

Design and Evaluation of NSAID Derivatives as AKR1C3 Inhibitors for Breast Cancer Treatment through Computer-Aided Drug Design and In Vitro Analysis

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
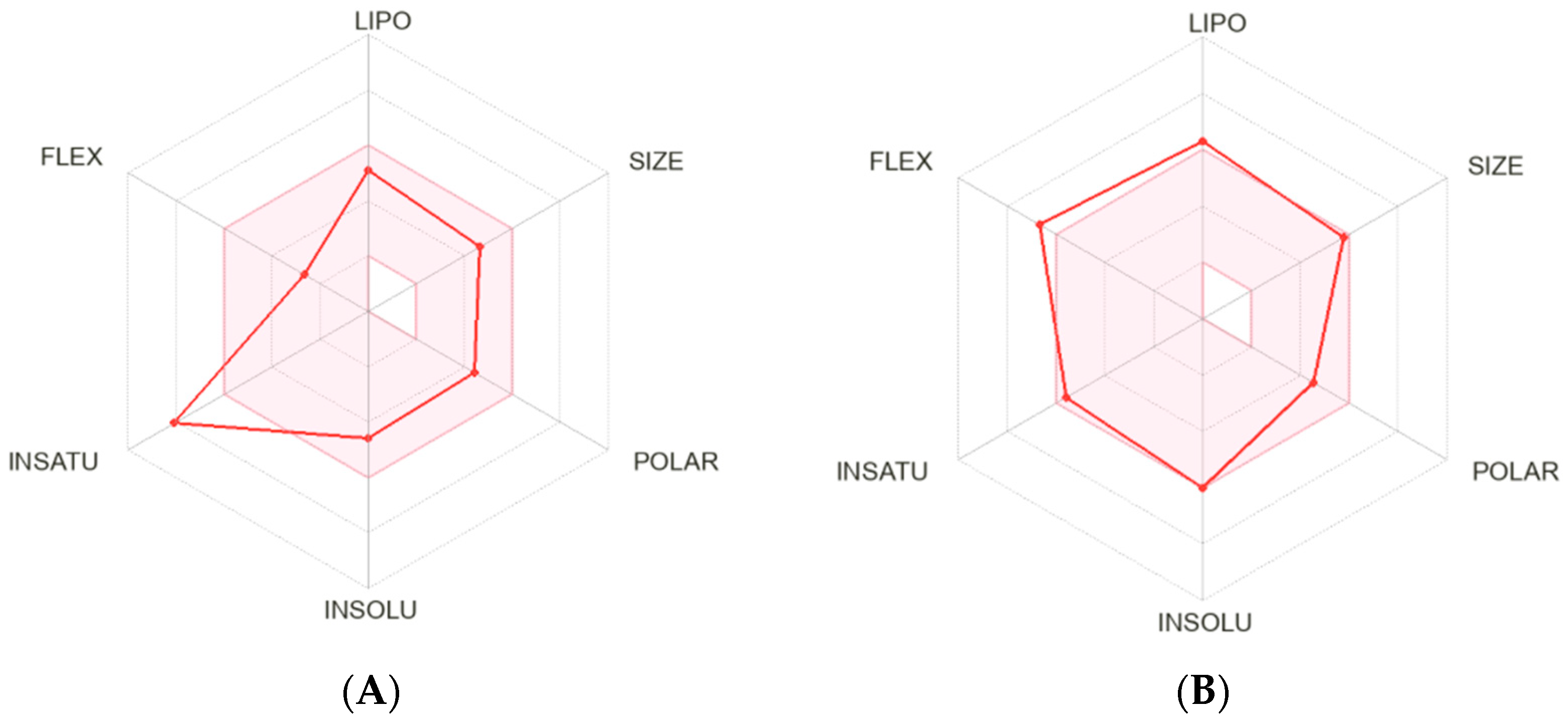
2.1. Bioinformatic
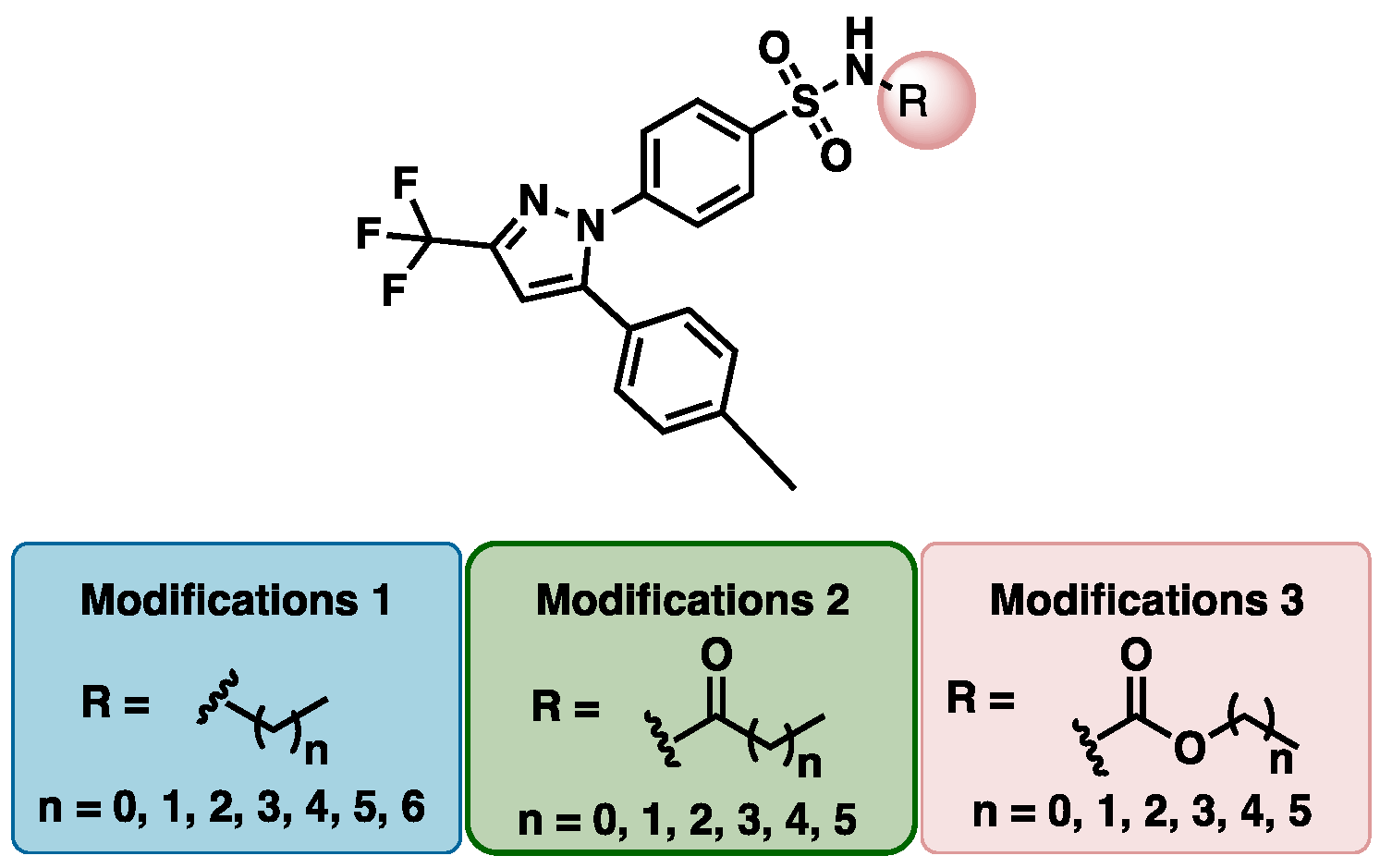
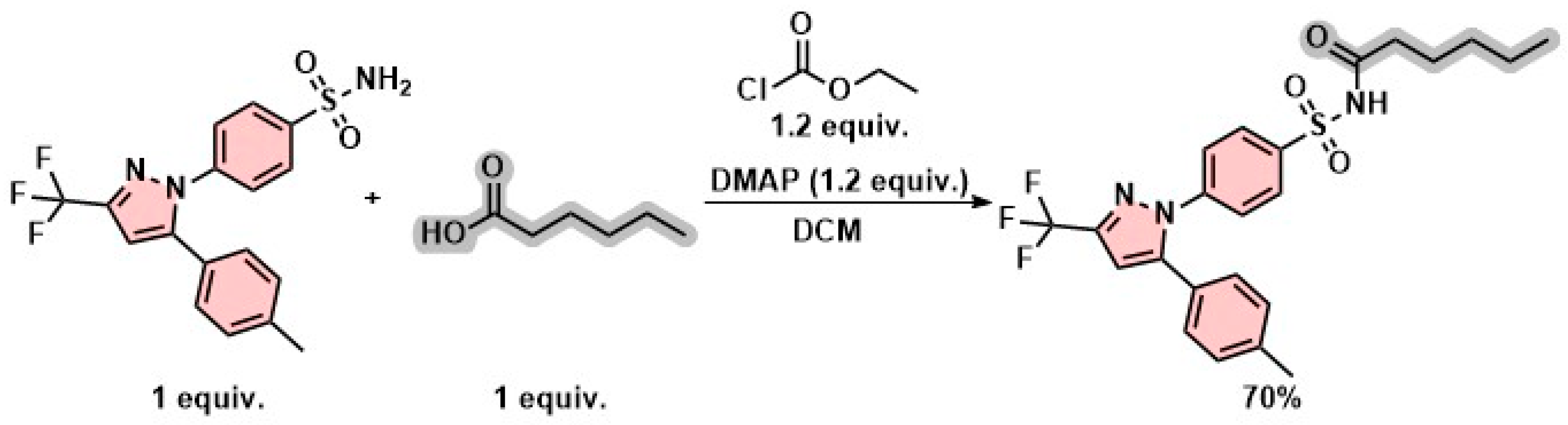
2.2. Synthesis and Characterization
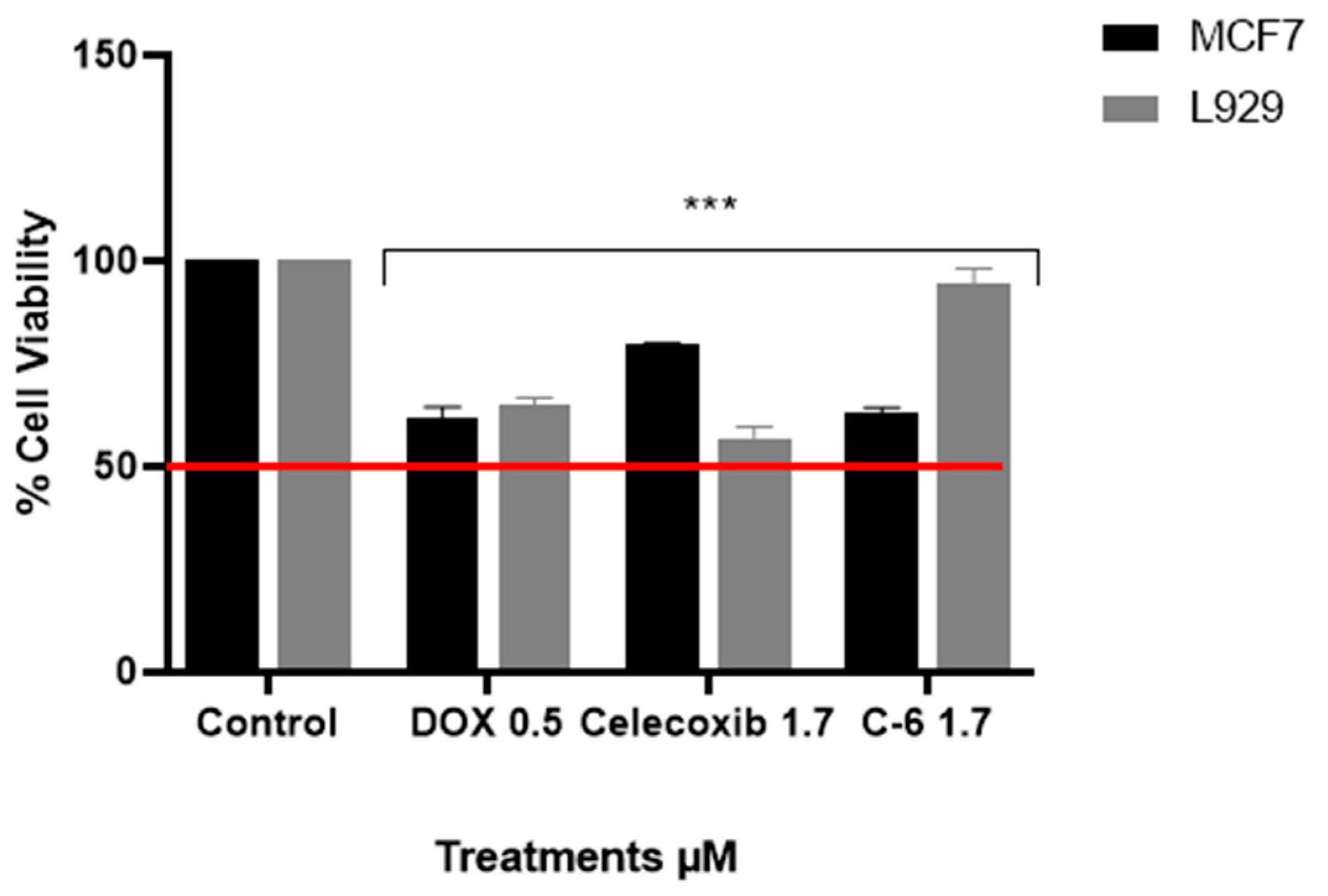
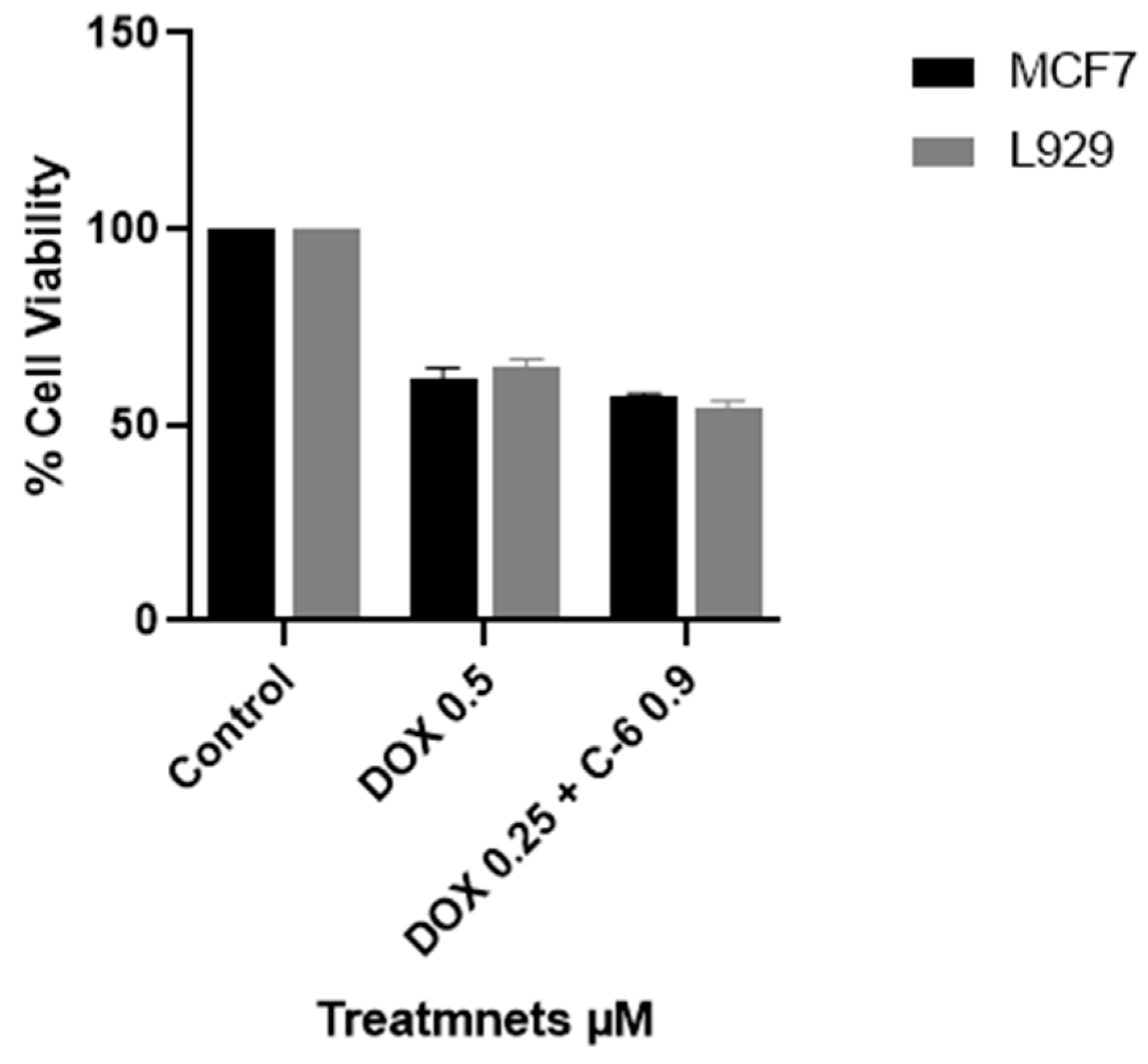
2.3. Viability Assay
3. Materials and Methods
3.1. Bioinformatics
3.1.1. Ligand-Based Virtual Screening (LBVS)
3.1.2. Structure-Based Virtual Screening (SBVS)
3.1.3. Toxicity and LogP Profile
3.2. Chemistry
Solubility and HPLC Method to C-6 and Celecoxib
3.3. Cell Lines and Culture Conditions
3.3.1. Treatments
3.3.2. Cytotoxicity Screening
3.3.3. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ji, X.; Lu, Y.; Tian, H.; Meng, X.; Wei, M.; Cho, W.C. Chemoresistance mechanisms of breast cancer and their countermeasures. Biomed. Pharmacother. 2019, 114, 108800–108810. [Google Scholar] [CrossRef]
- David, R.B.; Stephen, I.R.; Lawrence, M.S.; Yi, J.; Sridhar, G.; Trevor, M.P. Development of Nonsteroidal Anti-Inflammatory Drug Analogs and Steroid Carboxylates Selective for Human Aldo-Keto Reductase Isoforms: Potential Antineoplastic Agents That Work Independently of Cyclooxygenase Isozymes. Mol. Pharmacol. 2005, 67, 60–68. [Google Scholar]
- Liu, Y.; Chen, Y.; Jiang, J.; Chu, X.; Guo, Q.; Zhao, L.; Feng, F.; Liu, W.; Zhang, X.; He, S.; et al. Development of highly potent and specific AKR1C3 inhibitors to restore the chemosensitivity of drug-resistant breast cancer. Eur. J. Med. Chem. 2023, 247, 115013–115020. [Google Scholar] [CrossRef]
- Tołoczko-Iwaniuk, N.; Dziemiańczyk-Pakieła, D.; Nowaszewska, B.K.; Celińska-Janowicz, K.; Miltyk, W. Celecoxib in Cancer Therapy and Prevention—Review. Curr. Drug Targets 2019, 20, 302–315. [Google Scholar] [CrossRef]
- Yang, J.C.; Xu, P.; Ning, S.; Wasielewski, L.J.; Adomat, H.; Hwang, S.H.; Morisseau, C.; Gleave, M.; Corey, E.; Gao, A.C.; et al. Novel inhibition of AKR1C3 and androgen receptor axis by PTUPB synergizes enzalutamide treatment in advanced prostate cancer. Oncogene 2023, 42, 693–707. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Y.; Hang Liu1, J.Y.; Song, X.; Zhao, J.; He, N.; Zhou, C.J.; Wang, Y.; Huang, C.; Dong, Q. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E2 and down-regulating the Wnt pathway activity. Oncotarget 2017, 8, 115254–115269. [Google Scholar] [CrossRef]
- Khafaga, A.F.; Shamma, R.N.; Abdeen, A.; Barakat, A.M.; Noreldin, A.E.; Elzoghby, A.O.; Sallam, M.A. Celecoxib repurposing in cancer therapy: Molecular mechanisms and nanomedicine-based delivery technologies. Nanomedicine 2021, 16, 1691–1712. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Schwartzbaum, J.A. Celecoxib may be a viable treatment option for breast cancer patients not treated with chemotherapy. Front. Oncol. 2022, 12, 958308. [Google Scholar] [CrossRef] [PubMed]
- El-Haj, B.M.; Ahmed, S.B.M.; Garawi, M.A.; Ali, H.S. Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity. Molecules 2018, 23, 2119. [Google Scholar] [CrossRef]
- El-Shahat, M.; Salama, M.A.M.; El-Farargyj, A.F.; Ali, M.M.; Ahmed, D.M. Effective Pharmacophore for CDC25 Phosphatases Enzyme Inhibitors: Newly Synthesized Bromothiazolopyrimidine Derivatives. Bentham Sci. 2022, 21, 118–131. [Google Scholar]
- El-Sofany, W.I.; El-sayed, W.A.; Abd-Rabou, A.A.; El-Shahat, M. Synthesis of new imidazole-triazole-glycoside hybrids as anti-breast cancer candidates. J. Mol. Struct. 2022, 1270, 133942–133954. [Google Scholar] [CrossRef]
- Flefel, E.M.; El-Sofany, W.I.; Al-Harbi, R.A.K.; El-Shahat, M. Development of a Novel Series of Anticancer and Antidiabetic: Spirothiazolidines Analogs. Molecules 2019, 24, 2511. [Google Scholar] [CrossRef] [PubMed]
- Shamroukh, A.H.; El-Shahat, M.; Drabowicz, J.; Ali, M.M.; Rashad, A.E.; Ali, H.S. Anticancer evaluation of some newly synthesized N-nicotinonitrile derivative. Eur. J. Med. Chem. 2013, 69, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Moudgil, M.n.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef]
- Rizzuti, B.; Grande, F. Chapter 14—Virtual screening in drug discovery: A precious tool for a still-demanding challenge. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 309–327. [Google Scholar]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef]
- Lawless, M.S.; Waldman, M.; Fraczkiewicz, R.; Clark, R.D. Using Cheminformatics in Drug Discovery. In New Approaches to Drug Discovery; Nielsch, U., Fuhrmann, U., Jaroch, S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 139–168. [Google Scholar]
- Jaramillo, D.N.; Millán, D.; Guevara-Pulido, J. Design, synthesis and cytotoxic evaluation of a selective serotonin reuptake inhibitor (SSRI) by virtual screening. Eur. J. Pharm. Sci. 2023, 183, 106403–106412. [Google Scholar] [CrossRef]
- Guevara-Pulido, J.; Jiménez, R.A.; Morantes, S.J.; Jaramillo, D.N.; Acosta-Guzmán, P. Design, Synthesis, and Development of 4-[(7-Chloroquinoline-4-yl)amino]phenol as a Potential SARS-CoV-2 Mpro Inhibitor. ChemistrySelect 2022, 7, e202200125. [Google Scholar] [CrossRef]
- Zambrano, D.; Millán, D.; Guevara-Pulido, J. In silico design, synthesis and evaluation of a less toxic octinoxate alternative with suitable photoprotection properties. Eur. J. Pharm. Sci. 2023, 180, 106332–106340. [Google Scholar] [CrossRef] [PubMed]
- Byrns, M.C.; Jin, Y.; Penning, T.M. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): Overview and structural insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Aldo-Keto Reductase (AKR) 1C3 inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Prieto, M.; Niño, A.; Acosta-Guzmán, P.; Guevara-Pulido, J. Design and synthesis of a potential selective JAK-3 inhibitor for the treatment of rheumatoid arthritis using predictive QSAR models. Inform. Med. Unlocked 2024, 45, 101464–101476. [Google Scholar] [CrossRef]
- Pirela-Ocando, S.; Romero-Cabezas, A.; Guevara-Pulido, J. Construction of a predictive model for the design of triptamin analogues with potential activity in Parkinson’s and Alzheimer’s diseases. Inform. Med. Unlocked 2023, 43, 101413–101423. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Maunz, A.; Gütlein, M.; Rautenberg, M.; Vorgrimmler, D.; Gebele, D.; Helma, C. lazar: A modular predictive toxicology framework. Front. Pharmacol. 2013, 4, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Xing, E.; Wu, S.; Zhuang, T.; Li, P.-K.; Li, C.; Cheng, X. Computational modeling studies reveal the origin of the binding preference of 3-(3,4-di hydroisoquinolin-2(1H)-ylsulfonyl)benzoic acids for AKR1C3 over its isoforms. Protein Sci. 2022, 31, e4499. [Google Scholar] [CrossRef] [PubMed]
- Pippione, A.C.; Kilic-Kurt, Z.; Kovachka, S.; Sainas, S.; Rolando, B.; Denasio, E.; Pors, K.; Adinolfi, S.; Zonari, D.; Bagnati, R.; et al. New aldo-keto reductase 1C3 (AKR1C3) inhibitors based on the hydroxytriazole scaffold. Eur. J. Med. Chem. 2022, 237, 114366–114372. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. iLOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Byrns, M.C.; Duan, L.; Lee, S.H.; Blair, I.A.; Penning, T.M. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J. Steroid Biochem. Mol. Biol. 2010, 118, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Xu, F.; Xu, J.; Liu, L.; Chen, Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN Loss. Biomed. Pharmacother. 2015, 69, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17–42. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions, and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | LBVS Exp IC50 (µM) | Pred IC50 a (µM) | SBVS Affinity b (kcal/mol) | Lipophilicity Pred c |
|---|---|---|---|---|---|
| 1 | Naproxen | 0.5 | 0.6 | −8.6 | 2.76 |
| 2 | Diclofenac | 2.6 | 1.2 | −8.9 | 3.66 |
| 3 | Flurbiprofen | 7.8 | 4 | −9.3 | 3.59 |
| 4 | Lornoxicam | 0.7 | 0.6 | −8.7 | 1.50 |
| 5 | Mefenamic acid | 0.3 | 0.1 | −9.0 | 3.30 |
| 6 | Ibuprofen | 33.0 | 30 | −7.7 | 3.00 |
| 7 | Celecoxib | 5.2 | 2.3 | −10.4 | 3.40 d |
| 8 | Ketoprofen | 6.0 | 3.0 | −9.0 | 2.84 |
| 9 | Sulindac | 3.4 | 3.6 | −9.9 | 3.96 |
| 10 | Indomethacin | 2.3 | 0.4 | −9.4 | 3.63 |
| 11 | A1 | -- | 2.0 | −9.4 | 3.75 |
| 12 | A2 | -- | 2.2 | −9.3 | 4.03 |
| 13 | A3 | -- | 2.3 | −9.5 | 4.35 |
| 14 | A4 | -- | 2.2 | −9.6 | 4.74 |
| 15 | A5 | -- | 2.0 | −9.3 | 5.10 |
| 16 | A6 | -- | 2.1 | −9.4 | 5.42 |
| 17 | B1 | -- | 2.2 | −10.7 | 3.50 |
| 18 | B2 | -- | 2.3 | −10.9 | 3.82 |
| 19 | B3 | -- | 2.3 | −11.1 | 4.22 |
| 22 | B4 | -- | 2.5 | −11.5 | 4.55 |
| 23 | B5 | -- | 2.4 | −10.1 | 4.86 |
| 24 | B6 | -- | 2.4 | −10.1 | 5.14 |
| 25 | C2 | -- | 2.1 | −11.4 | 3.46 |
| 26 | C3 | -- | 1.9 | −11.3 | 3.76 |
| 27 | C4 | -- | 1.9 | −11.2 | 4.10 |
| 28 | C5 | -- | 1.8 | −11.1 | 4.46 |
| 29 | C6 | -- | 1.7 | −11.4 | 4.81 d |
| 30 | C7 | -- | 1.7 | −11.0 | 5.16 |
| Descriptor | Description |
|---|---|
| naAromAtom | Number of aromatic atoms |
| TopoPSA | Topological polar surface area |
| McGowan_Volume | Volume of a mole when the molecules are not in motion |
| Compound | Mutagenicity | Carcinogenicity in Rats | Carcinogenicity in Mice |
|---|---|---|---|
| Celecoxib | Non-mutagen | Negative | Positive |
| C-6 | Non-mutagen | Negative | Negative |
| C-7 | Non-mutagen | Negative | Negative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca-Benítez, V.; Acosta-Guzmán, P.; Sánchez, J.E.; Alarcón, Z.; Jiménez, R.A.; Guevara-Pulido, J. Design and Evaluation of NSAID Derivatives as AKR1C3 Inhibitors for Breast Cancer Treatment through Computer-Aided Drug Design and In Vitro Analysis. Molecules 2024, 29, 1802. https://doi.org/10.3390/molecules29081802
Fonseca-Benítez V, Acosta-Guzmán P, Sánchez JE, Alarcón Z, Jiménez RA, Guevara-Pulido J. Design and Evaluation of NSAID Derivatives as AKR1C3 Inhibitors for Breast Cancer Treatment through Computer-Aided Drug Design and In Vitro Analysis. Molecules. 2024; 29(8):1802. https://doi.org/10.3390/molecules29081802
Chicago/Turabian StyleFonseca-Benítez, Victoria, Paola Acosta-Guzmán, Juan Esteban Sánchez, Zaira Alarcón, Ronald Andrés Jiménez, and James Guevara-Pulido. 2024. "Design and Evaluation of NSAID Derivatives as AKR1C3 Inhibitors for Breast Cancer Treatment through Computer-Aided Drug Design and In Vitro Analysis" Molecules 29, no. 8: 1802. https://doi.org/10.3390/molecules29081802
APA StyleFonseca-Benítez, V., Acosta-Guzmán, P., Sánchez, J. E., Alarcón, Z., Jiménez, R. A., & Guevara-Pulido, J. (2024). Design and Evaluation of NSAID Derivatives as AKR1C3 Inhibitors for Breast Cancer Treatment through Computer-Aided Drug Design and In Vitro Analysis. Molecules, 29(8), 1802. https://doi.org/10.3390/molecules29081802