
NIMO: A Natural Product-Inspired Molecular Generative Model Based on Conditional Transformer

Abstract
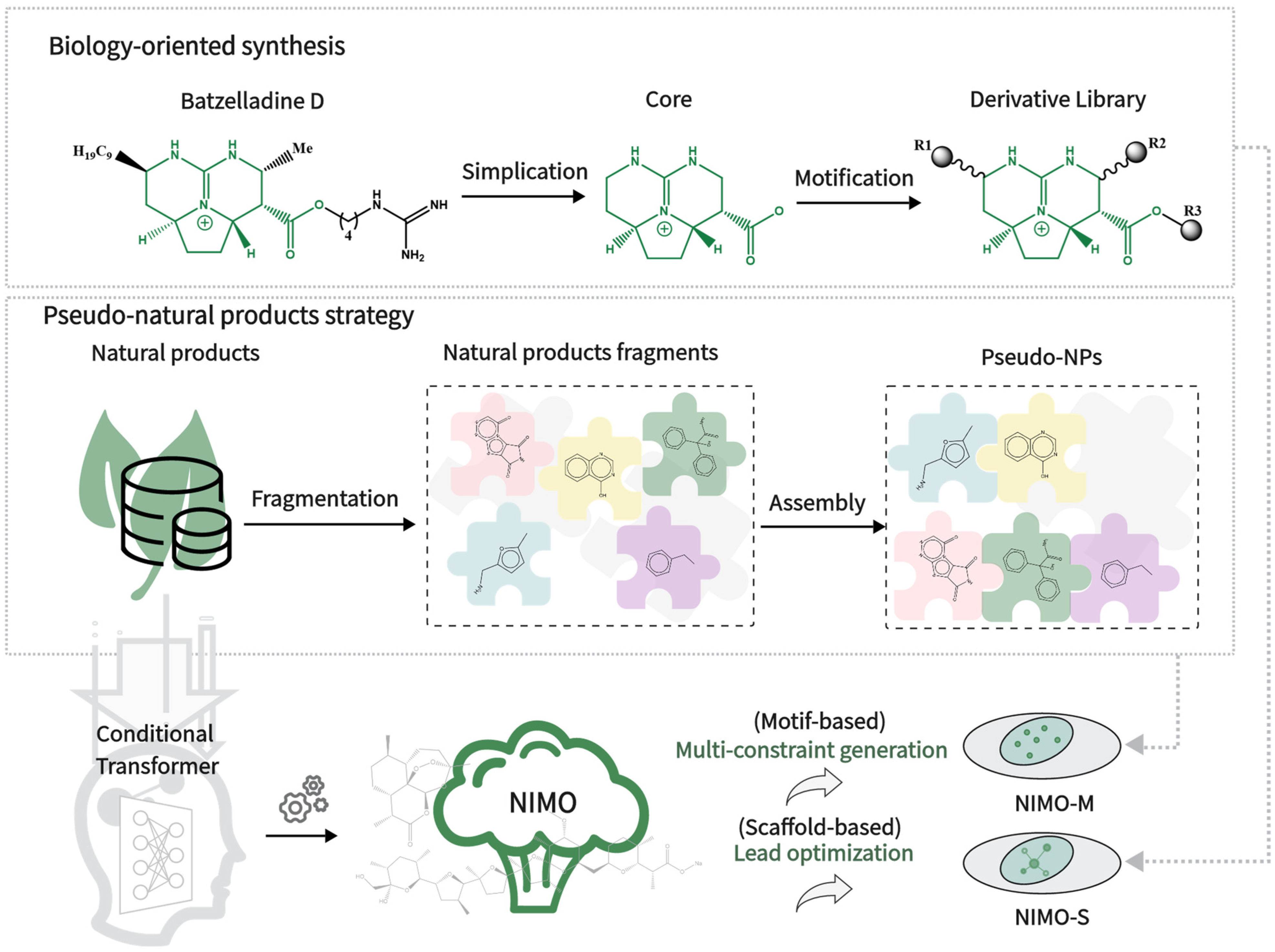
:1. Introduction
2. Results and Discussion
2.1. Model Evaluation
2.2. Terpenoid Generation
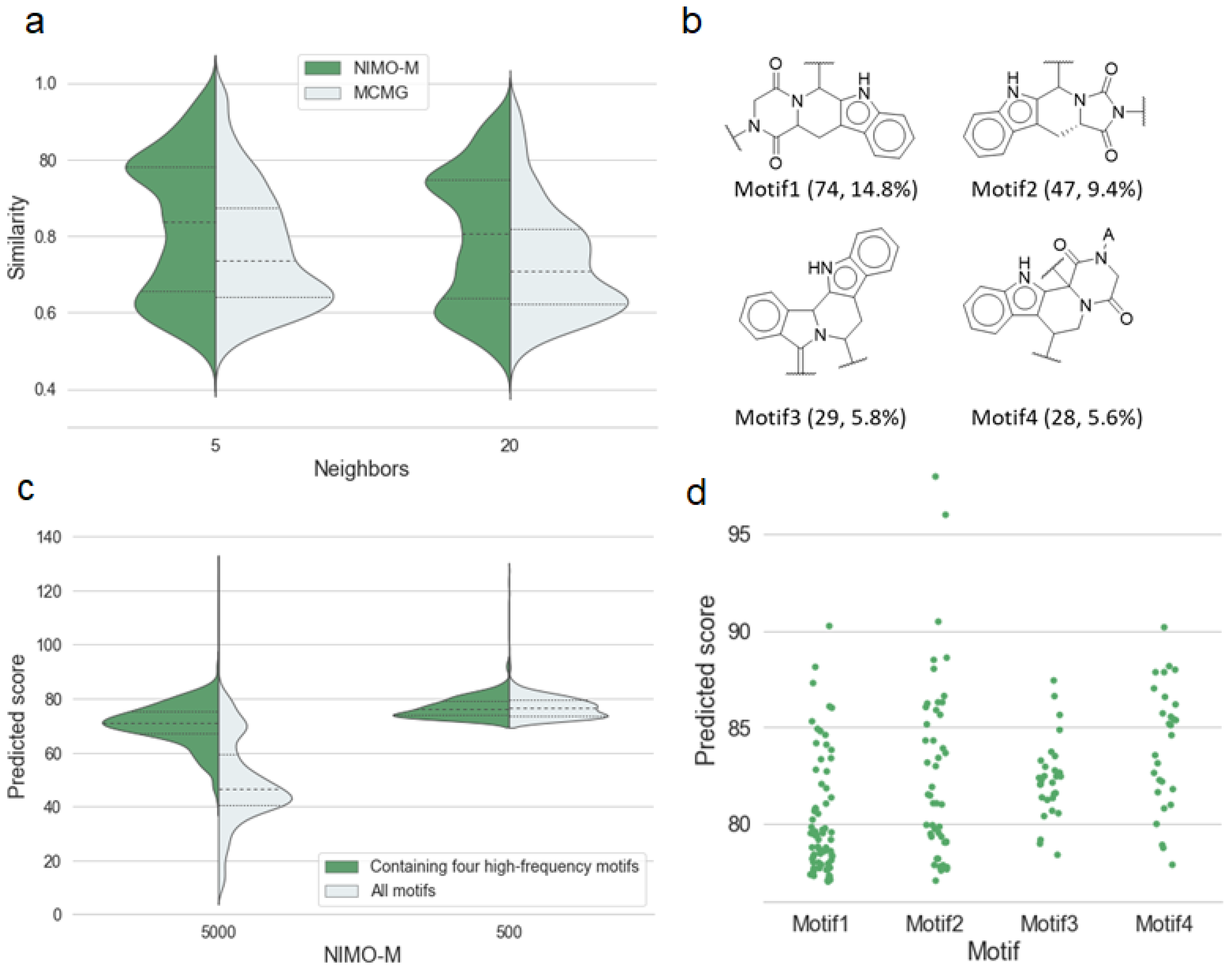
2.3. Antimalarial Activity-Oriented Molecular Generation
2.4. Pocket-Based Molecular Generation
2.5. Discussion
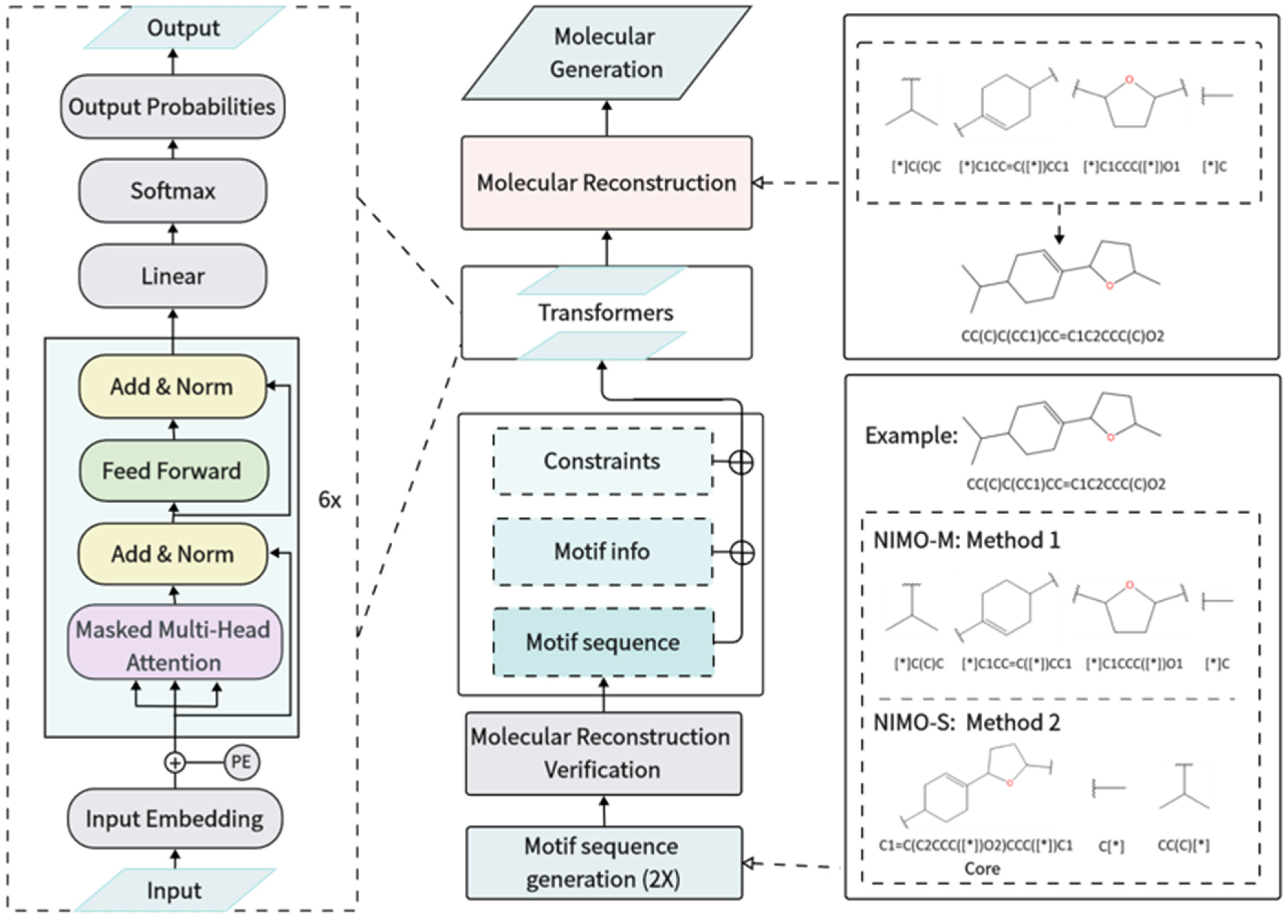
3. Methods
3.1. Data Preparation
3.2. Motif Sequence Generation
3.3. Molecular Reconstruction Verification
3.4. Model Architecture
4. Experiment Configuration
4.1. Datasets
4.1.1. COCONUT
4.1.2. TeroKIT
4.1.3. Anti-Malarial Experimental Activity Data Set
4.1.4. Antibacterial Dataset
4.2. Fragment Extraction
4.3. Evaluation Setting 1
4.4. Evaluation Setting 2
4.5. Evaluation Setting 3
4.6. Baseline Models
4.6.1. MCMG
4.6.2. QBMG
4.6.3. FBMG
4.7. NP-Likeness Score
4.8. NPClassifier
4.9. MAIP
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grigalunas, M.; Brakmann, S.; Waldmann, H. Chemical Evolution of Natural Product Structure. J. Am. Chem. Soc. 2022, 144, 3314–3329. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; International Natural Product Sciences Taskforce. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xue, X. Medicinal Chemistry Strategies for the Modification of Bioactive Natural Products. Molecules 2024, 29, 689. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Hernandez, A.L.; Sanchez-Cruz, N.; Medina-Franco, J.L. A Fragment Library of Natural Products and its Comparative Chemoinformatic Characterization. Mol. Inform. 2020, 39, e2000050. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, S.; Bon, R.S.; Kumar, K.; Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. Engl. 2011, 50, 10800–10826. [Google Scholar] [CrossRef] [PubMed]
- Gagare, S.; Patil, P.; Jain, A. Natural product-inspired strategies towards the discovery of novel bioactive molecules. Future J. Pharm. Sci. 2024, 10, 55. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- Bag, S.; Liu, J.; Patil, S.; Bonowski, J.; Koska, S.; Schölermann, B.; Zhang, R.; Wang, L.; Pahl, A.; Sievers, S. A divergent intermediate strategy yields biologically diverse pseudo-natural products. Nat. Chem. 2024, 1–14. [Google Scholar] [CrossRef]
- Nelson, A.; Karageorgis, G. Natural product-informed exploration of chemical space to enable bioactive molecular discovery. RSC Med. Chem. 2021, 12, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.H.; Zhou, F.F.; Sun, Y.H.; Li, Q.Z. Deconstructive and Divergent Synthesis of Bioactive Natural Products. Molecules 2023, 28, 6193. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.M. Dynamic combinatorial chemistry and virtual combinatorial libraries. In Essays in Contemporary Chemistry: From Molecular Structure towards Biology; Wiley Online Library: Hoboken, NJ, USA, 2001; pp. 307–326. [Google Scholar]
- Cheng, Y.; Gong, Y.; Liu, Y.; Song, B.; Zou, Q. Molecular design in drug discovery: A comprehensive review of deep generative models. Brief. Bioinform. 2021, 22, bbab344. [Google Scholar] [CrossRef] [PubMed]
- Fromer, J.C.; Coley, C.W. Computer-aided multi-objective optimization in small molecule discovery. Patterns 2023, 4, 100678. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Manica, M. Regression transformer enables concurrent sequence regression and generation for molecular language modelling. Nat. Mach. Intell. 2023, 5, 432–444. [Google Scholar] [CrossRef]
- Wang, J.; Hsieh, C.-Y.; Wang, M.; Wang, X.; Wu, Z.; Jiang, D.; Liao, B.; Zhang, X.; Yang, B.; He, Q.; et al. Multi-constraint molecular generation based on conditional transformer, knowledge distillation and reinforcement learning. Nat. Mach. Intell. 2021, 3, 914–922. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Proc. Syst. 2017, 30, 5998–6008. [Google Scholar]
- Zheng, S.; Yan, X.; Gu, Q.; Yang, Y.; Du, Y.; Lu, Y.; Xu, J. QBMG: Quasi-biogenic molecule generator with deep recurrent neural network. J. Cheminform. 2019, 11, 5. [Google Scholar] [CrossRef]
- Yoshikai, Y.; Mizuno, T.; Nemoto, S.; Kusuhara, H. Difficulty in chirality recognition for Transformer architectures learning chemical structures from string representations. Nat. Commun. 2024, 15, 1197. [Google Scholar] [CrossRef]
- Jin, W.; Barzilay, R.; Jaakkola, T. Hierarchical generation of molecular graphs using structural motifs. In Proceedings of the International Conference on Machine Learning, Virtual, 13–18 July 2020; pp. 4839–4848. [Google Scholar]
- Podda, M.; Bacciu, D.; Micheli, A. A Deep Generative Model for Fragment-Based Molecule Generation. Int. Conf. Artif. Intell. Stat. 2020, 108, 2240–2250. [Google Scholar]
- Ortholand, J.Y.; Ganesan, A. Natural products and combinatorial chemistry: Back to the future. Curr. Opin. Chem. Biol. 2004, 8, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Clark, R.L.; Mackay, S.P.; Johnston, B.F. Current strategies for drug discovery through natural products. Expert Opin. Drug Discov. 2010, 5, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mullowney, M.W.; Duncan, K.R.; Elsayed, S.S.; Garg, N.; van der Hooft, J.J.J.; Martin, N.I.; Meijer, D.; Terlouw, B.R.; Biermann, F.; Blin, K.; et al. Artificial intelligence for natural product drug discovery. Nat. Rev. Drug Discov. 2023, 22, 895–916. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715. [Google Scholar] [CrossRef]
- Jinsong, S.; Qifeng, J.; Xing, C.; Hao, Y.; Wang, L. Molecular fragmentation as a crucial step in the AI-based drug development pathway. Commun. Chem. 2024, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.H.; Cai, A.; Miret, S.; Malkomes, G.; Phielipp, M.; Aspuru-Guzik, A. Group SELFIES: A robust fragment-based molecular string representation. Digit. Discov. 2023, 2, 748–758. [Google Scholar] [CrossRef]
- Lim, J.; Hwang, S.-Y.; Moon, S.; Kim, S.; Kim, W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2020, 11, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, C.; Yang, R.; Zhao, S.; Li, F.; Li, X.; Chen, L.; Wan, X.; Liu, X.; Yang, T. Discovery of pyrazolo [3, 4-d] pyridazinone derivatives as selective DDR1 inhibitors via deep learning based design, synthesis, and biological evaluation. J. Med. Chem. 2021, 65, 103–119. [Google Scholar] [CrossRef]
- Seidel, T.; Wieder, O.; Garon, A.; Langer, T. Applications of the Pharmacophore Concept in Natural Product inspired Drug Design. Mol. Inform. 2020, 39, e2000059. [Google Scholar] [CrossRef]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008, 48, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Polykovskiy, D.; Zhebrak, A.; Sanchez-Lengeling, B.; Golovanov, S.; Tatanov, O.; Belyaev, S.; Kurbanov, R.; Artamonov, A.; Aladinskiy, V.; Veselov, M.; et al. Molecular Sets (MOSES): A Benchmarking Platform for Molecular Generation Models. Front. Pharmacol. 2020, 11, 565644. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A Deep Neural Network-Based Structural Classification Tool for Natural Products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuhmann, T. A Systematic Cheminformatics Analysis of Functional Groups Occurring in Natural Products. J. Nat. Prod. 2019, 82, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mercado, R.; Engkvist, O.; Chen, H. Comparative Study of Deep Generative Models on Chemical Space Coverage. J. Chem. Inf. Model. 2021, 61, 2572–2581. [Google Scholar] [CrossRef] [PubMed]
- Pahl, A.; Waldmann, H.; Kumar, K. Exploring Natural Product Fragments for Drug and Probe Discovery. Chimia 2017, 71, 653–660. [Google Scholar] [CrossRef]
- Hanna, J.N.; Bekono, B.D.; Owono, L.C.; Toze, F.A.; Mbah, J.A.; Günther, S.; Ntie-Kang, F. A chemoinformatic analysis of atoms, scaffolds and functional groups in natural products. Phys. Sci. Rev. 2023, 8, 1341–1365. [Google Scholar] [CrossRef]
- Vu, H.; Pedro, L.; Mak, T.; McCormick, B.; Rowley, J.; Liu, M.; Di Capua, A.; Williams-Noonan, B.; Pham, N.B.; Pouwer, R.; et al. Fragment-Based Screening of a Natural Product Library against 62 Potential Malaria Drug Targets Employing Native Mass Spectrometry. ACS Infect. Dis. 2018, 4, 431–444. [Google Scholar] [CrossRef]
- Godinez, W.J.; Ma, E.J.; Chao, A.T.; Pei, L.; Skewes-Cox, P.; Canham, S.M.; Jenkins, J.L.; Young, J.M.; Martin, E.J.; Guiguemde, W.A. Design of potent antimalarials with generative chemistry. Nat. Mach. Intell. 2022, 4, 180–186. [Google Scholar] [CrossRef]
- Bosc, N.; Felix, E.; Arcila, R.; Mendez, D.; Saunders, M.R.; Green, D.V.S.; Ochoada, J.; Shelat, A.A.; Martin, E.J.; Iyer, P.; et al. MAIP: A web service for predicting blood-stage malaria inhibitors. J. Cheminform. 2021, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Probst, D.; Reymond, J.L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Woodhead, A.J.; Erlanson, D.A.; de Esch, I.J.; Holvey, R.S.; Jahnke, W.; Pathuri, P. Fragment-to-Lead Medicinal Chemistry Publications in 2022. J. Med. Chem. 2024, 67, 2287–2304. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2022. [Google Scholar]
- Gargaro, A.R.; Soteriou, A.; Frenkiel, T.A.; Bauer, C.J.; Birdsall, B.; Polshakov, V.I.; Barsukov, I.L.; Roberts, G.C.; Feeney, J. The solution structure of the complex of Lactobacillus casei dihydrofolate reductase with methotrexate. J. Mol. Biol. 1998, 277, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Feeney, J.; Birdsall, B.; Kovalevskaya, N.V.; Smurnyy, Y.D.; Navarro Peran, E.M.; Polshakov, V.I. NMR structures of apo L. casei dihydrofolate reductase and its complexes with trimethoprim and NADPH: Contributions to positive cooperative binding from ligand-induced refolding, conformational changes, and interligand hydrophobic interactions. Biochemistry 2011, 50, 3609–3620. [Google Scholar] [CrossRef] [PubMed]
- Grigalunas, M.; Burhop, A.; Christoforow, A.; Waldmann, H. Pseudo-natural products and natural product-inspired methods in chemical biology and drug discovery. Curr. Opin. Chem. Biol. 2020, 56, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, S.; Tian, Y.; Zhang, Y.; Zhao, Y. Recent ring distortion reactions for diversifying complex natural products. Nat. Prod. Rep. 2022, 39, 1970–1992. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminform. 2021, 13, 2. [Google Scholar] [CrossRef]
- Zeng, T.; Liu, Z.; Zhuang, J.; Jiang, Y.; He, W.; Diao, H.; Lv, N.; Jian, Y.; Liang, D.; Qiu, Y.; et al. TeroKit: A Database-Driven Web Server for Terpenome Research. J. Chem. Inf. Model. 2020, 60, 2082–2090. [Google Scholar] [CrossRef]
- Zeng, T.; Chen, Y.; Jian, Y.; Zhang, F.; Wu, R. Chemotaxonomic investigation of plant terpenoids with an established database (TeroMOL). New Phytol. 2022, 235, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Verras, A.; Waller, C.L.; Gedeck, P.; Green, D.V.; Kogej, T.; Raichurkar, A.; Panda, M.; Shelat, A.A.; Clark, J.; Guy, R.K.; et al. Shared Consensus Machine Learning Models for Predicting Blood Stage Malaria Inhibition. J. Chem. Inf. Model. 2017, 57, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Degen, J.; Wegscheid-Gerlach, C.; Zaliani, A.; Rarey, M. On the art of compiling and using drug-like chemical fragment spaces. ChemMedChem 2008, 3, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Kim, Y.; Deng, Y.; Senellart, J.; Rush, A.M. Opennmt: Open-source toolkit for neural machine translation. arXiv 2017, arXiv:1701.02810. [Google Scholar]
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Greg Landrum 2013, 8, 5281. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | NIMO-M | NIMO-S | MCMG | QBMG | FBMG | |
|---|---|---|---|---|---|---|
| Conditional metrics | Validity | 94.5% | 99.3% | 95.0% | 94.5% | 42.9% |
| Uniqueness | 99.7% | 99.1% | 98.4% | 99.9% | 98.5% | |
| Novelty | 61.0% | 77.8% | 65.7% | 42.2% | 99.9% | |
| MOSES metrics | FCD↓ a | 3.71 | 11.2 | 4.52 | 19.2 | 6.11 |
| SNN↓ | 0.87 | 0.65 | 0.71 | 0.95 | 0.51 | |
| Frag↓ | 0.85 | 0.77 | 0.95 | 0.99 | 0.48 | |
| Scaf↓ | 0.67 | 0.83 | 0.65 | 0.66 | 0.57 | |
| IntDiv | 88.3% | 86.5% | 87.8% | 86.6% | 73.9% | |
| Novelty | 71.4% | 89.0% | 79.5% | 52.4% | 99.9% | |
| SAS↓ | 0.78 | 0.91 | 1.22 | 0.87 | 0.94 |
| Metrics | NIMO-S | NIMO-S’ | MCMG | QBMG | |
|---|---|---|---|---|---|
| Terpenoids | Success | 91.9% | 95.4% | 71.2% | 89.7% |
| Ring systems (RSs) a | Coverage | 27.5% | 29.8% | 28.1% | 8.3% |
| Recovery | 99.4% | 69.5% | 62.4% | 10.6% | |
| Functional groups (FGs) b | Coverage | 5.9% | 6.2% | 4.3% | 4.9% |
| Recovery | 93.2% | 89.7% | 58.1% | 47.1% |
| Train | NIMO-M | MCMG | NIMO-M’ | |
|---|---|---|---|---|
| Samples | 744,986 | 5000 | 5000 | 1000 |
| EF [50%] a | 20.07 | 46.82 | 44.99 | 68.22 |
| EF [10%] | 44.36 | 72.09 | 69.11 | 81.33 |
| EF [1%] | 80.4 | 81.97 | 89.17 | 92.21 |
| Active % | 10.0% | 55.9% | 52.1% | 85.5% |
| PDB | 2HMG (CHEMBL2902) | 1BO7 (CHEMBL5328) | ||
|---|---|---|---|---|
| Compounds | 1000 | 5000 | 1000 | 5000 |
| Predicted candidates | 15 | 82 | 93 | 294 |
| Docking score < native | 10 | 26 | 10 | 23 |
| RMSD < 2 | 10 | 65 | 48 | 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, X.; Zeng, T.; Chen, N.; Li, J.; Wu, R. NIMO: A Natural Product-Inspired Molecular Generative Model Based on Conditional Transformer. Molecules 2024, 29, 1867. https://doi.org/10.3390/molecules29081867
Shen X, Zeng T, Chen N, Li J, Wu R. NIMO: A Natural Product-Inspired Molecular Generative Model Based on Conditional Transformer. Molecules. 2024; 29(8):1867. https://doi.org/10.3390/molecules29081867
Chicago/Turabian StyleShen, Xiaojuan, Tao Zeng, Nianhang Chen, Jiabo Li, and Ruibo Wu. 2024. "NIMO: A Natural Product-Inspired Molecular Generative Model Based on Conditional Transformer" Molecules 29, no. 8: 1867. https://doi.org/10.3390/molecules29081867
APA StyleShen, X., Zeng, T., Chen, N., Li, J., & Wu, R. (2024). NIMO: A Natural Product-Inspired Molecular Generative Model Based on Conditional Transformer. Molecules, 29(8), 1867. https://doi.org/10.3390/molecules29081867