A Sweet Killer: Mesoporous Polysaccharide Confined Silver Nanoparticles for Antibacterial Applications

Abstract
:1. Introduction
2. Results and Discussion
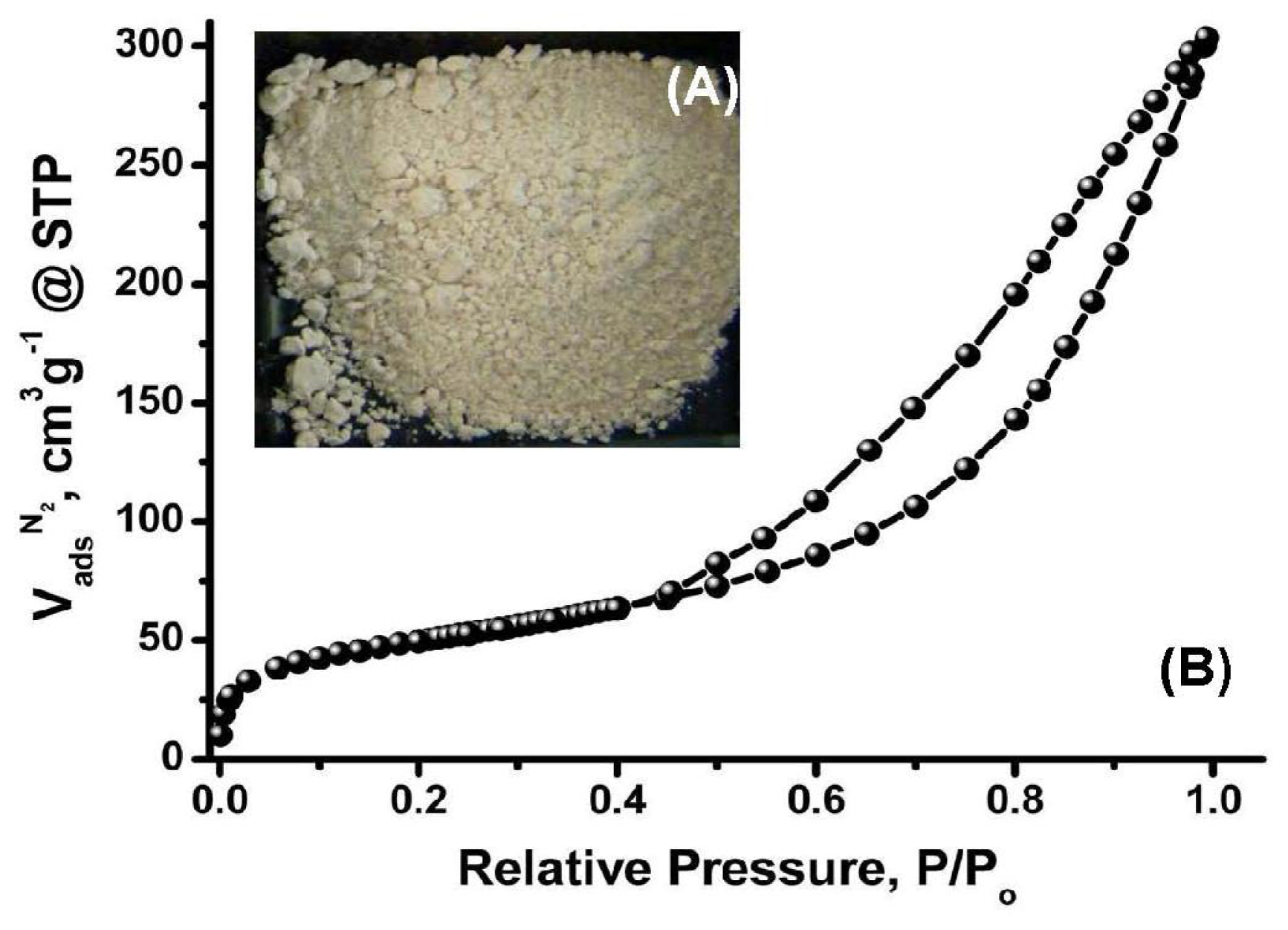
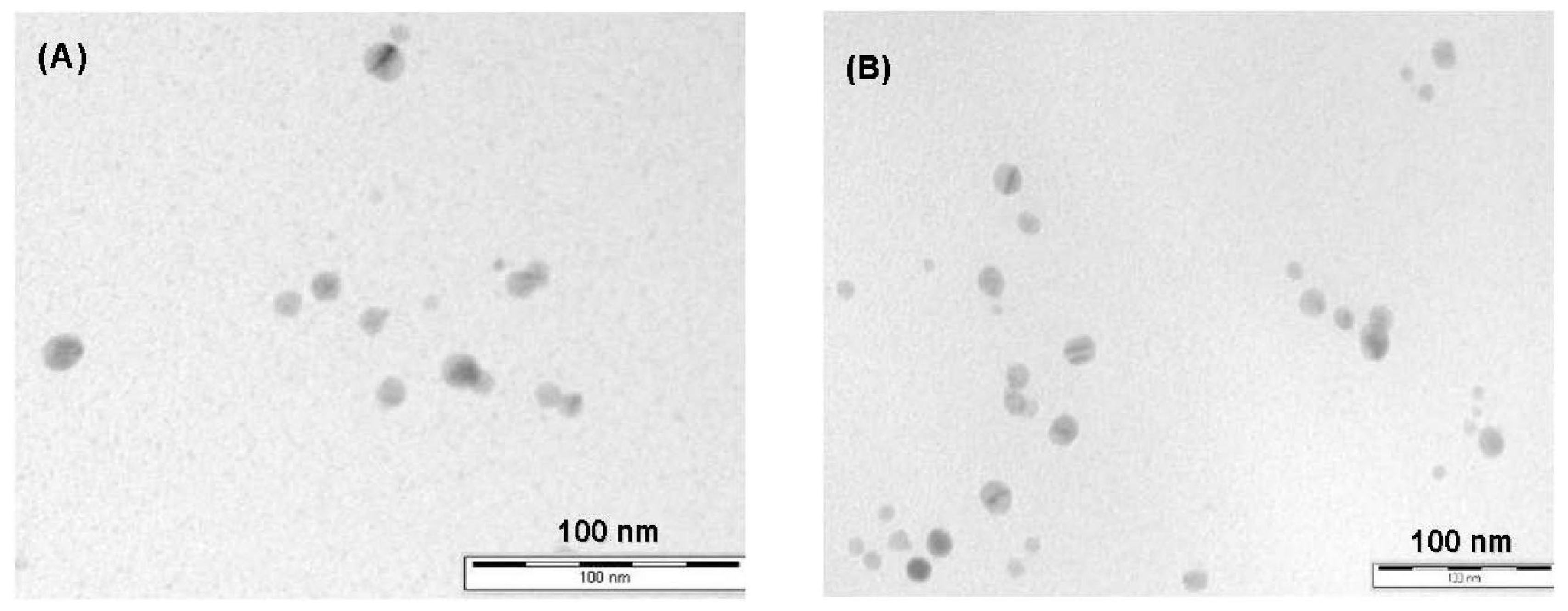
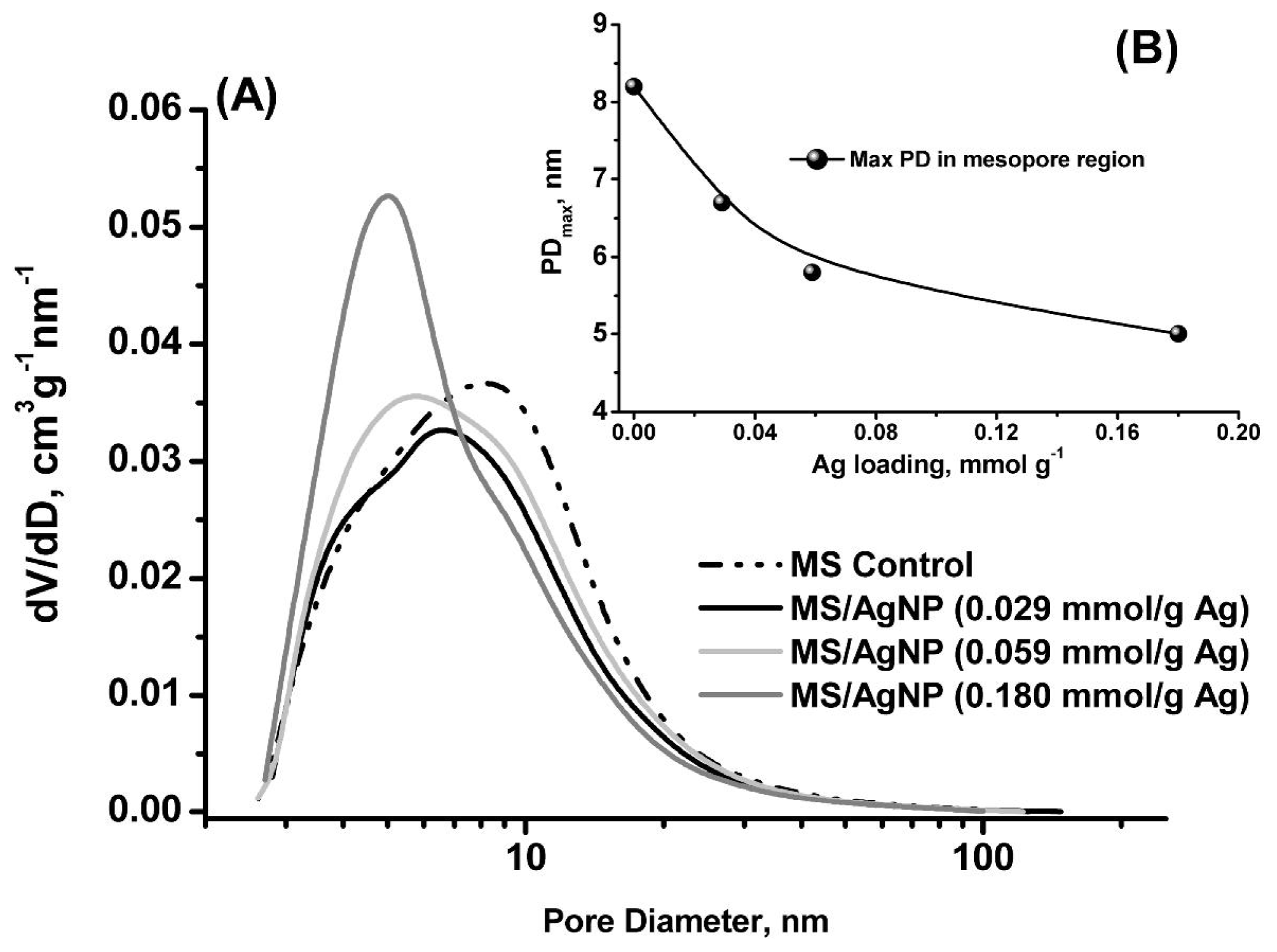
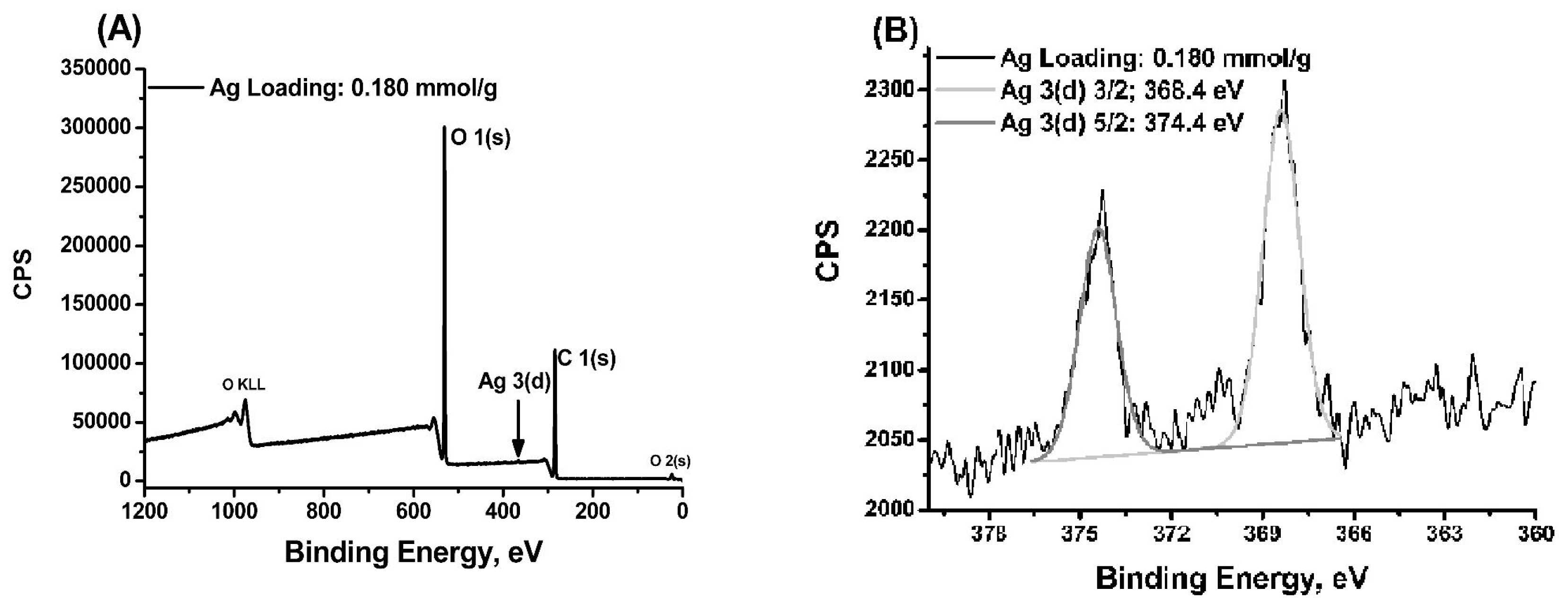
2.1. Characterisation of MS/AgNP Materials
2.2. Antimicrobial Activity of MS/AgNP Materials
3. Experimental Section
3.1. Materials and Chemicals
3.2. Preparation of Mesoporous Starch Supported Ag Nanoparticle Materials
3.3. Characterisation
3.4. Investigation of Antimicrobial Properties of Porous Starch Supported AgNP Materials
3.4.1. Procedure for Liquid Phase Growth Inhibition Studies
3.4.2. Procedure for Zone of Inhibition Plate Studies
4. Conclusions
Supplementary Information
ijms-12-05782-s001.pdfAcknowledgments
References
- Albrecht, MA; Evans, CW; Raston, CL. Green chemistry and the health implications of nanoparticles. Green Chem 2006, 8, 417–432. [Google Scholar]
- Anastas, PT; Warner, JC. Green Chemistry: Theory and Practice, 1st ed; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Anastas, PT; Kirchhoff, MM. Origins, current status, and future challenges of green chemistry. Account Chem Res 2002, 35, 686–694. [Google Scholar]
- Clark, JH; Budarin, V; Deswarte, FEI; Hardy, JJE; Kerton, FM; Hunt, AJ; Luque, R; Macquarrie, DJ; Milkowski, K; Rodriguez, A; et al. Green chemistry and the biorefinery: A partnership for a sustainable future. Green Chem 2006, 10, 853–860. [Google Scholar]
- White, RJ; Luque, R; Budarin, VL; Clark, JH; Macquarrie, DJ. Supported metal nanoparticles on porous materials. Methods and applications. Chem Soc Rev 2009, 2, 481–494. [Google Scholar]
- Badia, A; Gao, W; Singh, S; Demers, L; Cuccia, L; Reven, L. Structure and chain dynamics of alkanethiol-capped gold colloids. Langmuir 1996, 12, 1262–1269. [Google Scholar]
- Malinsky, MD; Kelly, KL; Schatz, GC; van Duyne, RP. Chain length dependence and sensing capabilities of the localized surface plasmon resonance of silver nanoparticles chemically modified with alkanethiol self-assembled monolayers. J Am Chem Soc 2001, 123, 1471–1482. [Google Scholar]
- Cooper, AI. Recent developments in materials synthesis and processing using supercritical CO2. Adv Mater 2001, 13, 1111–1114. [Google Scholar]
- Shah, PS; Husain, S; Johnston, KP; Korgel, BA. Role of steric stabilization on the arrested growth of silver nanocrystals in supercritical carbon dioxide. J Phys Chem B 2002, 106, 12178–12185. [Google Scholar]
- Morley, KS; Marr, PC; Webb, PB; Berry, AR; Allison, FJ; Moldovan, G; Brown, PD; Howdle, SM. Clean preparation of nanoparticulate metals in porous supports: a supercritical route. J Mat Chem 2002, 12, 1898–1905. [Google Scholar]
- Lewis, LN. Chemical catalysis by colloids and clusters. Chem Rev 1993, 93, 2693–2730. [Google Scholar]
- Aymonier, C; Schlotterbeck, U; Antonietti, L; Zacharias, P; Thomann, R; Tiller, JC; Mecking, S. Hybrids of silver nanoparticles with amphiphilic hyperbranched macromolecules exhibiting antimicrobial properties. Chem Commun 2002, 3018–3019. [Google Scholar]
- Maneerung, T; Tokura, S; Rujiravanit, R. Impregnation of silver nanoparticles into bacterial cellulose for antimicrobial wound dressing. Carbohyd Polym 2008, 72, 43–51. [Google Scholar]
- Wright, JB; Lam, K; Hansen, S; Burrell, S. Efficacy of topical silver against fungal burn wound pathogens. Am J Infect Control 1999, 27, 344–350. [Google Scholar]
- Sondi, I; Salopek-Sondi, B. Silver nanoparticles as antimicrobial agent: a case study on E-coli as a model for Gram-negative bacteria. J Colloid Interface Sci 2004, 275, 177–182. [Google Scholar]
- Morones, JR; Elechiguerra, JL; Camacho, A; Holt, K; Kouri, JB; Ramírez, JT; Yacaman, MJ. The bactericidal effect of silver nanoparticles. Nanotechnology 2005, 16, 2346–2353. [Google Scholar]
- Graham, D; Faulds, K; Smith, WE. Biosensing using silver nanoparticles and surface enhanced resonance Raman scattering. Chem Commun 2006, 4363–4371. [Google Scholar]
- Shao, M-W; Lu, L; Wang, H; Wang, S; Zhang, M-L; Ma, D-D-D; Lee, S-T. An ultrasensitive method: surface-enhanced Raman scattering of Ag nanoparticles from beta-silver vanadate and copper. Chem Commun 2008, 2310–2312. [Google Scholar]
- Sun, RWY; Chen, R; Chung, NPY; Ho, CM; Lin, CLC; Che, CM. Silver nanoparticles fabricated in Hepes buffer exhibit cytoprotective activities toward HIV-1 infected cells. Chem Commun 2005, 5059–5061. [Google Scholar]
- Vigneshwaran, N; Nachane, RP; Balasubramanya, RH; Varadarajan, PV. A novel one-pot ’green’ synthesis of stable silver nanoparticles using soluble starch. Carbohyd Res 2006, 341, 2012–2018. [Google Scholar]
- Stathatos, E; Lianos, P; Falaras, P. Photocatalytically deposited silver nanoparticles on mesoporous TiO2 films. Langmuir 2000, 16, 2398–2400. [Google Scholar]
- Hornebecq, V; Antonietti, M; Cardinal, T; Treguer-Delapierre, M. Stable silver nanoparticles immobilized in mesoporous silica. Chem Mater 2003, 15, 1993–1999. [Google Scholar]
- Xue, B; Chen, P; Hong, Q; Lin, J; Tan, KL. Growth of Pd, Pt, Ag and Au nanoparticles on carbon nanotubes. J Mat Chem 2001, 11, 2378–2381. [Google Scholar]
- Peng, X; Chen, J; Misewich, JA; Wong, SS. Carbon nanotube-nanocrystal heterostructures. Chem Soc Rev 2009, 38, 1076–1098. [Google Scholar]
- Raveendran, P; Fu, J; Wallen, SL. Completely "green" synthesis and stabilization of metal nanoparticles. J Am Chem Soc 2003, 125, 13940–13941. [Google Scholar]
- Konwarh, R; Karak, N; Sawian, CE; Baruah, S; Mandal, M. Effect of sonication and aging on the templating attribute of starch for “green” silver nanoparticles and their interactions at bio-interface. Carbohyd Polym 2011, 83, 1245–1252. [Google Scholar]
- Huang, H; Yang, X. Synthesis of polysaccharide-stabilized gold and silver nanoparticles: a green method. Carbohyd Res 2004, 339, 2627–2631. [Google Scholar]
- Laudenslager, MJ; Schiffman, JD; Schauer, CL. Carboxymethyl Chitosan as a Matrix Material for Platinum, Gold, and Silver Nanoparticles. Biomacromolecules 2008, 9, 2682–2685. [Google Scholar]
- Fu, X; Shen, Y; Jiang, X; Huang, D; Yan, Y. Chitosan derivatives with dual-antibacterial functional groups for antimicrobial finishing of cotton fabrics. Carbohyd Polym 2011, 85, 221–227. [Google Scholar]
- Son, WK; Youk, JH; Lee, TS; Park, WH. Preparation of antimicrobial ultrafine cellulose acetate fibers with silver nanoparticles. Macro Rap Commun 2004, 25, 1632–1637. [Google Scholar]
- Li, S-M; Jia, N; Maa, M-G; Zhang, Z; Liu, Q-H; Sun, R-C. Cellulose–silver nanocomposites: Microwave-assisted synthesis, characterization, their thermal stability, and antimicrobial property. Carbohyd Polym 2011, 86(2), 441–447. [Google Scholar]
- White, RJ; Budarin, VL; Clark, JH. Tuneable mesoporous materials from alpha-d-polysaccharides. ChemSusChem 2008, 1, 408–411. [Google Scholar]
- White, RJ; Budarin, VL; Clark, JH. Pectin-Derived Porous Materials. Chem Eur J 2010, 16, 1326–1335. [Google Scholar]
- Kelly, KL; Coronado, E; Zhao, LL; Schatz, GC. The optical properties of metal nanoparticles: The influence of size, shape, and dielectric environment. J Phys Chem B 2003, 107, 668–677. [Google Scholar]
- Budarin, V; Clark, JH; Deswarte, FEI; Hardy, JJE; Hunt, AJ; Kerton, FM. Delicious not siliceous: expanded carbohydrates as renewable separation media for column chromatography. Chem Commun 2005, 23, 2903–2905. [Google Scholar]
- Budarin, V; Clark, JH; Hardy, JJE; Luque, R; Milkowski, K; Tavener, SJ; Wilson, AJ. Starbons: New starch-derived mesoporous carbonaceous materials with tunable properties. Angew Chem Int Ed 2006, 45, 3782–3786. [Google Scholar]
- Budarin, VL; Clark, JH; Luque, R; Macquarrie, DJ; White, RJ. Palladium nanoparticles on polysaccharide-derived mesoporous materials and their catalytic performance in C-C coupling reactions. Green Chem 2008, 4, 382–387. [Google Scholar]
- Thiel, J; Pakstis, L; Buzby, S; Raffi, M; Ni, C; Pochan, DJ; Shah, SI. Antibacterial properties of silver-doped titania. Small 2007, 3, 799–803. [Google Scholar]
- Suh, MP; Moon, HR; Lee, EY; Jang, SY. A redox-active two-dimensional coordination polymer: Preparation of silver and gold nanoparticles and crystal dynamics on guest removal. J Am Chem Soc 2006, 128, 4710–4718. [Google Scholar]
- Pattabi, M; Rao, KM; Sainkar, S; Sastry, RM. Structural studies on silver cluster films deposited on softened PVP substrates. Thin Solid Films 1999, 338, 40–45. [Google Scholar]
- Canamares, MV; Garcia-Ramos, JV; Gomez-Varga, JD; Domingo, C; Sanchez-Cortes, S. Comparative study of the morphology, aggregation, adherence to glass, and surface-enhanced Raman scattering activity of silver nanoparticles prepared by chemical reduction of Ag+ using citrate and hydroxylamine. Langmuir 2005, 21, 8546–8553. [Google Scholar]
- Kapoor, S. Preparation, characterization, and surface modification of silver particles. Langmuir 1998, 14, 1021–1025. [Google Scholar]
- Budarin, VL; Clark, JH; Luque, R; Macquarrie, DJ; Milkowski, K; White, RJ. PCT Patent: Mesoporous Carbonaceous Materials, Preparation and the use thereof WO/2007/104798, 20 September 2007.
- Chimentão, RJ; Kirm, I; Medina, F; Rodríguez, X; Cesteros, Y; Salagre, P; Sueiras, JE. Different morphologies of silver nanoparticles as catalysts for the selective oxidation of styrene in the gas phase. Chem Commun 2004, 7, 846–847. [Google Scholar]
- Murugadoss, A; Goswami, P; Paul, A; Chattopadhyay, A. “Green” chitosan bound silver nanoparticles for selective C-C bond formation via in situ iodination of phenols. J Mol Cat A 2009, 304, 153–158. [Google Scholar]
- Brunauer, S; Emmett, PH; Teller, E. Adsorption of gases in multimolecular layers. J Am Chem Soc 1938, 60, 309–319. [Google Scholar]
- Barrett, EP; Joyner, LG; Halenda, PP. The determination of pore volume and area distributions in porous substances 1. Computations from Nitrogen Isotherms. J Am Chem Soc 1951, 73, 373–380. [Google Scholar]
- White, RJ; Antonio, C; Budarin, VL; Bergstrom, E; Thomas-Oates, J; Clark, JH. Polysaccharide-Derived Carbons for Polar Analyte Separations. Adv Funct Mat 2010, 20, 1834–1841. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ag Loading/mmol g−1 | aSBET/m2 g−1 | bVtotal/cm3 g−1 | bVmeso/cm3 g−1 | cVmicro/cm3 g−1 | bAPD/nm | bPDmax/nm |
|---|---|---|---|---|---|---|
| *0.000 | 170 | 0.53 | 0.50 | 0.008 | 9.6 | 8.2 |
| 0.029 | 152 | 0.47 | 0.44 | 0.007 | 8.8 | 6.7 |
| 0.059 | 161 | 0.49 | 0.45 | 0.007 | 9.0 | 5.8 |
| 0.180 | 175 | 0.47 | 0.45 | 0.008 | 7.7 | 5.0 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
White, R.J.; Budarin, V.L.; Moir, J.W.B.; Clark, J.H. A Sweet Killer: Mesoporous Polysaccharide Confined Silver Nanoparticles for Antibacterial Applications. Int. J. Mol. Sci. 2011, 12, 5782-5796. https://doi.org/10.3390/ijms12095782
White RJ, Budarin VL, Moir JWB, Clark JH. A Sweet Killer: Mesoporous Polysaccharide Confined Silver Nanoparticles for Antibacterial Applications. International Journal of Molecular Sciences. 2011; 12(9):5782-5796. https://doi.org/10.3390/ijms12095782
Chicago/Turabian StyleWhite, Robin J., Vitaly L. Budarin, James W.B. Moir, and James H. Clark. 2011. "A Sweet Killer: Mesoporous Polysaccharide Confined Silver Nanoparticles for Antibacterial Applications" International Journal of Molecular Sciences 12, no. 9: 5782-5796. https://doi.org/10.3390/ijms12095782
APA StyleWhite, R. J., Budarin, V. L., Moir, J. W. B., & Clark, J. H. (2011). A Sweet Killer: Mesoporous Polysaccharide Confined Silver Nanoparticles for Antibacterial Applications. International Journal of Molecular Sciences, 12(9), 5782-5796. https://doi.org/10.3390/ijms12095782