | DMAP | 59 |
| 14 e | fac-Ir(ppy)3 | K2CO3 | 54 |
| 15 f | fac-Ir(ppy)3 | K2CO3 | 39 |
| 16 g | fac-Ir(ppy)3 | K2CO3 | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Bao, Y.; Yan, S.; Wang, J.; Zhang, Y.; Li, G. Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules 2024, 29, 782. https://doi.org/10.3390/molecules29040782
Chen D, Bao Y, Yan S, Wang J, Zhang Y, Li G. Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules. 2024; 29(4):782. https://doi.org/10.3390/molecules29040782
Chicago/Turabian StyleChen, Daixiang, Yu Bao, Shenghu Yan, Jiayin Wang, Yue Zhang, and Guigen Li. 2024. "Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane" Molecules 29, no. 4: 782. https://doi.org/10.3390/molecules29040782
APA StyleChen, D., Bao, Y., Yan, S., Wang, J., Zhang, Y., & Li, G. (2024). Photocatalytic Multicomponent Annulation of Amide-Anchored 1,7-Diynes Enabled by Deconstruction of Bromotrichloromethane. Molecules, 29(4), 782. https://doi.org/10.3390/molecules29040782