The Creation and Physiological Relevance of Divergent Hydroxylation Patterns in the Flavonoid Pathway

Abstract
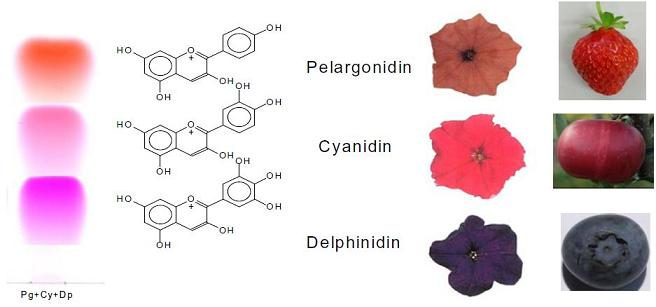
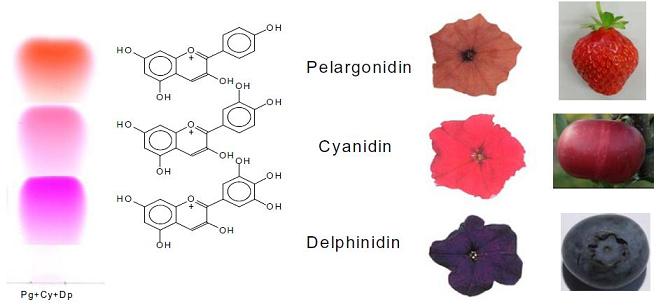
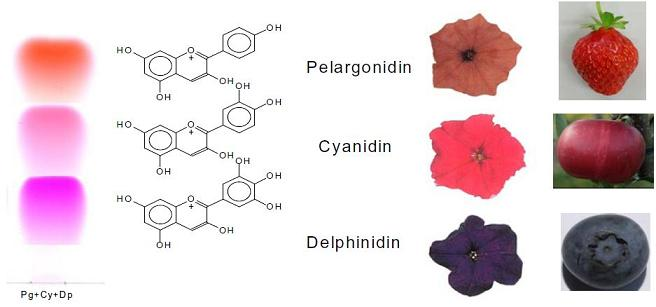
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
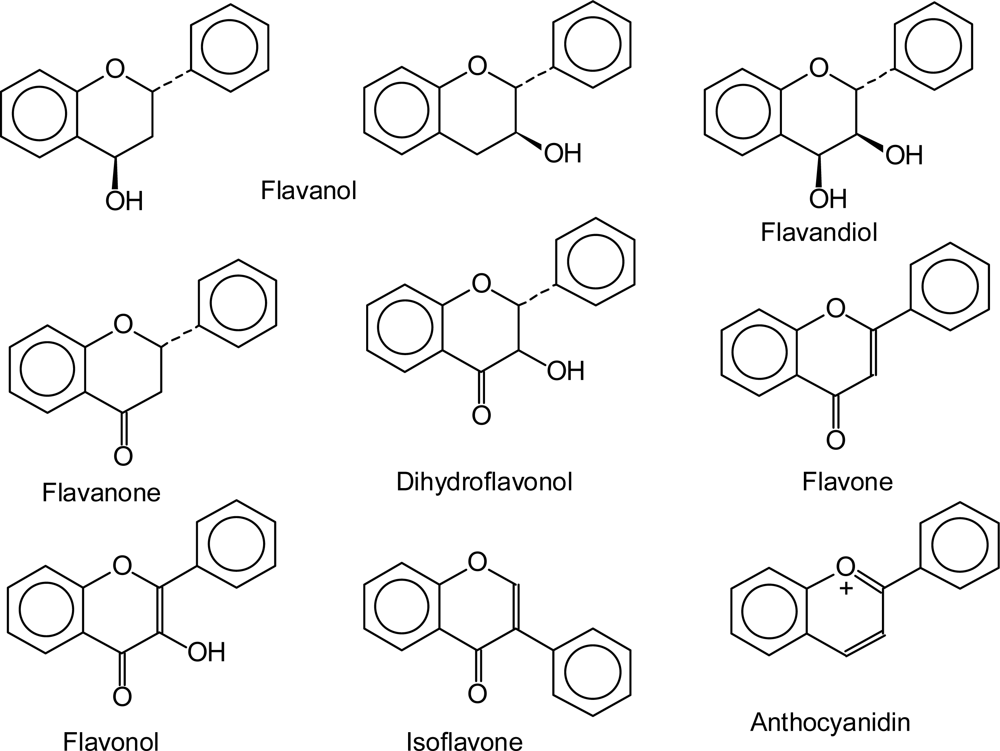
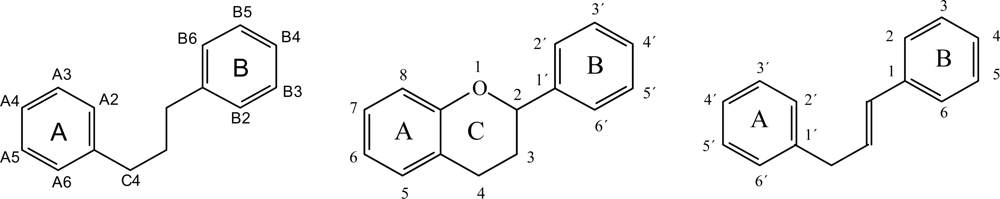
2. Flavonoids: Structures and Physiological Functions
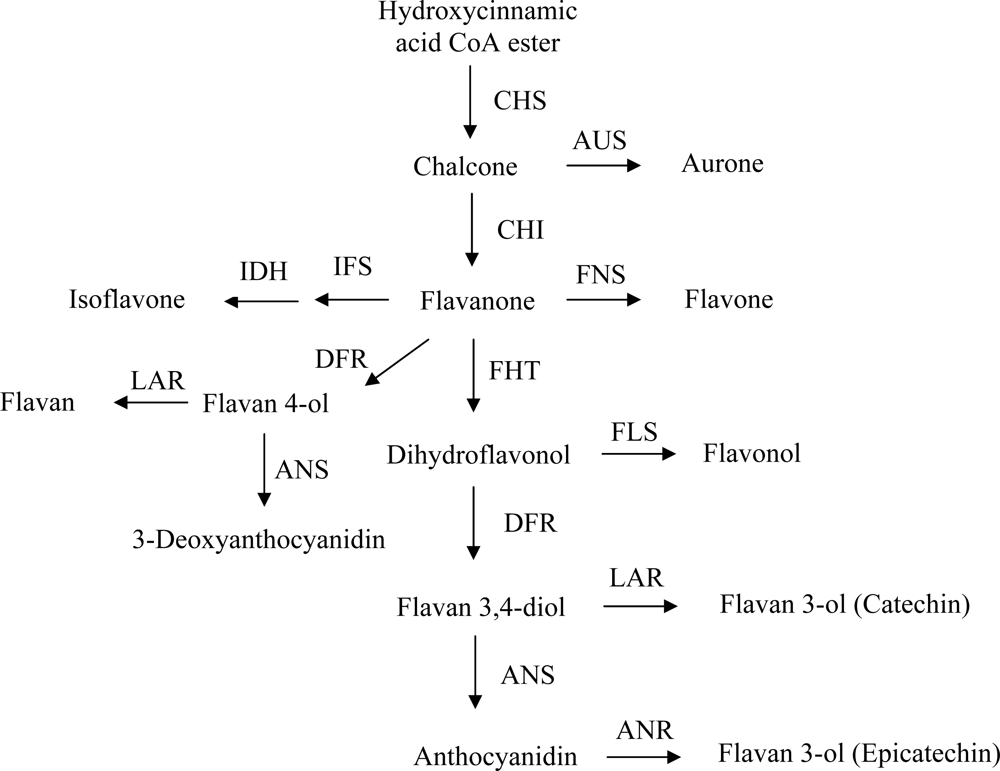
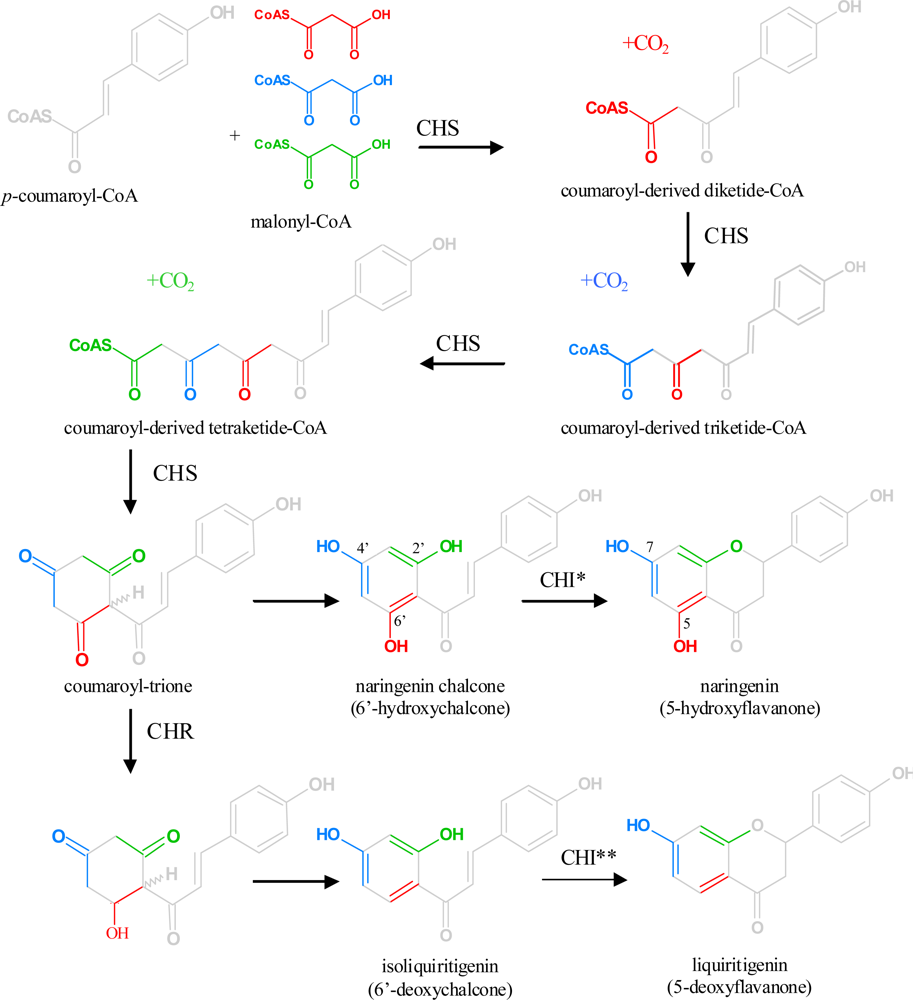
3. Overview of the Flavonoid Pathway
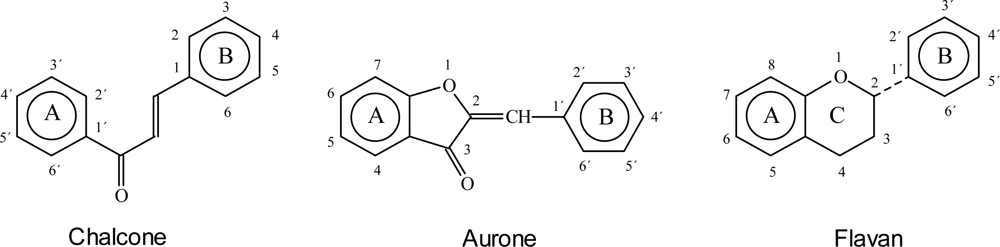
4. The Creation of the Hydroxylation Patterns of Flavonoids
- The kind and position of hydroxylation in the naturally occurring C6-C3 compounds is similar to that found in the C6(B)-C3-fragment of the naturally-occurring flavonoids, and is usually different from that of the C6(A)-C3-fragments;
- Hydroxylation (or a C-O-bond respectively) is always found at A-2, and in the great majority of cases, at A-2,4,6, respectively. With a few exceptions, hydroxylation always occurs at least at both A-2 and A-4;
- Hydroxylation is known to occur at all possible positions in A; flavonoids are known which are A-2,3,4,5,6-hydroxylated;
- The greatest number of compounds are hydroxylated at B-4 with B-3,4 and B-3,4,5 also common;
- Hydroxylation at B-2 is known, but there is no example of B-2,6 hydroxylation;
- The state of oxidation of the C-chain can vary; and
- The C-4 carbon atom is, in most cases, present as a carbonyl group.
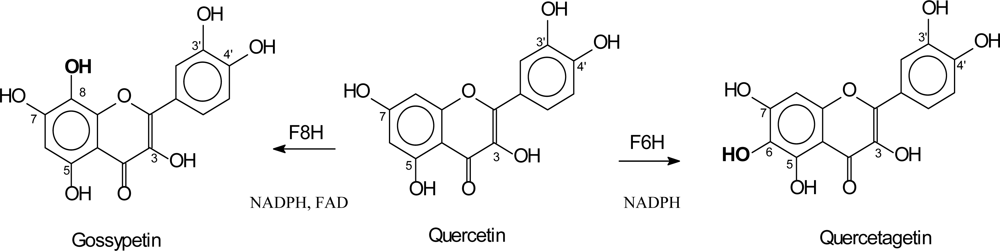
4.1. Introduction of Hydroxyl Groups in the Flavonoid A-ring
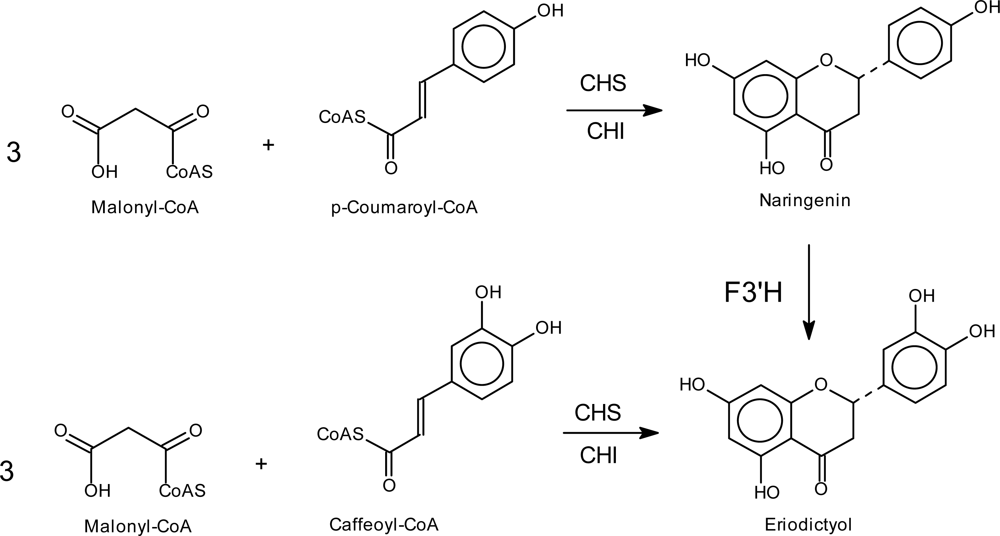
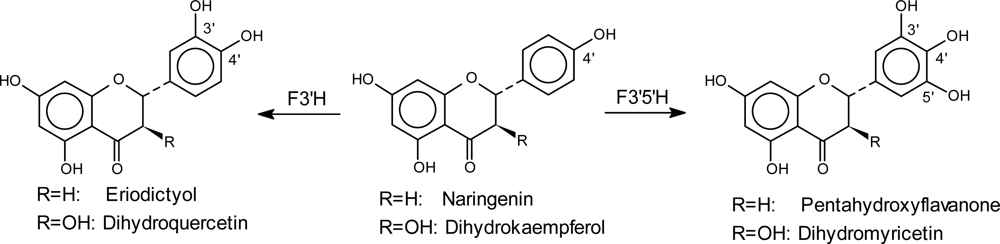
4.2. Introduction of Hydroxyl Groups in the Flavonoid B-ring
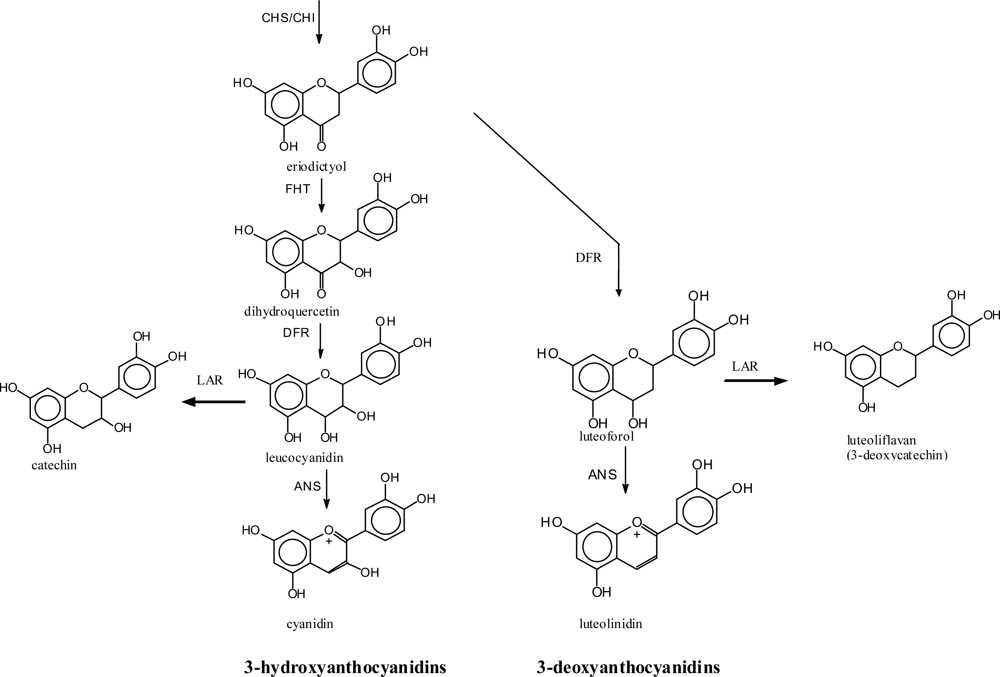
4.3. Introduction of Hydroxyl Groups into the C-ring of Flavonoids
5. Structure-Activity Relationships (SARs)
5.1. Relationship between the Structure and Light Absorbance of Flavonoids
5.2. Relationship between the Structure and Antioxidant Activity of Flavonoids
5.3. Relationship between the Structure and Antiinfective Effects
6. Outlook
Acknowledgments
References and Notes
- Stafford, HA. Flavonoid Metabolism; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Harborne, JB; Baxter, H. A Handbook of the Natural Flavonoids; Wiley: Chichester, UK, 1999. [Google Scholar]
- Forkmann, G; Heller, W. Biosynthesis of flavonoids. In Comprehensive Natural Products Chemistry; Barton, D, Nakanishi, K, Meth-Cohn, O, Eds.; Elsevier: Oxford, UK, 1999; Volume 1, pp. 713–748. [Google Scholar]
- Harborne, JB. Comparative Biochemistry of the Flavonoids; Academic Press: New York, NY, USA, 1967. [Google Scholar]
- Bohm, B. The minor flavonoids. Flavonoids 1994, 387–440. [Google Scholar]
- Bird, AE; Marshall, AC. Structure of chlorflavonin. J. Chem. Soc. Perkin Trans 1969, 18, 2418–2420. [Google Scholar]
- Bohm, B. Chalcones, aurones and dihydrochalcones. In The Flavonoids; Harborne, JB, Marbry, TJ, Marbry, H, Eds.; Chapman Hall: London, UK, 1975; Chapter 9, pp. 460–472. [Google Scholar]
- Gottlieb, OR. Flavonols. In The Flavonoids; Harborne, JB, Marbry, TJ, Marbry, H, Eds.; Chapman Hall: London, UK, 1975; pp. 296–375. [Google Scholar]
- Iwashina, T. The structure and distribution of the flavonoids in plants. J. Plant Res 2000, 113, 287–299. [Google Scholar]
- Markham, KR. Flavonoids: Chemistry, Biochemistry and Applications; Andersen, OM, Markham, KR, Eds.; CRC-Taylor & Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar]
- Winkel-Shirley, B. Flavonoid biosynthesis: “New” functions for an “old” way. Trends P. Sci 1996, 1, 377–382. [Google Scholar]
- Harborne, JB; Williams, CA. Advances in flavonoid research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar]
- Schultze, M; Kondorosi, E; Ratet, P; Buire, M; Kondorosi, A. Cell and molecular biology of rhizobium-plant interactions. Int. Rev. Cytol 1996, 156, 1–75. [Google Scholar]
- Harborne, JB. Introduction to Ecological Biochemistry, 3rd ed; Academic Press: London, UK, 1988. [Google Scholar]
- Haslam, E. Plant polyphenols (syn Vegetable Tannins) and chemical defense-a reappraisal. J. Chem. Ecol 1988, 14, 1789–1805. [Google Scholar]
- Brown, DE; Rashotte, AM; Murphy, AS; Normanly, J; Tague, BW; Peer, WA; Taiz, L; Muday, GK. Flavonoids act as negative regulators of auxin transport in vivo in Arabidopsis. Plant Physiol 2001, 126, 524–535. [Google Scholar]
- Besseau, S; Hoffmann, L; Geoffroy, P; Lapierre, C; Pollet, B; Legrand, M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell 2007, 19, 148–162. [Google Scholar]
- Mueller, LA; Walbot, V. Models for vacuolar sequestration of anthocyanins. Rec. Adv. Phytochem 2001, 35, 297–312. [Google Scholar]
- Kitamura, S. Transport of flavonoids: from cytosolic synthesis to vacuolar accumulation in grotewolde. In The Science of Flavonoids; Springer Verlag: New York, NY, USA, 2006; pp. 123–146. [Google Scholar]
- Buer, CS; Muday, GK. The transparent testa4 mutation prevents flavonoid synthesis and alters auxin transport and the response of Arabidopsis roots to gravity and light. Plant Cell 2004, 16, 1191–1205. [Google Scholar]
- Matern, U; Reichenbach, C; Heller, W. Efficient uptake of flavonoids into parsley (Petroselinum hortense) vacuoles requires acylated glycosides. Planta 1986, 167, 183–189. [Google Scholar]
- Hopp, W; Seitz, HU. The uptake of acylated anthocyanininto isolated vacuoles from a cell suspension culture of Daucus carota. Planta 1987, 170, 74–85. [Google Scholar]
- Polster, J; Dithmar, H; Burgemeister, R; Friedemann, G; Feucht, W. Flavonoids in plant nuclei: detection by laser microdissection and pressure catapulting (LMPC), in vivo staining, and uv-visible spectroscopic titration. Physiol. Planta 2006, 128, 163–174. [Google Scholar]
- Feucht, W; Dithmar, H; Polster, J. Variation of the nuclear, subnuclear and chromosomal flavanol deposition in hemlock and rye. Int. J. Mol. Sci 2007, 8, 635–650. [Google Scholar]
- Herrmann, K. Inhaltsstoffe von Obst und Gemüse. Verlag Eugen Ulmer, 2001. [Google Scholar]
- Amilot-Carlin, M-J; Tourniaire, F; Margotat, A. Flavonoids in food and wine. Acta Hort (ISHS) 2008, 744, 107–116. [Google Scholar]
- Viscupičová, J; Ondrejovič, M; Šturdik, E. Biovailability and metabolism of flavonoids. J. Food Nutr. Res 2008, 47, 151–162. [Google Scholar]
- Arroo, RRJ; Androutsopoulos, V; Beresford, K; Ruparelia, K; Surichan, S; Wilsher, N; Potter, GA. Phytoestrogens as natural prodrugs in cancer prevention: dietary flavonoids. Phytochem. Rev 2009, 8, 375–386. [Google Scholar]
- Benavente-Garcia, O; Castillo, J. Update on uses and properties of citrus flavonoids: new findings in anticancer, cardiovascular, and anti-inflammatory activity. J. Agr. Food Chem 2008, 56, 6185–6205. [Google Scholar]
- Grassi, D; Desideri, G; Croce, G; Tiberti, S; Aggio, A; Ferri, C. Flavonoids, vascular function and cardiovascular protection. Curr. Pharm. Design 2009, 15, 1072–1084. [Google Scholar]
- Orsolic, N; Basic, I. Honey bee products and their polyphenolic compounds in treatment of diabetes. Rec. Prog. Med. Plants 2008, 22, 443–540. [Google Scholar]
- Dajas, F; Arredondo, F; Echeverry, C; Ferreira, M; Morquio, A; Rivera, F. Flavonoids and the brain: Evidences and putative mechanisms for a protective capacity. Curr. Neuropharmacol 2005, 3, 193–205. [Google Scholar]
- Horvathova, K; Vachalkova, A; Novotny, L. Flavonoids as chemoprotective agents in civilization diseases. Neoplasma 2001, 48, 435–441. [Google Scholar]
- Liu, RH. Health benefits of fruit and vegetables are from additive and synergistic combinations of phytochemicals. Am. J. Clin. Nutr 2003, 78, 517–520. [Google Scholar]
- Cushnie, TPT; Lamb, AJ. Antimicrobial activity of flavonoids. Int. J. Antimicrob. Ag 2005, 26, 343–356. [Google Scholar]
- Paganga, G; Rice-Evans, CA. The identification of flavonoids as glycosides in human plasma. FEBS Lett 1997, 401, 78–82. [Google Scholar]
- Erlund, I; Kosonen, T; Alfthan, G; Maenpaa, J; Perttunen, K; Kenraali, J; Parantainen, J; Aro, A. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy voluteers. Eur. J. Clin. Pharmacol 2000, 56, 545–553. [Google Scholar]
- Graefe, EU; Wittig, J; Mueller, S; Riethling, AK; Uehleke, B; Drewelow, B; Pforte, H; Jacobasch, G; Derendorf, H; Veit, M. Pharmacokinetics and bioavailability of quercetin glycosides in hmans. J. Clin. Pharmacol 2001, 41, 492–499. [Google Scholar]
- Goldberg, DM; Yan, J; Soleas, GJ. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clin. Biochem 2003, 36, 79–87. [Google Scholar]
- Treutter, D. Managing phenol contents in crop plants by phytochemical farming and breeding. Int J Mol Sci 2009. [Google Scholar]
- Kreuzaler, F; Hahlbrock, K. Enzymatic synthesis of an aromatic compound in higher plants. Formation of naringenin (5,7,4′-trihydroxyflavanone) from p-coumaryl coenzyme A and malonyl coenzyme A. FEBS Lett 1972, 28, 69–72. [Google Scholar]
- Pfeiffer, J; Kuehnel, C; Brandt, J; Duy, D; Punyasiri, PAN; Forkmann, G; Fischer, TC. Biosynthesis of flavan 3-ols by leucoanthocyanidin 4-reductases and anthocyanidin reductases in leaves of grape (Vitis vinifera L.), apple (Malus x domestica Borkh.) and other crops. Plant Physiol. Biochem 2006, 44, 323–334. [Google Scholar]
- Dixon, RA; Xie, DY; Sharma, SB. Proanthocyanidins—a final frontier in flavonoid research? New Phytol 2005, 165, 9–28. [Google Scholar]
- Stafford, HA. Possible multienzyme complexes regulating the formation of C6-C3 phenolic compounds and lignins in higher plants. Rec. Adv. Phytochem 1974, 8, 53–79. [Google Scholar]
- Hrazdina, G. Compartmentation in aromatic metabolism. In Phenolic Metabolism in Plants; Stafford, HA, Ibrahim, RK, Eds.; Plenum Press: New York, NY, USA, 1992; pp. 1–23. [Google Scholar]
- Winkel-Shirley, B. Evidence for enzyme complexes in the phenylpropanoid and flavonoid pathways. Physiol. Plant 1999, 107, 142–149. [Google Scholar]
- Ono, E; Hatayama, M; Isono, Y; Sato, T; Watanabe, R; Yonekura-Sakakibara, K; Fukuchi-Mizutani, M; Tanaka, Y; Kusumi, T; Nishino, T; Nakayama, T. Localization of a flavonoid biosynthetic polyphenol oxidase in vacuoles. Plant J 2006, 45, 133–143. [Google Scholar]
- Saslowsky, DE; Warek, U; Winkel, BSJ. Nuclear localization of flavonoid enzymes in Arabidopsis. J. Biol. Chem 2005, 280, 23735–23740. [Google Scholar]
- Geissman, TA; Hinreiner, E. Theories of the biogenesis of flavonoid compounds. Bot. Rev 1952, 18, 77–164. [Google Scholar]
- Ullrich, R; Hofrichter, M. Enzymatic hydroxylation of aromatic componds. Cell. Mol. Life Sci 2007, 64, 271–293. [Google Scholar]
- Strack, D; Schliemann, W. Bifunctional polyphenol oxidases: novel functions in plant pigment biosynthesis. Angew. Chem. Int. Edit 2001, 40, 3791–3794. [Google Scholar]
- Werck-Reichhart, D; Bark, S; Paquette, S. Cytochromes. The Arabidopsis Book. Somerville, SR, Meyerowitz, EM, Eds.;
- Kreuzaler, F; Hahlbrock, K. Enzymic synthesis of an aromatic ring from acetate units. Partial purification and some properties of flavanone synthase from cell-suspension cultures of Petroselinum hortense. Eur. J. Biochem 1975, 56, 205–213. [Google Scholar]
- Schröder, G; Brown, JWS; Schröder, J. Molecular analysis of resveratrol synthase. cDNA, genomic clones and relationship with chalcone synthase. Eur. J. Biochem 1988, 172, 161–169. [Google Scholar]
- Forkmann, G; Dangelmayr, B. Genetic control of chalcone isomerase activity in flowers of Dianthus caryophyllus. Biochem. Genetics 1980, 18, 519–527. [Google Scholar]
- Bomati, EK; Austin, MB; Bowman, ME; Dixon, RA; Noel, JP. Structural elucidation of chalcone reductase and implications for deoxychalcone biosynthesis. J. Biol. Chem 2005, 280, 30496–30503. [Google Scholar]
- Fischer, TC; Halbwirth, H; Meisel, B; Stich, K; Forkmann, G. Molecular cloning, substrate specificity of the functionally expressed dihydroflavonol 4-reductases from Malus domestica and Pyrus communis cultivars and the consequences for flavonoid metabolism. Arch. Biochem. Biophys 2003, 412, 223–230. [Google Scholar]
- Halbwirth, H; Kahl, S; Jaeger, W; Reznicek, G; Forkmann, G; Stich, K. Synthesis of (14C)-labeled 5-deoxyflavonoids and their application in the study of dihydroflavonol/leucoanthocyanidin interconversion by dihydroflavonol 4-reductase. Plant Sci 2006a, 170, 587–595. [Google Scholar]
- Halbwirth, H; Fischer, TC; Schlangen, K; Rademacher, W; Schleifer, KJ; Forkmann, G; Stich, K. Screening for inhibitors of 2-oxoglutarate-dependent dioxygenases: Flavanone 3ß-hydroxylase and flavonol synthase. Plant Sci 2006b, 171, 194–205. [Google Scholar]
- Anzellotti, D; Ibrahim, RK. Novel flavonol 2-oxoglutarate dependent dioxygenase: affinity purification, characterization, and kinetic properties. Arch. Biochem. Biophys 2000, 382, 161–172. [Google Scholar]
- Anzellotti, D; Ibrahim, RK. Molecular characterization and functional expression of flavonol 6-hydroxylase. BMC Plant Biol 2004, 4. [Google Scholar]
- Halbwirth, H; Forkmann, G; Stich, K. The A-ring specific hydroxylation of flavonols in position 6 in Tagetes sp. is catalyzed by a cytochrome P450 dependent monooxygenase. Plant Sci 2004, 167, 129–131. [Google Scholar]
- Schlangen, K; Miosic, S; Castro, A; Freudmann, K; Luczkiewicz, M; Vitzthum, F; Schwab, W; Gamsjäger, S; Musso, M; Halbwirth, H. Formation of UV-honey guides in Rudbeckia hirta. Phytochemistry 2009a, 70, 889–898. [Google Scholar]
- Latunde-Dada, AO; Cabello-Hurtado, F; Czittrich, N; Didierjean, L; Schopfer, C; Hertkorn, N; Werck-Reichhart, D; Ebel, J. Flavonoid 6-hydroxylase from soybean (Glycine max L.), a novel plant P-450 monooxygenase. J. Biol. Chem 2001, 276, 1688–1695. [Google Scholar]
- Halbwirth, H; Stich, K. An NADPH and FAD dependent enzyme catalyzes hydroxylation of flavonoids in position 8. Phytochemistry 2006c, 67, 1080–1087. [Google Scholar]
- Hrazdina, G; Wagner, GJ. Metabolic pathways as enzyme complexes: evidence for the synthesis of phenylpropanoids and flavonoids on membrane associated enzyme complexes. Arch. Biochem. Biophys 1985, 237, 88–100. [Google Scholar]
- Werck-Reichhart, D. Cytochromes P450 in phenylpropanoid metabolism. Drug Metabol Drug Interact 1995, 221–243. [Google Scholar]
- Christensen, AB; Gregersen, PL; Schröder, J; Collinge, DB. A chalcone synthase with an unusual substrate preference is expressed in barley leaves in response to UV light and pathogen attack. Plant Mol. Biol 1998, 37, 849–857. [Google Scholar]
- Stotz, G; Spribille, R; Forkmann, G. Flavonoid biosynthesis in flowers of Verbena hybrida. J. Plant Physiol 1984, 116, 173–183. [Google Scholar]
- Fritsch, H; Grisebach, H. Biosynthesis of cyanidin an cell cultures of Haplopappus gracilis. Phytochemistry 1975, 14, 2437–2442. [Google Scholar]
- Hagmann, ML; Heller, W; Grisebach, H. Induction and characterization of a microsomal flavonoid 3′-hydroxylase from parsley cell cultures. Eur. J. Biochem 1983, 134, 547–554. [Google Scholar]
- Brugliera, F; Barri-Rewell, G; Holton, TA; Mason, JG. Isolation and characterization of a flavonoid 3′- hydroxylase cDNA clone corresponding to the Ht1 locus of petunia hybrida. Plant J 1999, 19, 441–451. [Google Scholar]
- Wimmer, G; Halbwirth, H; Wurst, F; Forkmann, G; Stich, K. Enzymatic hydroxylation of 6′-deoxychalcones with protein preparations from petals of Dahlia variabilis. Phytochemistry 1998, 47, 1013–1016. [Google Scholar]
- Schlangen, K; Miosic, S; Topuz, F; Muster, G; Marosits, T; Seitz, C; Halbwirth, H. Chalcone 3-hydroxylation is not a general property of flavonoid 3′-hydroxylase. Plant Sci 2009b, 177, 97–102. [Google Scholar]
- Schlangen, K; Miosic, S; Halbwirth, H. Allelic variants from Dahlia variabilis encode flavonoid 3′-hydroxylases with functional differences in chalcone 3-hydroxylase activity. Arch Biochem Biophys 2009. [Google Scholar]
- Halbwirth, H; Schlangen, K; Stich, K. Chalcon 3 hydroxylase. Österreichische Patentanmeldung 2009. [Google Scholar]
- Menting, JGT; Scopes, RK; Stevenson, TW. Characterization of a flavonoid 3′,5′-hydroxylase in microsomal membrane fraction of Petunia hybrida flowers. Plant Physiol 1994, 106, 633–642. [Google Scholar]
- de Vetten, N; Ter, HJ; Van Schaik, H-P; de Boer, A; Mol, J; Koes, R. A cytochrome b5 is required for full activity of flavonoid 3′,5′-hydroxylase, a cytochrome P450 involved in the formation of blue flower colors. Proc. Natl. Acad. Sci. USA 1999, 96, 778–783. [Google Scholar]
- Stotz, G; Forkmann, G. Hydroxylation of the B-ring of flavonoids in the 3′- and 5′-position with enzyme extracts from flowers of Verbena hybrida. Z. Naturforsch. C 1982, 37C, 19–23. [Google Scholar]
- Seitz, C; Eder, C; Deiml, B; Kellner, S; Martens, S; Forkmann, G. Cloning, functional identification and sequence analysis of flavonoid 3′-hydroxylase and flavonoid 3′,5′-hydroxylase cDNAs reveals independent evolution of flavonoid 3′,5′-hydroxylase in the Asteraceae family. Plant Mol. Biol 2006, 61, 365–381. [Google Scholar]
- Harborne, JB. The rare flavone isoetin as a yellow flower pigment in Heywoodiella oligocephala and in other cichoriae. Phytochemistry 1978, 17, 915–917. [Google Scholar]
- Harborne, JB. Revised structures for three isoetin glycosides, yellow flower pigments in Heywoodiella oligocephala. Phytochemistry 1991, 30, 1677–1678. [Google Scholar]
- Britsch, L; Grisebach, H. Purification and characterization of (2S)-flavanone 3-hydroxylase from Petunia hybrida. Eur. J. Biochem 1986, 156, 569–577. [Google Scholar]
- Britsch, L. Purification of flavanone 3-hydroxylase from Petunia hybrida: antibody preparation and characterization of a chemogenetically defined mutant. Arch. Biochem. Biophys 1990, 276, 348–354. [Google Scholar]
- Lukacin, R; Urbanke, C; Groning, I; Matern, U. The monomeric polypeptide comprises the functional flavanone 3beta-hydroxylase from Petunia hybrida. FEBS Lett 2000, 467, 353–358. [Google Scholar]
- Halbwirth, H; Martens, S; Wienand, U; Forkmann, G; Stich, K. Biochemical formation of anthocyanins in silk tissue of Zea mays. Plant Science 2003, 164, 489–495. [Google Scholar]
- Dedio, J; Saedler, H; Forkmann, G. Molecular cloning of the flavanone 3b-hydroxylase gene (FHT) from carnation (Dianthus caryophyllus) and analysis of stable and unstable FHT mutants. Theor. Appl. Genet 1995, 90, 611–617. [Google Scholar]
- Tyrach, A; Horn, W. Inheritance of flower color and flavonoid pigments in Gerbera. Plant Breeding 1997, 116, 377–381. [Google Scholar]
- Winefield, C; Lewis, DH; Swinny, EE; Zhang, H; Arathoona, HS; Fischer, TC; Halbwirth, H; Stich, K; Gosch, C; Forkmann, G; Davies, KM. Investigation of the biosynthesis of 3-deoxyanthocyanins in Sinningia cardinalis. Physiol. Plantarum 2005, 124, 419–430. [Google Scholar]
- Halbwirth, H; Muster, G; Stich, K. Unraveling the Biochemical Base of Dahlia Flower Coloration. Nat. Prod. Comm 2008, 3, 1259–1266. [Google Scholar]
- Lo, SC; Nicholson, RL. Reduction of light-induced anthocyanin accumulation in inoculated sorghum mesocotyls. Implications for a compensatory role in the defense response. Plant Physiol 1998, 116, 979–989. [Google Scholar]
- Boddu, J; Svabek, C; Sekhon, R; Gevens, A; Nicholson, RL; Jones, AD; Pedersen, JF; Gustine, DL; Chopra, S. Expression of a putative flavonoid 3′-hydroxylase in sorghum mesocotyles synthesizing 3-deoxyanthocyanidin phytoalexins. Physiol. Mol. Plant Pathol 2004, 65, 101–113. [Google Scholar]
- Forkmann, G. Control of pigmentation in natural and transgenic plants. Curr. Opin. Biotech 1993, 4, 159–165. [Google Scholar]
- Zuker, A; Shklarman, E; Scovel, G; Ben-Meir, H; Ovadis, M; Neta-Sharir, I; Ben-Yephet, Y; Weiss, D; Watad, A; Vainstein, A. Genetic engineering of agronomic and ornamental traits in carnation. Acta Hort 2001, 560, 91–94. [Google Scholar]
- Schlangen, K; Halbwirth, H; Fischer, TC; Flachowsky, H; Treutter, D; Hanke, MV; Stich, K. Breeding for fire blight resistance in apple trees. J. Biotechnol 2007, 131, 34–35. [Google Scholar]
- Fischer, D; Stich, K; Britsch, L; Grisebach, H. Purification and characterization of (+)dihydroflavonol (3-hydroxyflavanone) 4-reductase from flowers of Dahlia variabilis. Arch. Biochem. Biophys 1988, 264, 40–47. [Google Scholar]
- Martens, S; Teeri, T; Forkmann, G. Heterologous expression of dihydroflavonol 4-reductases from various plants. FEBS Lett 2002, 531, 453–458. [Google Scholar]
- Petit, P; Granier, T; d’Estaintot, BL; Manigand, C; Bathany, K; Schmitter, J-M; Lauvergeat, V; Hamdi, S; Gallois, B. Crystal structure of grape dihydroflavonol 4-reductase, a key enzyme in flavonoid biosynthesis. J. Mol. Biol 2007, 368, 1345–1357. [Google Scholar]
- Stich, K; Forkmann, G. Biosynthesis of 3-deoxyanthocyanins with flower extracts from Sinningia cardinalis. Phytochemistry 1988, 27, 785–789. [Google Scholar]
- Gosch, C; Puhl, I; Halbwirth, H; Schlangen, K; Römmelt, S; Andreotti, C; Costa, G; Fischer, TC; Treutter, D; Stich, K; Forkmann, G. Effect of prohexadione-Ca on various fruit crops: flavonoid composition and substrate specificity of their dihydroflavonol 4-reductases. Eur. J. Hort. Sci 2003, 68, 144–151. [Google Scholar]
- Schlangen, K; Gosch, C; Römmelt, S; Knott, J; Fischer, TC; Treutter, D; Forkmann, G; Stich, K; Halbwirth, H. Can Prohexadione-Ca induce antimicrobial flavonoids in rose? Eur. J. Hort. Sci 2003, 68, 137–143. [Google Scholar]
- Schlangen, K; Halbwirth, H; Peterek, S; Gosch, C; Ringl, A; Fischer, TC; Treutter, D; Forkmann, G; Kopp, B; Stich, K. Transient Induction of Antimicrobial 3-Deoxyflavonoids does not affect Pharmacological Compounds in Hawthorn. Nat. Prod. Comm 2008, 3, 1245–1250. [Google Scholar]
- Firuzi, O; Lacanna, A; Petrucci, R; Marrosu, G; Saso, L. Evaluation of the antioxidant activity of flavonoids by “ferric reducing antioxidant power” assay and cyclic voltammetry. BBA-Gen. Subj 2005, 1721, 174–184. [Google Scholar]
- Melidou, M; Riganakos, K; Galaris, D. Protection against nuclear DNA damage offered by flavonoids in cells exposed to hydrogen peroxide: The role of iron chelation. Free Radical Bio. Med 2005, 39, 1591–1600. [Google Scholar]
- Plochmann, K; Korte, G; Koutsilieri, E; Richling, E; Riederer, P; Rethwilm, A; Schreier, P; Scheller, C. Structure-activity relationships of flavonoid-induced cytotoxicity on human leukemia cells. Arch. Biochem. Biophys 2007, 460, 1–9. [Google Scholar]
- Snijman, PW; Swanevelder, S; Joubert, E; Green, IR; Gelderblom, WCA. The antimutagenic activity of the major flavonoids of rooibos (Aspalathus linearis): Some dose-response effects on mutagen activation-flavonoid interactions. Mutat. Res.- Gen. Tox. En 2007, 631, 111–123. [Google Scholar]
- Ichimatsu, D; Nomura, M; Nakamura, S; Moritani, S; Yokogawa, K; Kobayashi, S; Nishioka, T; Miyamoto, K. Structure-activity relationship of flavonoids for inhibition of epidermal growth factor-induced transformation of JB6 Cl 41 cells. Mol. Carcinogen 2007, 46, 436–445. [Google Scholar]
- Xu, YC; Leung, SWS; Yeung, DKY; Hu, LH; Chen, GH; Che, CM; Man, RYK. Structure-activity relationships of flavonoids for vascular relaxation in porcine coronary artery. Phytochemistry 2007, 68, 1179–1188. [Google Scholar]
- Kato, A; Nasu, N; Takebayashi, K; Adachi, I; Minami, Y; Sanae, F; Asano, N; Watson, AA; Nash, RJ. Structure-activity relationships of flavonoids as potential inhibitors of glycogen phosphorylase. J. Agr. Food Chem 2008, 56, 4469–4473. [Google Scholar]
- Liu, A-L; Wang, H-D; Lee, SMY; Wang, Y-T; Du, G-H. Structure -activity relationship of flavonoids as influenza virus neuraminidase inhibitors and their in vitro anti-viral activities. Bioorgan. Med. Chem 2008, 16, 7141–7147. [Google Scholar]
- Cholbi, MR; Paya, M; Alcaraz, MJ. Inhibitory effect of phenolic compounds on CCI4-induced microsomal lipid peroxidation. Experientia 1991, 47, 195–199. [Google Scholar]
- Sadeghipour, M; Terreux, R; Phipps, J. Flavonoids and tyrosine nitration: structure-activity relationship correlation with enthalpy of formation. Toxicol. Vitro 2005, 19, 155–165. [Google Scholar]
- Woodman, OL; Meeker, WF; Boujaoude, M. Vasorelaxant and Antioxidant Activity of Flavonols and Flavones: Structure-Activity Relationships. J. Cardiovasc. Phar 2005, 46, 302–309. [Google Scholar]
- Brunet, S; Hoste, H. Monomers of condensed tannins affect the larval exsheathment of parasitic nematodes of ruminants. J. Agr. Food Chem 2006, 54, 7481–7487. [Google Scholar]
- Kinjo, J; Hitoshi, M; Tsuchihashi, R; Korematsu, Y; Miyakoshi, M; Murakami, T; Niiho, D; Mizutani, K; Tanaka, T; Nonaka, G; Nohara, T; Okawa, M; Okabe, H. Hepatoprotective constituents in plants 15: protective effects of natural-occurring flavonoids and miscellaneous phenolic compounds as determined in an HepG2 cell cytotoxicity assay. J. Nat. Med 2006, 60, 36–41. [Google Scholar]
- Brunet, S; Jackson, F; Hoste, H. Effects of sainfoin (Onobrychis viciifolia) extract and monomers of condensed tannins on the association of abomasal nematode larvae with fundic explants. Int. J. Parasitol 2008, 38, 783–790. [Google Scholar]
- Marbry, TJ; Markham, KR; Thomas, MB. The Systematic Identification of Flavonoids; Springer Verlag: Berlin-Heidelberg-New York, NY, USA, 1970. [Google Scholar]
- Eisner, T; Silberglied, RE; Aneshansley, D; Carrel, JF; Howlans, HC. Ultraviolet video-viewing: the television camera as an insect eye. Science 1969, 166, 1172–1174. [Google Scholar]
- Thompson, WR; Meinwald, J; Aneshansley, D; Eisner, T. Flavonols: Responsible for ultraviolet absorption in nectar guide of flower. Science 1972, 177, 528–530. [Google Scholar]
- Silberglied, RE. Communication in the ultraviolet. Annu. Rev. Ecol. Syst 1979, 10, 373–398. [Google Scholar]
- McCrea, KD; Levy, M. Photographic visualization of floral colors as perceived by honeybee pollinators. Am. J. Bot 1983, 70, 369–375. [Google Scholar]
- Dyer, AG. The reflection of near ultraviolet radiation from flowers of Australian Native Plants. Aust. J. Bot 1996, 44, 473–488. [Google Scholar]
- Indsto, JO; Weston, PH; Clements, MA; Dyer, AG; Batley, M; Whelan, RJ. Pollination of Diuris maculate (Orchidaceae) by male Trichocolletes venustus bees. Aust. J. Bot 2006, 54, 669–679. [Google Scholar]
- Heim, KE; Tagliaferro, AR; Bobilya, DJ. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem 2002, 13, 572–584. [Google Scholar]
- Bors, W; Michel, C; Stettmaier, K. Structure-activity relationships governing antioxidant capacities of plant polyphenols. Methods Enzymol 2001, 335, 166–180. [Google Scholar]
- van Acker, SABE; van den Berg, DJ; Tromp, MNJL; Griffioen, DH; van Bennekom, WP; van der Vijgh, WJF; Bast, A. Structural aspects of antioxidant activity of flavonoids. Free Radic. Biol. Med 1996a, 20, 331–342. [Google Scholar]
- Bors, W; Heller, W; Michael, C; Saran, M. Flavonoids as antioxidants: determination of radical-scavenging efficiencies. Methods Enzymol 1990, 186, 343–355. [Google Scholar]
- Hider, RC; Liu, ZD; Khodr, HH. Metal chelation of polyphenols. Methods Enzymol 2001, 335, 190–203. [Google Scholar]
- Rice-Evans, CA; Miller, NJ; Paganda, G. Antioxidant properties of phenolic compounds. Trends Plant Sci 1997, 2, 152–159. [Google Scholar]
- Kim, D; Park, J; Kim, J; Han, C; Yoon, J; Kim, N; Seo, J; Lee, C. Flavonoids ac mushroom tyrosinase inhibitors: a fluorescence quenching study. J. Agric. Food Chem 2006, 54, 935–941. [Google Scholar]
- Pannala, AS; Chan, TS; O’Brien, PJ; Rice-Evans, CA. Flavonoid B-Ring Chemistry and Antioxodant Activity: Fast Reaction Kinetics. Biochem. Bioph. Res. Co 2001, 282, 1161–1168. [Google Scholar]
- Rahimuddin, SA; Khoja, SM; Zuhair, MM; Howell, NK; Brown, JE. Inhibition of lipid peroxidation in UVA-treated skin fibroblasts by luteolin and its glucosides. Eur. J. Lipid Sci. Tech 2007, 109, 647–655. [Google Scholar]
- Sim, G-S; Lee, B-C; Cho, HS; Lee, JW; Kim, J-H; Lee, D-H; Kim, J-H; Pyo, H-B; Moon, DC; Oh, K-W; Yun, YP; Hong, JT. Structure activity relationship of antioxidative property of flavonoids and inhibitory effect on matrix metalloproteinase activity in UVA-irradiated human dermal fibroblast. Arch. Pharm. Res 2007, 30, 290–298. [Google Scholar]
- van Acker, SABE; de Groot, MJ; van den Berg, DJ; Tromp, MNJL; den Kelder, GD-O; van der Vijgh, WJF; Bast, A. A quantum chemical Explanation of the antioxidant activity of flavonoids. Chem. Res. Toxicol 1996b, 9, 1305–1312. [Google Scholar]
- Cao, G; Sofic, E; Prior, RL. Antioxidant and prooxidant behaviour of flavonoids: structure-activity relationships. Free Rad. Biol. Med 1997, 22, 749–760. [Google Scholar]
- Ren, J; Meng, S; Lekka, CE; Kaxiras, E. Complexation of flavonoids with iron: structure and optical signatures. J. Phys. Chem. B 2008, 112, 1845–1850. [Google Scholar]
- Kostyuk, VA; Potapovich, AI; Strigunova, EN; Kostyuk, TV; Afanas’ev, IB. Experimental evidence that flavonoid metal complexes may act as mimics of superoxide dismutase. Arch. Biochem. Biophys 2004, 428, 204–208. [Google Scholar]
- Malesev, D; Kuntic, V. Investigation of metal-flavonoid chelates and the determination of flavonoids via metal-flavonoid complexing reactions. J. Serb. Chem. Soc 2007, 72, 921–939. [Google Scholar]
- Tsuchiya, H; Sato, M; Miyazaki, T; Fujiwara, S; Tanigaki, S; Ohyama, M; Tanaka, T; Iinuma, M. Comparative study on the antibacterial activity of phytochemical flavanones against methicillin-resistant Staphylococcus aureus. J. Ethnopharmacol 1996, 50, 27–34. [Google Scholar]
- Laks, PE; Pruner, MS. Flavonoid biocides: Structure/activity relations of flavonoid phytoalexin analogs. Phytochemistry 1988, 28, 87–91. [Google Scholar]
- Elstner, EF; Oßwald, W; Schneider, I. Phytopathologie Allgemeine und biochemische Grundlagen; Spektrum Akademischer Verlag: Heidelberg-Berlin, Oxford, UK, 1996. [Google Scholar]
- Snyder, BA; Nicholson, RL. Synthesis of phytoalexins in sorghum as a site-specific response to fungal ingress. Science 1990, 248, 1637–1639. [Google Scholar]
- Lo, SK; De Verdier, K; Nicholson, RL. Accumulation of 3-deoxyanthocyanidin phytoalexins and resistance to Colletotrichum sublineolum in Sorghum. Physiol. Mol. Plant Path 1999, 55, 263–273. [Google Scholar]
- Lo, C; Coolbaugh, RC; Nicholson, RL. Molecular characterization and in silico expression analysis of a chalcone synthase gene family in Sorghum bicolor. Physiol. Mol. Plant Path 2002, 61, 179–188. [Google Scholar] [Green Version]
- Nielsen, KA; Gotfredsen, CH; Buch-Pedersen, MJ; Ammitzboll, H; Mattsson, O; Duus, JO; Nicholson, RL. Inclusions of flavonoid 3 -deoxyanthocyanidins in Sorghum bicolor self-organize into spherical structures. Physiol. Mol. Plant Path 2004, 65, 187–196. [Google Scholar]
- Weiergang, I; Hipskind, JD; Nicholson, RL. Synthesis of 3-deoxyanthocyanidin phytoalexins in sorghum occurs independent of light. Physiol. Mol. Plant Path 1996, 49, 377–388. [Google Scholar]
- Wharton, PS; Nicholson, RL. Temporal synthesis and radiolabelling of the sorghum 3-deoxyanthocyanidin phytoalexins and the anthocyanin, cyanidin 3-dimalonyl glucoside. New Phytol 2000, 145, 457–469. [Google Scholar]
- Chopra, S; Gevens, A; Svabek, C; Wood, KV; Peterson, T; Nicholson, RL. Excision of the Candystripe1 transposon from a hyper-mutable Y1-cs allele shows that the sorghum Y1 gene controls the biosynthesis of both 3-deoxyanthocyanidin phytoalexins and phlobaphene pigments. Physiol. Mol. Plant Pathol 2002, 60, 321–330. [Google Scholar]
- Shih, C-H; Chu, IK; Yip, WK; Lo, C. Differential expression of two flavonoid 3′-hydroxylase cDNAs involved in biosynthesis of anthocyanin pigments and 3-deoxyanthocyanidin phytoalexins in sorghum. Plant Cell Physiol 2006, 47, 1412–1419. [Google Scholar]
- Ramesh, N; Viswanathan, MB; Saraswathy, A; Brindha, P; Balakrishna, K; Lakshmanaperumalsamy, P. Antibacterial activity of luteoforol from Bridelia crenulata. Fitoterapia 2001, 72, 409–411. [Google Scholar]
- Spinelli, F; Speakman, J-B; Rademacher, W; Halbwirth, H; Stich, K; Costa, G. Luteoforol, a flavan 4-ol, is induced in pome fruits by prohexadione-calcium and shows phytoalexin-like properties against Erwinia amylovora and other plant pathogens. Eur. J. Plant Path 2005, 112, 133–142. [Google Scholar]
- Bell, AC. Host Plant Resistance To Fire Blight (Erwinia amylovora) in the Rosaceae Subfamily Maloideae. North Carolina State University: Raleigh, NC, USA, 2004.
- Roemmelt, S; Peterek, S; Treutter, D; Rademacher, W; Speakman, JB; Bazzi, C; Tortoreto, L; Sponza, G; Costa, G; Andreotti, C; Halbwirth, H; Zimmermann, N; Stich, K; Forkmann, G. Alteration of phenylpropanoid biosynthesis of fruit trees as a tool for enhancement of fire blight resistance. Acta Hort 2002, 590, 477–484. [Google Scholar]















© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Halbwirth, H. The Creation and Physiological Relevance of Divergent Hydroxylation Patterns in the Flavonoid Pathway. Int. J. Mol. Sci. 2010, 11, 595-621. https://doi.org/10.3390/ijms11020595
Halbwirth H. The Creation and Physiological Relevance of Divergent Hydroxylation Patterns in the Flavonoid Pathway. International Journal of Molecular Sciences. 2010; 11(2):595-621. https://doi.org/10.3390/ijms11020595
Chicago/Turabian StyleHalbwirth, Heidi. 2010. "The Creation and Physiological Relevance of Divergent Hydroxylation Patterns in the Flavonoid Pathway" International Journal of Molecular Sciences 11, no. 2: 595-621. https://doi.org/10.3390/ijms11020595
APA StyleHalbwirth, H. (2010). The Creation and Physiological Relevance of Divergent Hydroxylation Patterns in the Flavonoid Pathway. International Journal of Molecular Sciences, 11(2), 595-621. https://doi.org/10.3390/ijms11020595