3.1. Structural Features
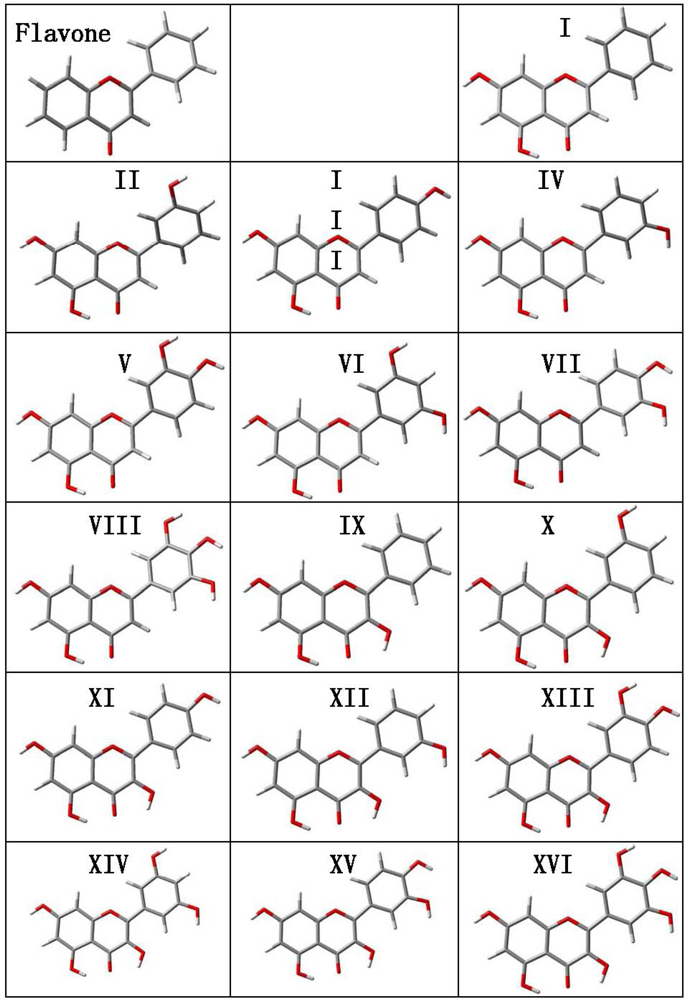
Structural optimizations were carried out for flavone and the 16 derived flavonoids with the calculated structures reported in
Figure 2 and the main molecular parameters in
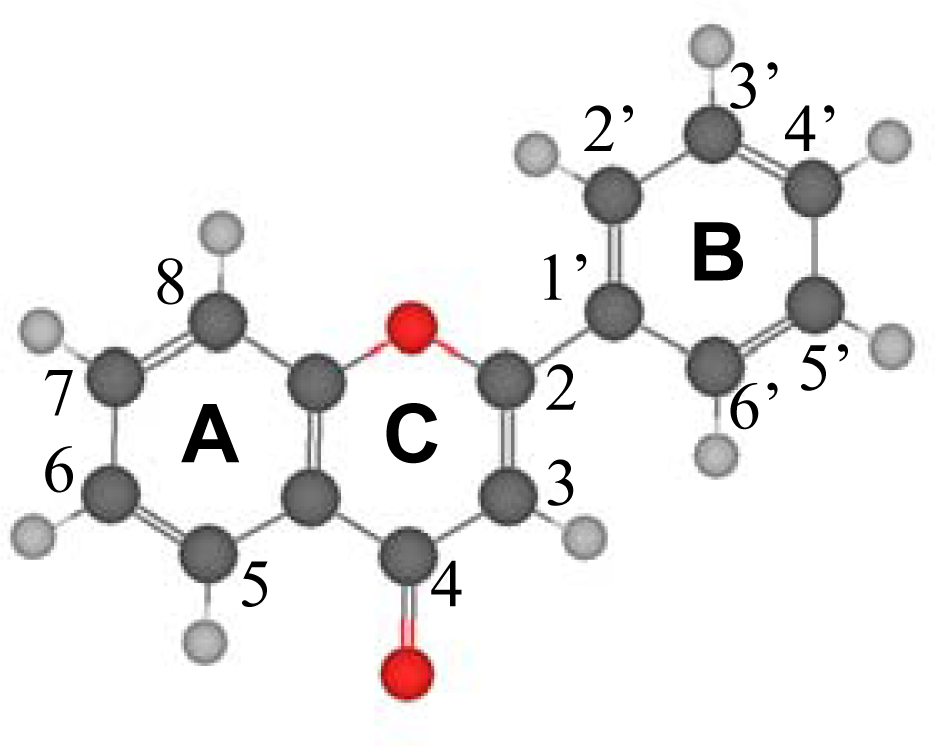
Table 2. Several questions rise from the reported gas phase results. The first one is the planarity of the studied molecules. Reported results show that the dihedral angle between the catechol ring (B ring in
Figure 1) and the plane formed by A and C rings is around 20° for flavone and those flavonoids without an hydroxyl group in position 3 of C ring (molecules I to VIII), whereas for those molecules with an hydroxyl group in potion 3, molecules IX to XVI, the dihedral angle is 0° and the three rings, A, B and C, are coplanar. This may be justified considering that the presence of an hydroxyl group in position 3 leads to the development of an intramolecular hydrogen bonding with the hydrogen in position 6′ of B ring. The development of an effective interaction between these positions forces the coplanarity of both rings, and thus, all flavonoids with an hydroxyl group in position 3 on ring C should be planar. Moreover, the π-delocalization from the C ring toward the catechol B-ring will be more effective with this planar structure forced by the 3-OH group. These results are in agreement with the previous findings of van Acker
et al. [
19,
36] using the HF/STO-3G approach, whereas other literature results obtained at low theoretical levels (HF/6-31G(d) [
37,
38] or AM1 [
39,
40]) report non-planarity for flavonoids with 3-OH groups such as quercetin. These low level theoretical calculations underestimate the effect rising from the rings coplanarity, such as the π-delocalization [
37], and thus planar structures obtained in this work should be more reliable. Available X-ray crystal structures lead to molecules almost planar, as is the case for quercetin, for which a 5° experimental angle between cathecol and AC rings is reported [
41]. Although packing effects rise in solid phases that are not present in gas phase calculations such as those reported in this work, these crystal results show that even in solid phases, close to planar structures are obtained, thus reinforcing our gas phase conclusions.
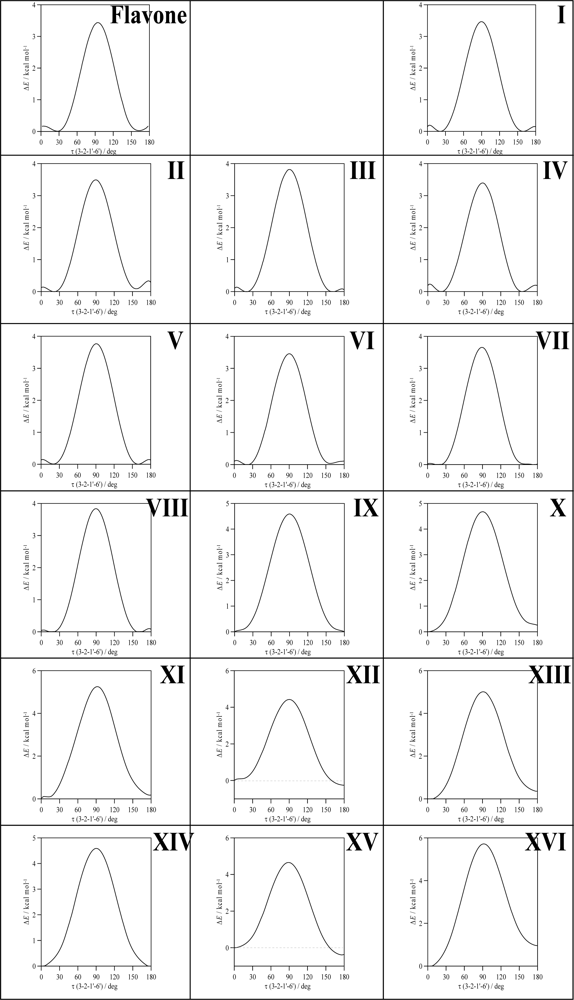
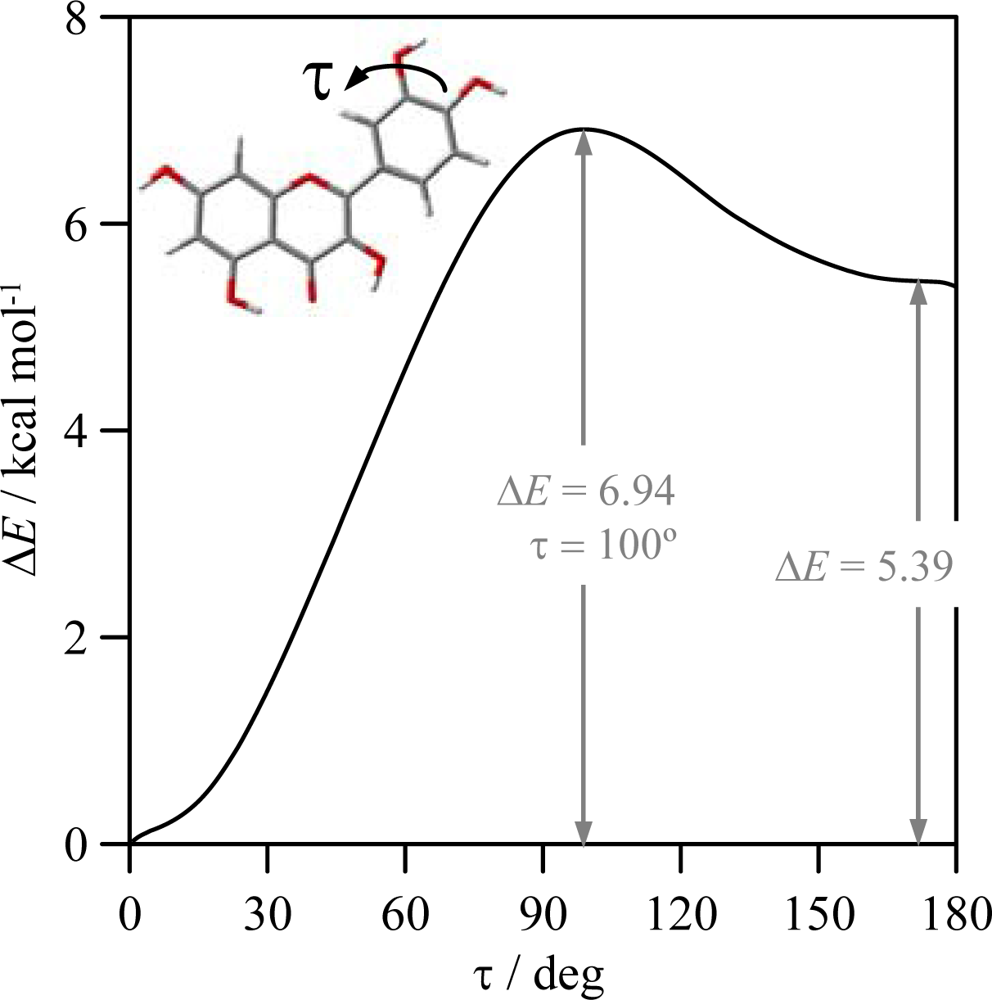
To clarify the question of molecular planarity, relaxed potential energy scans were carried out for the (3-2-1’-6’),
Figure 1, dihedral angle, for flavone and the 16 studied flavonoids in which the dihedral angle was changed in 10° steps and the remaining molecular parameters were optimized for each fixed dihedral angle. The results are reported in
Figure 3 for the whole torsional profiles, and in
Table 3, the maxima of the torsional barriers are reported.
All the molecules show a remarkable torsional barrier with maxima for a dihedral angle of 90°, as is to be expected, because the conjugation stabilizing effect for this angle is null. The torsional barriers are different for flavone and molecules I–VIII to those of molecules IX–XVI. For flavone and molecules I–VIII, minima appear at dihedrals around 20°, as we have reported in the previous paragraph, with small maxima for 0° and 180°, whereas for molecules IX–XVI minima are clearly defined for 0° dihedrals. Moreover, the maxima of the torsional barrier for flavone and I–VIII molecules are in the 3.4–3.8 kcal mol
−1 range, whereas, for the molecules IX–XVI, they are in the 4.4–5.7 kcal mol
−1 range,
Table 2. These facts show that two factors contribute to the structure of the studied molecules:
i) the conjugation between rings and
ii) the hydrogen bonding between 3-OH and 6′-H. The first factor is the only one present for flavone and molecules I–VIII, and thus, because of the almost null energetic barrier ongoing from 0° dihedral to 20° dihedral (∼0.2 kcal mol
−1), the 20° dihedral structure should be preferred because in this case the repulsions between hydrogens in 3 and 6′ are minimized, and at the same time the conjugation between rings is maintained. For molecules IX–XVI, the presence of the hydrogen bonding between 3-OH and 6′-H leads to an additional contribution that increases the rotational barrier, and thus the hydrogen bonding through this position may be confirmed and it should be the responsibility of the planarity of molecules IX–XVI. The additional energy contribution rising from the 3-OH/6’-H hydrogen bonding may be quantified as the difference of the torsional barrier maxima reported in
Table 3, that is to say, comparing molecule II with molecule X, molecule III with molecule XI and so on; then, this contribution is around 1 kcal mol
−1, except for compound XVI (myricetin) for which it is 1.87 kcal mol
−1. The remaining parameters reported in
Table 2 show very weak variations for the studied compounds. 7-OH group interatomic distance is unaffected by the changes in the molecular structure studied in this work. 3-OH group is not affected by the presence of OH groups in the cathecol ring, and, thus, its interaction with the keto group in C-ring and the 6′-H in catechol ring, does not change with the presence of hydroxyl groups in this B-ring. Likewise, hydroxyl groups in the 3′, 4′ and 5′ positions on B ring does not change remarkably with the presence/absence of neighboring hydroxyl groups. Hence, the interaction between C and B rings is done through conjugation, for molecules without 3-OH group, and through this effect and the interaction between 3-OH and 6′-H sites, for molecules with a 3-OH group.
In
Table 2, gas phase dipole moments are also reported, flavonoids without 3-OH groups are more polar (moments in the 4–5 D range) than those with 3-OH groups (moments in the 2–3 D range). The knowledge of flavonoids dipole moment is important to understand their biological activity because the interaction of these molecules with biomolecules is developed through hydrogen bonding of the hydroxyl sites and through van der Waals forces [
42]. Nonetheless, dipole moment is strongly dependent on very weak structural changes, for example molecule XVI (myricetin) has a dipole moment of 1.50 D for the structure reported in
Figure 2, in this molecule, hydroxyl groups in the B ring are clockwise oriented, then if these groups are counter clockwise oriented (the energy difference between both structures is only −0.86 kcal mol
−1) the dipole moment increases to 5.14 D This is in agreement with literature results at a lower theoretical level [
22]. As a rule, flavonoid conformations with hydroxyl groups counter clockwise oriented in the B ring show a remarkably larger dipole moment than those clockwise oriented, and the energy difference between both orientations is very low, but favorable to clockwise orientations. Moreover, the orientation of the 7-OH group leads also to important variations of dipole moment. For molecule XVI (myricetin), the counter clockwise oriented 7-OH group leads to 1.50 D whereas clockwise oriented leads to 5.10 D. Considering that the energy difference between both conformations is just −0.55 kcal mol
−1, changes in dipole moments through reorientations through 7-OH positions may be easily produced at very low energy cost. Therefore, to analyze the rule of dipolar forces on the flavonoid/biomolecule interactions, the exact configuration rising from the geometry and characteristics of the interaction site should be analyzed [
22]. Nevertheless, these molecules may lead to remarkable dipole – dipole interactions with the biomolecules’ specific sites through changes in their conformations leading to remarkable increases in their dipole moments.
3.2. Intramolecular Hydrogen Bonding. AIM and NBO Analysis
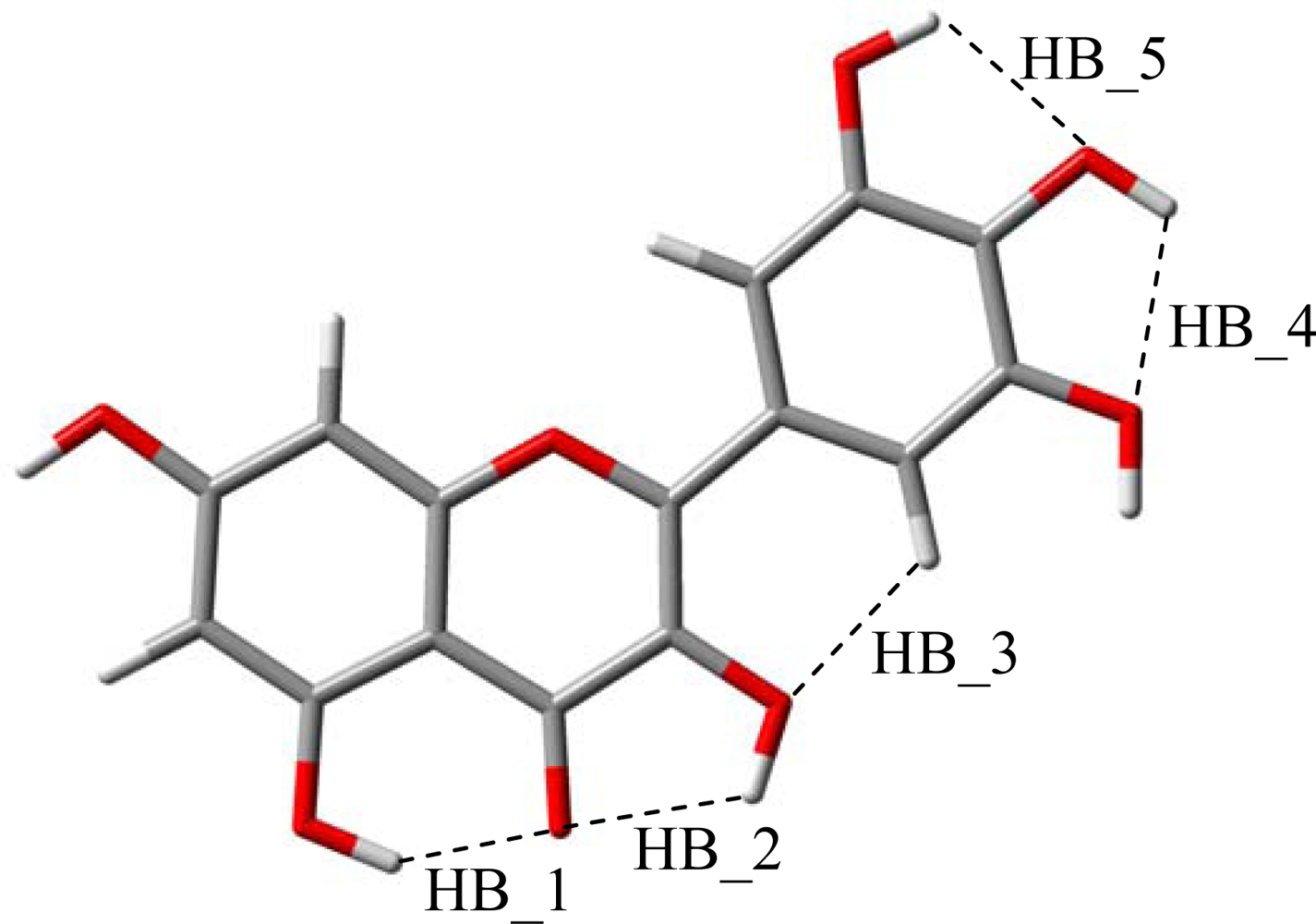
The reported results for the studied flavonoids show that up to five intramolecular hydrogen bonds are possible, HB_1 to HB_5,
Figure 4. In this section, the characteristics of these hydrogen bonds will be analyzed. The strength of these bonds may be quantified through relaxed potential energy scans for hydroxyl groups involved in each interaction. In
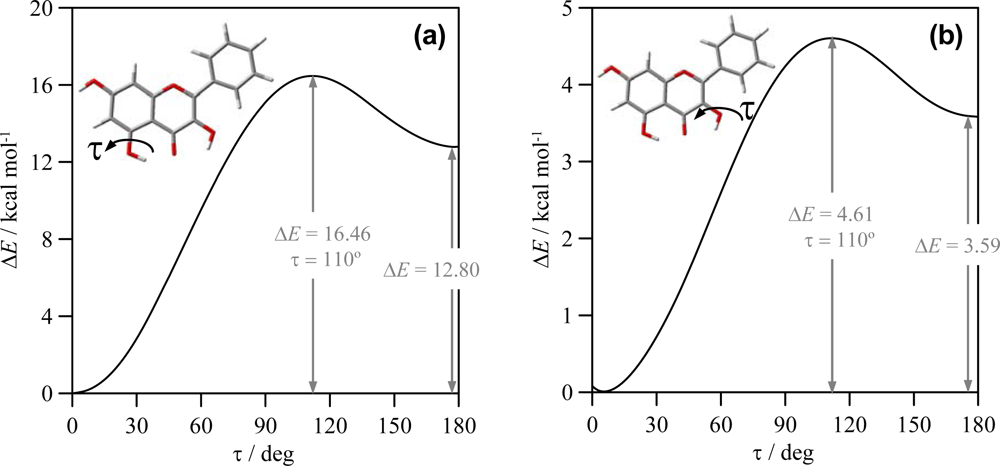
Figure 5, these torsional profiles are reported for HB_1 and HB_2 in molecule IX (galangin).
The results reported in
Figure 5a for HB_1 show that this H-bond is remarkably strong, the energy difference between conformers with and without HB_1 is 12.80 kcal mol
−1 and the interconversion of both structures evolves through a 16.46 kcal mol
−1 torsional barrier. Previously reported results at AM1 theoretical level [
39] reported a destabilization energy of 6.04 kcal mol
−1 for quercetin because of the absence of HB_1: as it has been reported in the previous section, HB_1 is almost unaffected by the presence of hydroxyl groups in cathecol ring, and thus the strength of HB_1 should be almost constant for the studied compounds. Therefore, AM1 results reported [
39] remarkably underestimated the strength of HB_1, and, considering also the results reported in the previous section, AM1 literature results for these compounds should be taken with caution, and only high level computations should be considered as reliable. Torsional analysis of HB_2 is reported in
Figure 5b; this bond is also strong, although weaker than HB_1, and the interconversion of conformers with and without HB_2 evolves though a remarkable barrier. albeit lower than for HB_1. AM1 results in Ref. [
39] reported a 3.65 kcal mol
−1 energetic difference for the HB_2 in quercetin, which is in surprising agreement with the 3.58 kcal mol
−1 reported in this work. The weaker character of HB_2 is in agreement with the shorter O-H interatomic distance for the OH group in position 3 than for the group in position 5,
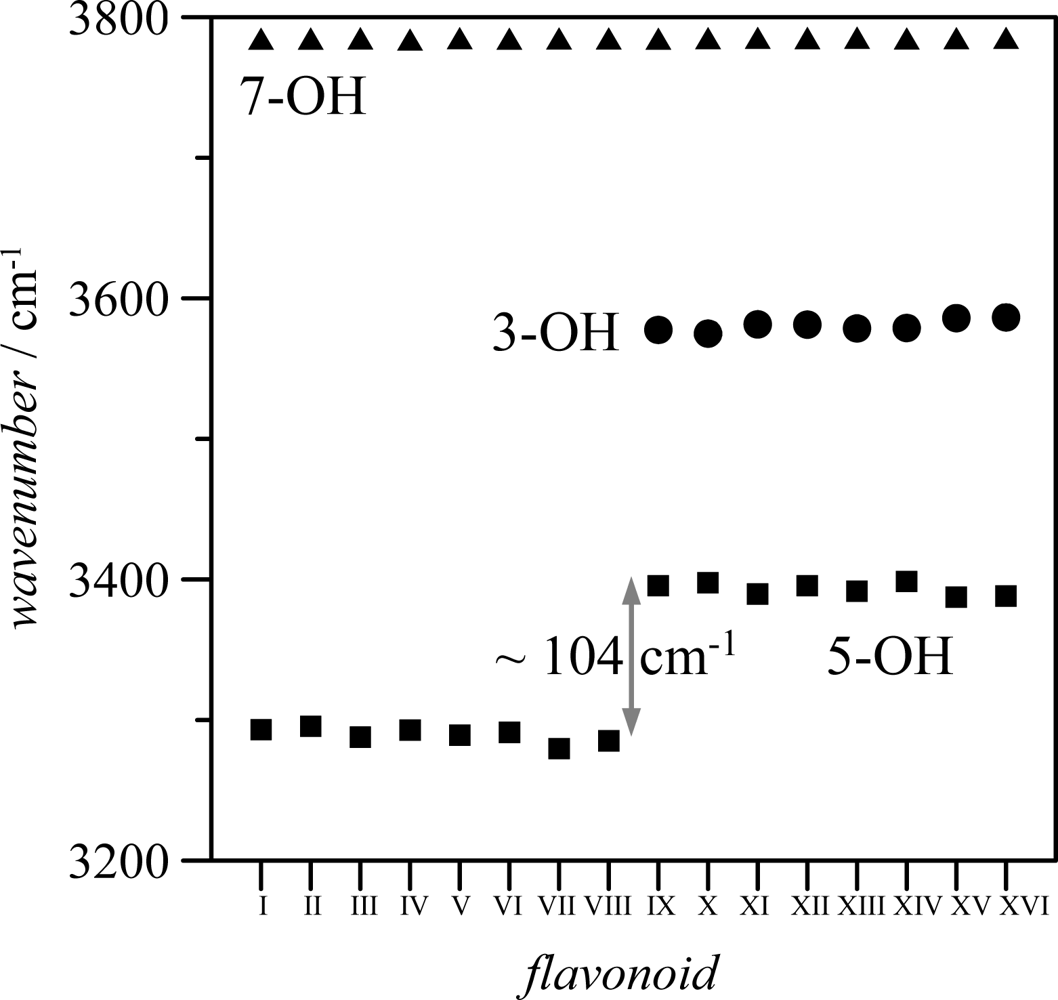
Table 2; the longer the OH interatomic distance, the stronger the hydrogen bonding. Moreover, the interaction of 3-OH with 6′-H, should lead to a weakening of 3-OH interaction with the keto group oxygen to allow the interaction with the 6′-H atom, at least for one of the lonely pairs of the 3-OH oxygen. Stretching vibrational frequencies of 3-OH and 5-OH groups also show remarkable differences between both groups, for molecule IX these frequencies appear at 3577. 5 and 3395.6 cm
−1 (unscaled values), respectively, pointing to a stronger hydrogen bonding for 5-OH group. This behavior is confirmed by the calculated values of hydroxyl stretching vibrational frequencies reported in
Figure 6 for 3-OH, 5-OH and 7-OH groups in the 16 studied flavonoids.
The frequency order is 7-OH > 3-OH > 5-OH. The almost constant values of 7-OH frequencies (∼3785 cm−1 for the 16 flavonoids) discard any hydrogen bonding through this position. The larger values of frequencies for 3-OH in comparison with 5-OH group point to a stronger hydrogen bonding between keto group oxygen and 5-OH for all the studied flavonoids; nevertheless, the introduction of an hydroxyl group in position 3 also weakens 5-OH interaction with keto groups, as it may be inferred from the blue-shifting of 5-OH vibrational frequencies for molecules IX–XVI in comparison with molecules I–VIII. Therefore, the main effect on hydrogen bonding in the studied molecules is the presence or absence of hydroxyl group in position 3, because the hydroxyl groups in B-ring do not affect remarkably the interactions in A and C rings. Stretching vibrations for the hydroxyl groups in B ring are of 3,750–3,800 cm−1 (unscaled values) for groups in 3′, 4′ and 5′, for all the studied compounds, and thus, in the same range as 7-OH vibrations, discarding strong hydrogen bonding for these hydroxyl groups.
The strength of HB_3 was analyzed in the previous section through the torsional profiles of (3-2-1’-6’) dihedrals, this hydrogen bonding has a strength of around 1 kcal mol
−1, and thus, it is clearly weaker than HB_1 and HB_2. Finally, possible hydrogen bonding between OH groups in the catechol moiety (HB_4 and HB_5), are analyzed in
Figure 7, in which results of a relaxed potential energy scan in quercetin is reported. Torsional profile reported in
Figure 7 shows that interactions between neighbour hydroxyl groups in B-ring are clearly weaker than HB_1 but stronger than HB_2 and HB_3.
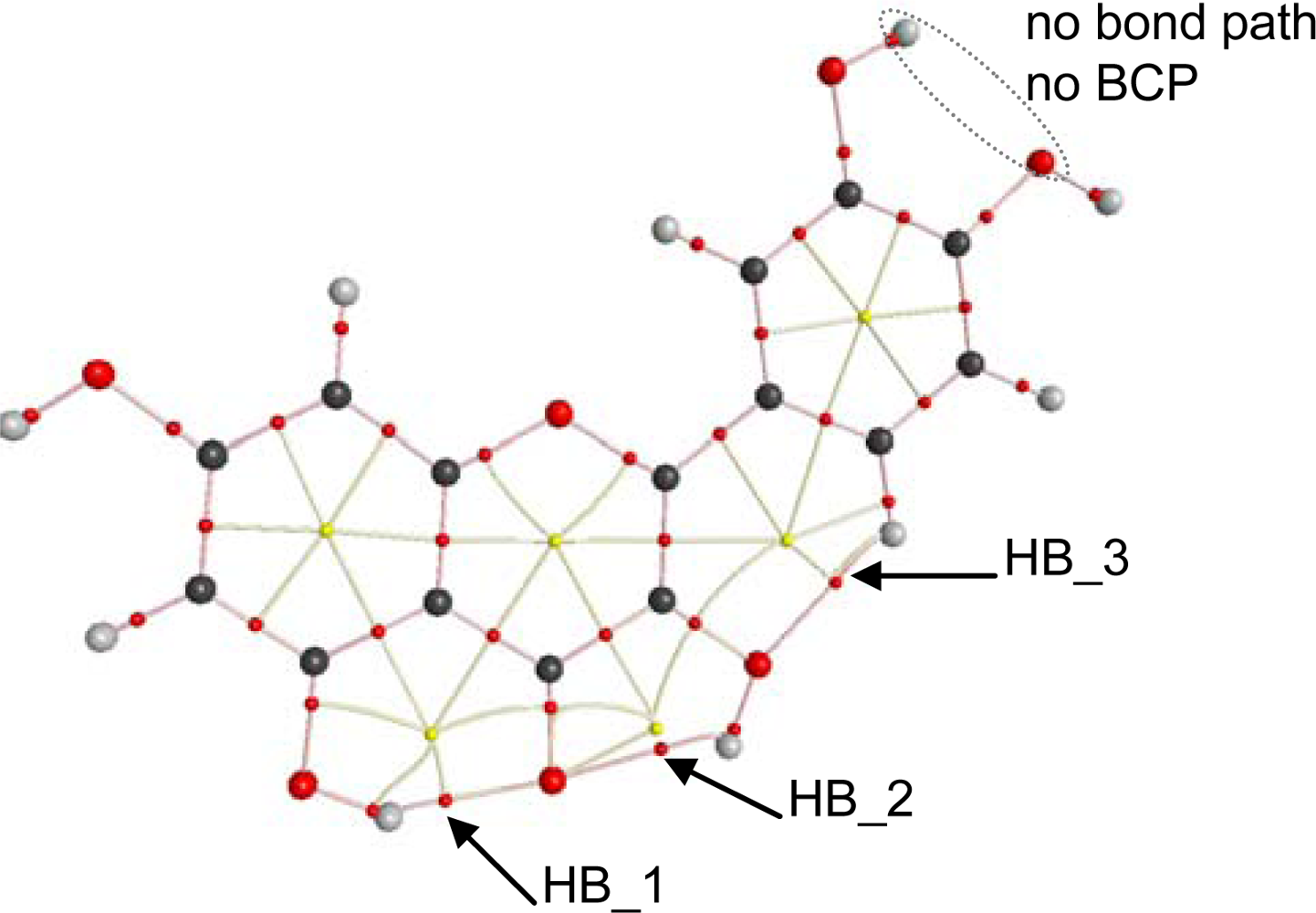
To get a deeper insight into the characteristics of intramolecular hydrogen bonding, AIM and NBO analysis were carried out for the optimized structures reported in
Figure 2. The first evidence of hydrogen bonding according to the AIM approach is the existence of a bond path between two atoms and the existence of a bond critical point, BCP, in the middle of the path [
44,
45]. AIM results keep the Lewis bonding scheme for all the studied flavonoids, moreover, BCPs are found for HB_1, HB_2 and HB_3 but not for proposed HB_4 and HB_5,
Table 4, as it is reported in
Figure 8 for quercetin. These results are very surprising for HB_4 and HB_5 interactions, considering the behavior reported in
Figure 7 pointing to a remarkable interaction through these positions, clearly stronger than those for HB_2 and HB_3 sites. These results for HB_4 and HB_5 interactions will be clarified in the next sections using both AIM and NBO results.
BCPs are not exactly placed in the middle of the bond paths for HB_1 to HB_3; instead they are shifted toward the hydrogen atoms involved in each hydrogen bond for all the studied flavonoids. A second AIM criterion to define and hydrogen bond considers that electron density at BCP, ρ
BCP, and the Laplacian of electron density at BCP, ∇
2 ρBCP, must be within the 0.002–0.035 and 0.024–0.139 ranges, respectively (both in atomic units) [
43,
44]. This second criterion is fulfilled for HB_1, HB_2 and HB_3 in the 16 studied flavonoids,
Table 4, and the ordering of these two properties is HB_1 > HB_2 > HB_3. Thus, this should be the strength ordering of these hydrogen bonds, which is in agreement with the results reported from torsional barriers. The main effect on ρ
BCP, and ∇
2 ρBCP values is the presence/absence of 3-OH group, and thus, for each hydrogen bond, these properties do not change from flavonoid I up to VIII (without 3-OH group) and from IX up to XVI (with 3-OH group), and are clearly different for these two groups of flavonoids. Moreover, ρ
BCP, and ∇
2 ρBCP for HB_1 decrease with the presence of 3-OH group, and thus, this hydrogen bond is weakened by the presence of this neighboring group. Diagonalization of the Hessian of the electron density yields three eigenvalues, λ
1 < λ
2 < λ
3; the ellipticity, ɛ, is defined as λ
1/λ
2 − 1 and measures the extent to which charge is preferentially accumulated; these properties are reported in
Table 5 for quercetin and luteolin, and similar results are obtained for the remaining studied flavonoids. Ellipticity is much larger for HB_2 than for HB_1, confirming that HB_2 is weaker, because large ɛ shows structural (topological) instability, and therefore a hydrogen bond that can easily be ruptured. The presence of a neighbor 3-OH group does not remarkably affect the ɛ value of HB_1,
Table 5, in which results for luteolin (without 3-OH group) and quercetin (with 3-OH group) are reported. Ellipticity for HB_3 is low, even lower than for HB_1. Nevertheless, λ
1 and λ
2 values for HB_3 are around three times lower, in absolute value, than those for HB_1,
Table 5, and thus, a lower concentration of charge density in HB_3, and therefore a weaker hydrogen bond, may be inferred. The larger values of λ
1 and λ
2 for HB_1 in flavonoids without 3-OH (luteolin in
Table 5) point also to a lowering of charge density upon 3-OH group addition. λ
3 parameter shows how easily the BCP can be moved along the bond path [
45], and thus the larger this parameter, the stronger the hydrogen bond. Therefore, the lower values of λ
3 also point to a weakening of HB_1 when 3-OH is added and to the weaker character of HB_3 in comparison with HB_1 and HB_2,
Table 5. Another criterion for structural stability is the distance between a BCP and a ring critical point, RCP; RCP is close to BCP for HB_2, whereas the distance between BCP and the closer RCP is larger for HB_1 and HB_3,
Figure 8, and thus, a greater instability for HB_2 may be inferred.
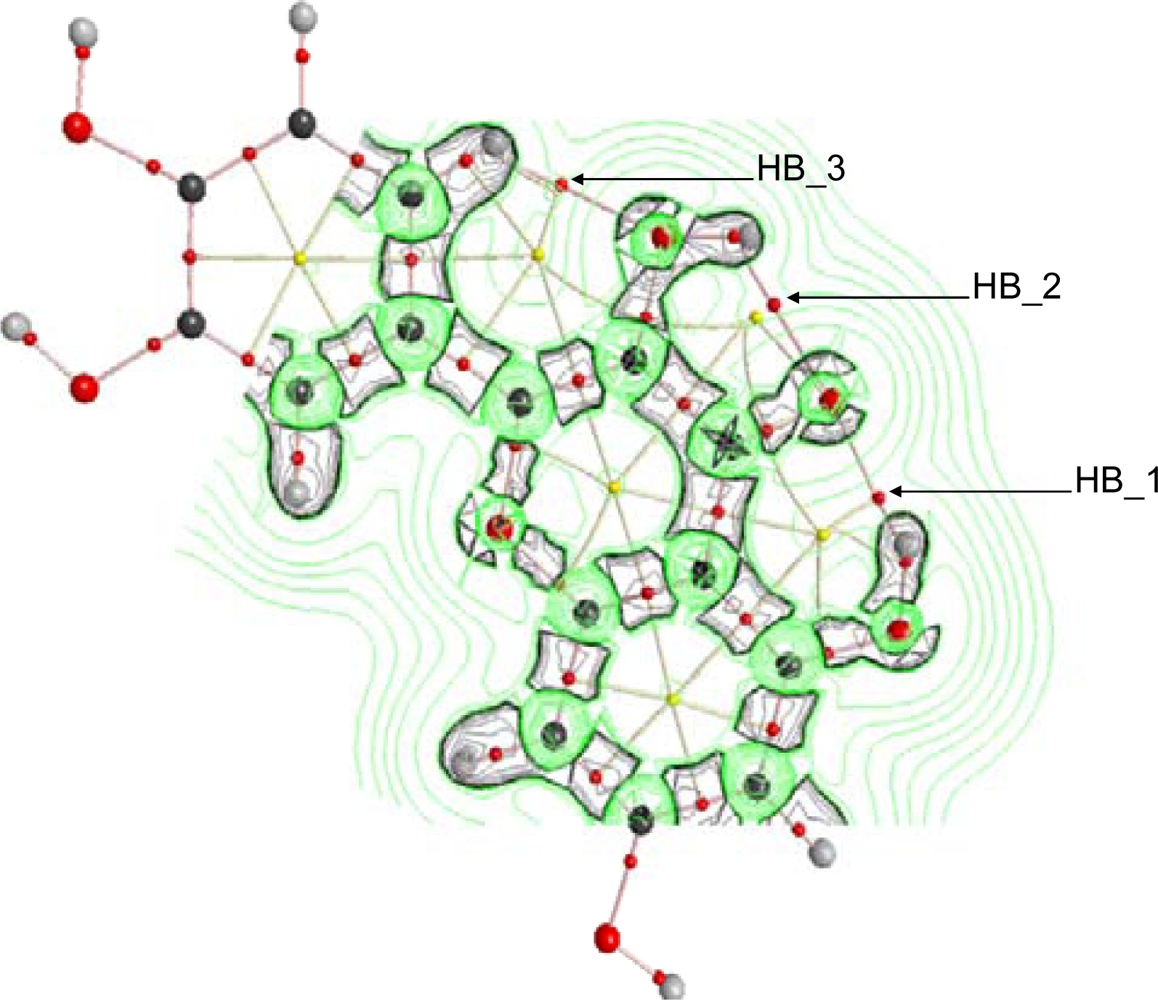
The behavior of ∇
2 ρ in the vicinity of HB_1, HB_2 and HB_3 is reported in
Figure 9 for quercetin, although similar trends are obtained for flavonoids IX – XVI. ∇
2 ρis the sum of eigenvalues λ
i, with ∇
2 ρ < 0 values showing internuclear charge concentration corresponding to the so-called shared interactions (covalent bonds), whereas ∇
2 ρ > 0 shows charge depletion corresponding to the so-called closed shell interactions (hydrogen bonding, ionic bonds and van der waals interactions). Valence shell charge concentration, VSCC, is not polarized for all the hydrogens involved in HB_1, HB_2 and HB_3, and thus, hydrogen bonds with purely electrostatic nature may be inferred. Contours plots of ∇
2 ρ are slightly different in the region of HB_1 compared with HB_2, with larger charge depletion for HB_1, and thus pointing again to a stronger HB_1 hydrogen bond in comparison with HB_2.
The energetic properties of BCP associated with hydrogen bonding also provide valuable information about these interactions according to the AIM approach. Kinetic energy density,
G, is always positive, and its ratio to electron density,
G/ρBCP, may be used to define the character of the interaction, being larger than 1.0 for closed shell and less than 1.0 for shared interactions [
33]; this property is related to the ionic character of the interaction [
46]. Potential energy density (or virial field),
V, that is always negative, is related to the covalency of the interaction [
46] and with hydrogen bonding strength through several empirical relationships [
47,
48]. It should be remarked the relationship between
V,
G and ∇
2 ρ through the local form of the virial theorem [
33]. Moreover, total energy density,
H, the sum of
G and
V, should be positive for closed shell interactions, such as hydrogen bonding, indicating that kinetics energy density dominates the potential energy density, and negative for shared interactions [
33].
G and
H values are reported in
Table 5 for quercetin and luteolin. The ratios
G/ρBCP are lower than 1.0 for HB_1, HB_2 and HB_3, although for closed shell interactions as these ones they should be larger than 1.0 [
33]; nevertheless, values slightly lower than 1.0 have been previously reported for hydrogen bonding in other compounds [
46].
H values are negative for HB_1, corresponding to strong hydrogen bonds, and slightly positive for HB_2 and HB_3.
H for BCP in HB_1 decreases on going from luteolin to quercetin, and so
G does, this is in agreement with the weakening of HB_1 upon addition of 3-OH group, the larger the
G values, the stronger the hydrogen bonding. HB_2 and HB_3 show lower
G values in comparison with HB_1, and thus they are weaker hydrogen bonds.
NBO results are reported in
Table 6 and valuable information on the energy of studied hydrogen bonds may be inferred from them. Moreover, charge delocalization from A to B rings via C one (through the C2–C3 double bond) can be also analyzed using the NBO approach. Results reported in
Figure 10 show a remarkably non-Lewis percentage for Quercetin, the population of antibonding orbitals, BD*, is also important and shows how the charge delocalization between A and B rings is done through the double bond in C ring. Analogous results are obtained for all the studied flavonoids, with a % non–Lewis character in the 2.6–2.9 range, and thus, an effective charge delocalization is obtained for all the studied molecules, including those slightly non–planar (I–VIII).
Hydrogen bonding may be analyzed considering a charge transfer between the donor and acceptor atoms. Therefore, hydrogen bonding arising from keto oxygen/hydroxyl hydrogen interactions, HB_1 and HB_2, and from hydroxyl/hydroxyl interactions, HB_4 and HB_5, should be determined by the hyperconjugation induced charge transfer between the corresponding oxygen electron lonely pairs (donor) and the antibonding orbitals for the hydroxyl bond (acceptor), nO→σ*O–H. For HB_3, hyperconjugation will be carried out between lonely pairs in hydroxyl oxygen and orbitals for hydrogen atom in position 6′. Three main properties are considered to analyze hydrogen bonding in the studied molecules according to the NBO method: i) second order perturbation energy, E(2), ii) energy difference among the donor and the acceptor, ΔE, and iii) Fock matrix element between the donor and acceptor, Fij. In agreement with the results reported in previous sections using different approaches, the main effect on NBO parameters for the studied hydrogen bonds is the absence/presence of a 3-OH group.
Properties of HB_1 do not change remarkably on going from flavonoids I to VIII, without 3-OH group, and do change remarkably upon 3-OH addition, flavonoids IX to XVI, especially for the interaction between the second oxygen pair. Properties of HB_1 also do no change by the presence of OH groups in the B ring. Hyperconjugation in HB_1 rises from two different keto oxygen lone pairs, pairs 1 and 2;
E(2) for pair 2 is remarkably larger than for pair 1 because of the lower energy difference and the orbital overlapping between donor and acceptor orbitals, which is reflected in the larger
Fij value for pair 2. The large value of
E(2) should be the molecular origin of the remarkable strength for HB_1. Nevertheless, HB_1 strength decreases upon 3-OH addition, the properties for interaction through pair 1 do not change when 3-OH group is added, but for pair 2 they change remarkably: the energy difference between donor and acceptor is the same both with and without 3-OH but the Fock matrix element decreases. Therefore, the molecular origin of the weakening of HB_1 upon 3-OH presence is the loss of orbital overlapping ability between donor and acceptor that hinders the hyperconjugation induced charge transfer between the second pair in keto oxygen and OH in position 5 antibonding orbitals. For HB_2, NBO results show that it is clearly weaker than HB_1, with stronger interaction through pair 2 but with
E(2) values lower than for HB_1 for both pairs. The reason for this weaker character of HB_2 stands in the poorer orbital overlapping between donor and acceptor for both pairs, lower
Fij values, in spite of the equal values of
ΔE for HB_2 and HB_1. In
Table 7, the main properties of molecular orbitals involved in HB_1 for flavonoid V (luteolin) and HB_1 and HB_2 for flavonoid XIII (quercetin) are reported. The main differences between orbitals corresponding to lonely pairs 1 and 2 (LP-1 and LP-2) in 4-
O oxygen, for both flavonoids, are:
i) LP-1 has a remarkably lower energy, which hinders hyperconjugation induced charge transfer with antibonding orbitals in 5-OH and 3-OH positions and
ii) LP-1 has a mixed s/p character, whereas LP-2 is an almost purely p orbital. The
Fij value is larger for transferences from LP-2, and thus the increasing p character of donor orbital seems to favour the efficiency of charge transference. The remarkably lower
Fij value for the transference from LP-2 toward 3-OH orbital in comparison with that toward 5-OH, which leads to the lower strength of HB_2, can not be justified by the energies of acceptor OH orbital because the eigenvalues for 3-OH and 5-OH are almost equal, and thus only the very subtle differences in the s/p characters of the OH acceptor orbitals could justify the weaker character of HB_2. The weakening of HB_1 upon 3-OH addition may be justified comparing the changes in donor/acceptor orbital for flavonoids V and XIII reported in
Table 7. The energy of LP-2 orbital does not change and the s character decreases upon 3-OH addition. The energy of 5-OH acceptor orbital does not change also with the presence of 3-OH group and only very subtle differences in the s/p character rises in quercetin in comparison with luteolin. Therefore, these subtle changes in the characteristics of the donor/acceptor orbitals involved in HB_1 upon 3-OH addition may justify the remarkable decrease of HB_1 with the presence of neighbor 3-OH group and HB_2.
HB_3 hydrogen bonding is produced only through the interaction of pair 1 in 3-OH oxygen with 6′-H, this interaction is weak because of the large energy difference and the poor orbital overlapping between the acceptor and donor orbitals. HB_4 and HB_5 were not considered as true H-bonds from the AIM viewpoint, because of the absence of BCPs, in spite of the remarkable large energy stabilization reported in
Figure 7. Results reported in
Table 6 using NBO method are in full agreement with those obtained with AIM, donor/acceptor interactions for these hydrogen bonds are carried out only through pair 1 in the corresponding hydroxyl oxygen, and these interactions are not very effective mainly because of the poor orbital overlap between donor/acceptor orbitals, as the very low
Fij values show. Moreover, results using AIM and NBO approaches are in full agreement with geometrical parameters reported in
Tables 4 and
6 together with vibrational frequencies reported in
Figure 6 and along the text:
The interatomic O-H distances are 5-OH (HB_1) > 3-OH (HB_2) > 3′-OH, 4′-OH, 5′-OH (HB_4 and HB_5),
Table 4. Moreover, these distances for 3′-OH, 4′-OH, 5′-OH are almost equal to those for 7-OH, for which no hydrogen bonding is present.
Vibrational frequencies of OH groups are 5-OH (HB_1) < 3-OH (HB_2) < 3′-OH, 4′-OH, 5′-OH (HB_4 and HB_5),
Table 4. Moreover, these frequencies for 3′-OH, 4′-OH, 5′-OH are almost equal to those for 7-OH, for which no hydrogen bonding is present.
The donor/acceptor distances are HB_1 < HB_2 < HB_4, HB_5,
Table 4.
The donor/acceptor angles are HB_1 > HB_2 > HB_4, HB_5,
Table 4.
NBO results,
Table 6, shows
E(2) and
Fij values HB_1 > HB_2 > HB_4, HB_5.
Therefore, all the results shows that the strength of the interactions is HB_1 > HB_2 > HB4, HB5. Nevertheless, results reported in
Figure 7 show a remarkable torsional barrier and energy stabilization for HB_4 and HB_5. This apparent contradictory results could be explained considering the values of the interaction angles reported in
Table 4, that show the lower values for HB_4 and HB_5, and thus leading to a poor donor/acceptor overlapping (low
E(2) and
Fij values), the absence of BCPs, and interatomic distances and angles pointing to weak interactions.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}