Fabrication, Modeling and Characterization of Multi-Crosslinked Methacrylate Copolymeric Nanoparticles for Oral Drug Delivery

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Modeling, Changes in pH and Absorbance of Poly-Lipo Nanoparticles during Fabrication
2.2. Size and Surface Charge Analyses of Poly-Lipo Nanoparticles
2.3. Fourier Transform Infra-Red (FTIR) Spectroscopy of the Poly-Lipo Nanoparticles
2.4. Surface Morphology of the Poly-Lipo Nanoparticles
2.5. Drug Loading Efficiency of Poly-Lipo Nanoparticles
2.6. Direct Compression of Poly-Lipo Nanoparticles into Tablet Matrices
2.7. In vitro Drug Release Studies
2.8. Polymeric Nanoparticles Improve Mechanical Strength
2.9. Magnetic Resonance Imaging
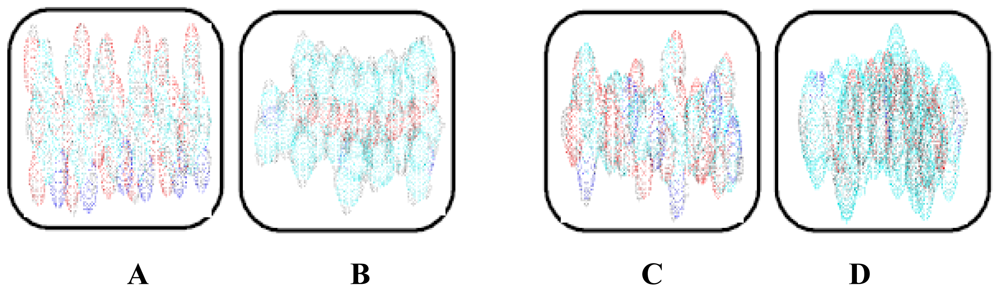
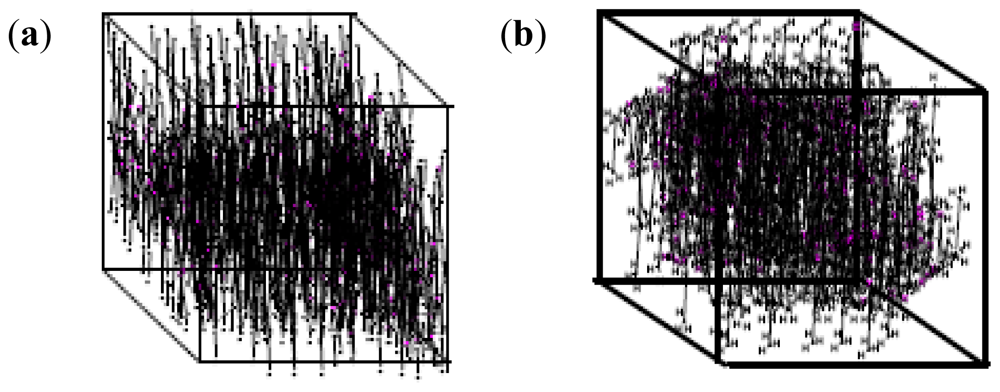
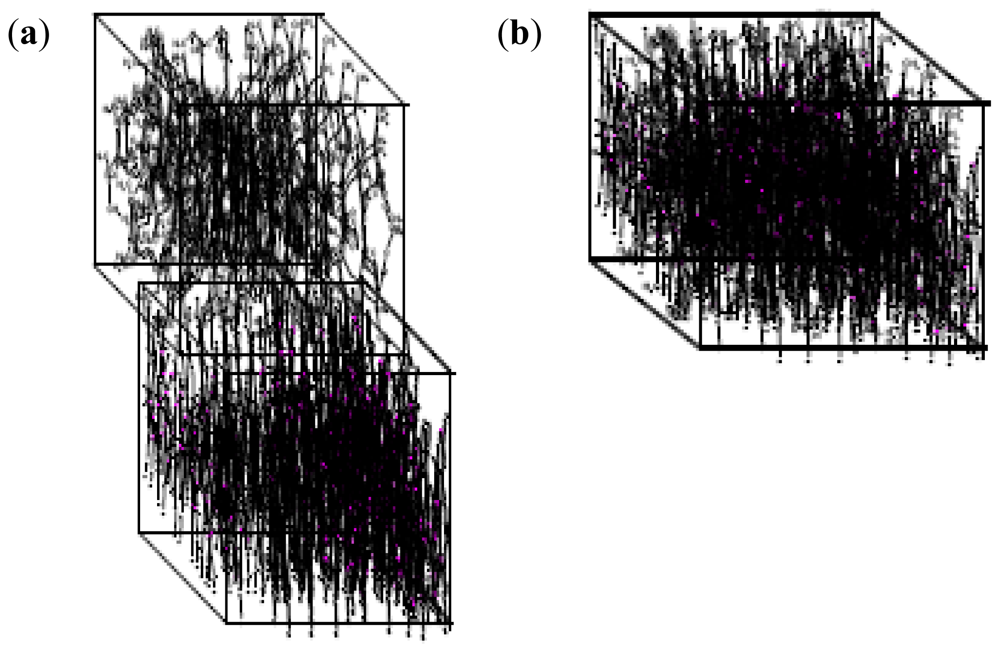
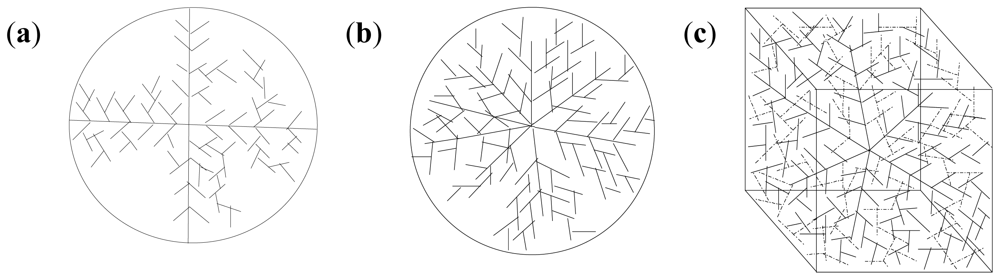
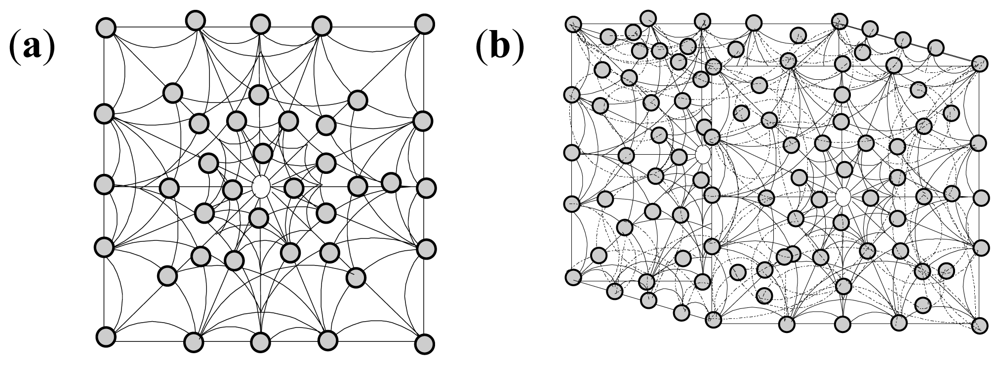
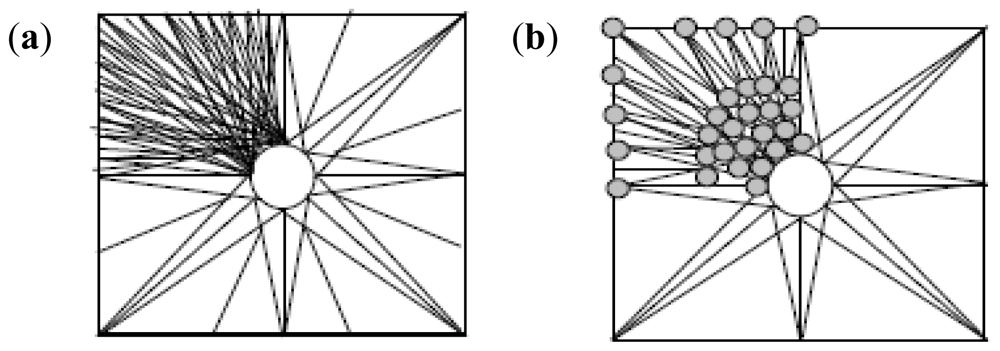
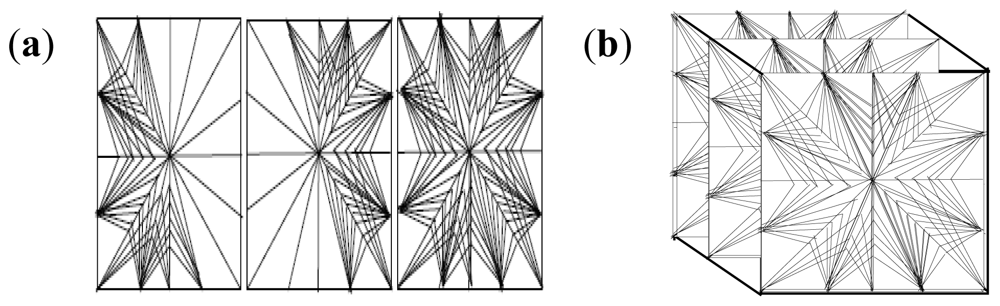
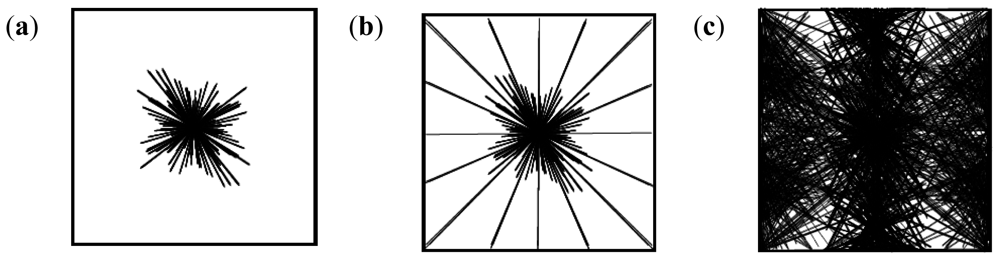
2.10. Molecular Mechanics Assisted Model Building and Energy Refinements
2.10.1. Molecular Mechanics Energy Relationship (MMER) Analysis
2.10.2. Energy-Minimizations Involving Crosslinked-Polymer Morphologies
3. Experimental Section
3.1. Materials
3.2. Nanofabrication of Polymer-Lipid Nanoparticles
3.3. Determination of pH and Absorbance Changes During Fabrication
3.4. Computational Modeling of the Fabrication of Multi-Crosslinked Nanoparticles
3.5. Size and Surface Charge Analyses of the Poly-Lipo Nanoparticles
3.6. Fourier Transform Infra-Red (FTIR) Spectroscopy of the Poly-Lipo Nanoparticles
3.7. Microscopical Analysis of the Levodopa-Loaded Poly-Lipo Nanoparticles
3.8. Determination of Drug Loading and Drug Entrapment Efficiency of the Poly-Lipo Nanoparticles
3.9. Direct Compression of the Poly-Lipo Nanoparticles
3.9.1. Exclusive Direct Compression of the Poly-Lipo Nanoparticles
3.9.2. Incorporation of Poly-Lipo Nanoparticles into an Interpolymeric Blend
3.10. In vitro Drug Release Studies
3.10.1. Drug Release Studies of Nanosuspension Employing Dialysis Technique
3.10.2. Drug Release Studies of the Compressed Matrices
3.11. Magnetic Resonance Imaging of Mechanical Behavior
3.12. Static Lattice Atomistic Simulations
4. Conclusions
References
- Thomas, VH; Bhattachar, S; Hitchingham, L; Zocharski, P; Naath, M; Surendran, N; Stoner, CL; El-Kattan, A. The road map to oral bioavailability: An industrial perspective. Expert Opin Drug Metab Toxicol 2006, 2, 591–608. [Google Scholar]
- Martinez, MN; Amidon, GL. A mechanistic approach to understanding the factors affecting drug absorption: A review of fundamentals. J Clin Pharmacol 2002, 42, 620–643. [Google Scholar]
- Varma, MVS; Kaushal, AM; Garg, A; Garg, S. Factors affecting mechanism and kinetics of drug release from matrix-based oral controlled drug delivery systems. Am J Drug Deliv 2004, 2, 43–57. [Google Scholar]
- Hurst, S; Loi, C; Brodfuehrer, J; El-Kattan, A. Impact of physiological, physicochemical and biopharmaceutical factors in absorption and metabolism mechanisms on the drug oral bioavailability of rats and humans. Expert Opin Drug Metab Toxicol 2007, 3, 469–489. [Google Scholar]
- Rasenack, N; Müller, BW. Micron-size drug particles: Common and novel micronization techniques. Pharm Dev Technol 2004, 9, 1–13. [Google Scholar]
- Thanos, CG; Liu, Z; Reineke, J; Edwards, E; Mathiowitz, E. Improving relative bioavailability of dicumarol by reducing particle size and adding the adhesive poly(Fumaric-Co-Sebacic) anhydride. Pharm Res 2003, 20, 1093–1100. [Google Scholar]
- Chan, OH; Schmid, HL; Stilgenbauer, LA; Howson, W; Horwell, DC; Stewart, BH. Evaluation of a targeted prodrug strategy to enhance oral absorption of poorly water-soluble compounds. Pharm Res 1998, 15, 1012–1018. [Google Scholar]
- Gomez-Orellana, I. Strategies to improve oral drug bioavailability. Expert Opin Drug Deliv 2005, 2, 419–433. [Google Scholar]
- Baudy, RB; Butera, JA; Abou-Gharbia, M; Chen, H; Harrison, B; Jain, U; Magolda, R; Sze, JY; Brandt, MR; Cummons, TA; et al. Prodrugs of perzinfotel with improved oral bioavailability. J Med Chem 2009, 52, 771–778. [Google Scholar]
- Gwak, H; Choi, J; Choi, H. Enhanced bioavailability of piroxicam via salt formation with ethanolamines. Int J Pharm 2005, 297, 156–161. [Google Scholar]
- Thanou, M; Verhoef, JC; Junginger, HE. Oral drug absorption enhancement by chitosan and its derivatives. Adv Drug Deliv Rev 2001, 52, 117–126. [Google Scholar]
- Wong, SM; Kellaway, IW; Murdan, S. Enhancement of the dissolution rate and oral absorption of a poorly water soluble drug by formation of surfactant-containing microparticles. Int J Pharm 2006, 317, 61–68. [Google Scholar]
- Leuner, C; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 2000, 50, 47–60. [Google Scholar]
- Vasconcelos, T; Sarmento, B; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today 2007, 12, 1068–1075. [Google Scholar]
- Sant, VP; Smith, D; Leroux, J. Enhancement of oral bioavailability of poorly water-soluble drugs by poly(ethylene glycol)-block-poly(alkyl acrylate-co-methacrylic acid) self-assemblies. J Control Release 2005, 104, 289–300. [Google Scholar]
- Morishita, M; Matsuzawa, A; Takayama, K; Isowa, K; Nagai, T. Improving insulin enteral absorption using water-in-oil-in-water emulsion. Int J Pharm 1998, 172, 189–198. [Google Scholar]
- Dollo, G; Le Corre, P; Guérin, A; Chevanne, F; Burgot, JL; Leverge, R. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability of poorly soluble drugs. Eur J Pharm Sci 2003, 19, 273–280. [Google Scholar]
- Araya, H; Tomita, M; Hayashi, M. The novel formulation design of o/w microemulsion for improving the gastrointestinal absorption of poorly water soluble compounds. Int J Pharm 2005, 305, 61–74. [Google Scholar]
- Constantinides, PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res 1995, 12, 1561–1572. [Google Scholar]
- Kuo, F; Subramanian, B; Kotyla, T; Wilson, TA; Yoganathan, S; Nicolosi, RJ. Nanoemulsions of an anti-oxidant synergy formulation containing gamma tocopherol have enhanced bioavailability and anti-inflammatory properties. Int J Pharm 2008, 363, 206–213. [Google Scholar]
- Yin, Y; Cui, F; Mu, C; Choi, M; Kim, JS; Chung, S; Shim, C; Kim, D. Docetaxel microemulsion for enhanced oral bioavailability: Preparation and in vitro and in vivo evaluation. J Control Release 2009, 140, 86–94. [Google Scholar]
- Gershanik, T; Benita, S. Positively charged self-emulsifying oil formulation for improving oral bioavailability of progesterone. Pharm Dev Technol 1996, 1, 147–157. [Google Scholar]
- Kang, BK; Lee, JS; Chon, SK; Jeong, SY; Yuk, SH; Khang, G; Lee, HB; Cho, SH. Development of self-microemulsifying drug delivery systems (smedds) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm 2004, 274, 65–73. [Google Scholar]
- Carrier, RL; Miller, LA; Ahmed, I. The utility of cyclodextrins for enhancing oral bioavailability. J Control Release 2007, 123, 78–99. [Google Scholar]
- Reddy, M; Rehana, T; Ramakrishna, S; Chowdary, K; Diwan, P. B-cyclodextrin complexes of celecoxib: Molecular-modeling, characterization, and dissolution studies. AAPS J 2004, 6, 68–76. [Google Scholar]
- Song, Y; Clizbe, L; Bhakta, C; Teng, W; Li, W; Wong, P; Huang, B; Sinha, U; Park, G; Reed, A; et al. Substituted acrylamides as factor xa inhibitors: Improving bioavailability by p1 modification. Bioorg Med Chem Lett 2002, 12, 2043–2046. [Google Scholar]
- Biron, E; Chatterjee, J; Ovadia, O; Langenegger, D; Brueggen, J; Hoyer, D; Schmid, H; Jelinek, R; Gilon, C; Hoffman, A; et al. Improving oral bioavailability of peptides by multiple n-methylation: somatostatin analogues13. Angew Chem Int Ed 2008, 47, 2595–2599. [Google Scholar]
- Hu, L; Tang, X; Cui, F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J Pharm Pharmacol 2004, 56, 1527–1535. [Google Scholar]
- Kesisoglou, F; Panmai, S; Wu, Y. Nanosizing–oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev 2007, 59, 631–644. [Google Scholar]
- Hans, ML; Lowman, AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci 2002, 6, 319–327. [Google Scholar]
- Sahoo, SK; Labhasetwar, V. Nanotech approaches to drug delivery and imaging. Drug Discov Today 2003, 8, 1112–1120. [Google Scholar]
- Aliabadi, HM; Lavasanifar, A. Polymeric micelles for drug delivery. Expert Opin Drug Deliv 2006, 3, 139–162. [Google Scholar]
- Almeida, AJ; Souto, E. Solid lipid nanoparticles as a drug delivery system for peptides and proteins. Adv Drug Deliv Rev 2007, 59, 478–490. [Google Scholar]
- Bali, V; Ali, M; Ali, J. Study of surfactant combinations and development of a novel nanoemulsion for minimising variations in bioavailability of ezetimibe. Colloids Surf B 2010, 76, 410–420. [Google Scholar]
- Drulis-Kawa, Z; Dorotkiewicz-Jach, A. Liposomes as delivery systems for antibiotics. Int J Pharm 2010, 387, 187–198. [Google Scholar]
- Gillies, ER; Fréchet, JMJ. Dendrimers and dendritic polymers in drug delivery. Drug Discov Today 2005, 10, 35–43. [Google Scholar]
- Kumari, A; Yadav, SK; Yadav, SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B 2010, 75, 1–18. [Google Scholar]
- Jin, S; Ye, K. Nanoparticle-mediated drug delivery and gene therapy. Biotechnol Prog 2007, 23, 32–41. [Google Scholar]
- Mora-Huertas, CE; Fessi, H; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int J Pharm 2010, 385, 113–142. [Google Scholar]
- Shaikh, J; Ankola, DD; Beniwal, V; Singh, D; Kumar, MNVR. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci 2009, 37, 223–230. [Google Scholar]
- El-Shabouri, MH. Positively charged nanoparticles for improving the oral bioavailability of Cyclosporin-A. Int J Pharm 2002, 249, 101–108. [Google Scholar]
- Italia, J; Yahya, M; Singh, D; Ravi Kumar, M. Biodegradable nanoparticles improve oral bioavailability of amphotericin b and show reduced nephrotoxicity compared to intravenous Fungizone®. Pharm Res 2009, 26, 1324–1331. [Google Scholar]
- Pan, Y; Li, Y; Zhao, H; Zheng, J; Xu, H; Wei, G; Hao, J; Cui, F. Bioadhesive polysaccharide in protein delivery system: chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int J Pharm 2002, 249, 139–147. [Google Scholar]
- Sarmento, B; Ribeiro, A; Veiga, F; Ferreira, D; Neufeld, R. Oral bioavailability of insulin contained in polysaccharide nanoparticles. Biomacromolecules 2007, 8, 3054–3060. [Google Scholar]
- Leroux, J; Cozens, RM; Roesel, JL; Galli, B; Doelker, E; Gurny, R. PH-sensitive nanoparticles: An effective means to improve the oral delivery of hiv-1 protease inhibitors in dogs. Pharm Res 1996, 13, 485–487. [Google Scholar]
- Kawashima, Y; Yamamoto, H; Takeuchi, H; Kuno, Y. Mucoadhesive dl-lactide/glycolide copolymer nanospheres coated with chitosan to improve oral delivery of elcatonin. Pharm Dev Technol 2000, 5, 77–85. [Google Scholar]
- Mu, L; Feng, SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J Control Release 2003, 86, 33–48. [Google Scholar]
- Mittal, G; Sahana, DK; Bhardwaj, V; Ravi Kumar, MNV. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Control Release 2007, 119, 77–85. [Google Scholar]
- Garcia-Fuentes, M; Torres, D; Alonso, MJ. New surface-modified lipid nanoparticles as delivery vehicles for salmon calcitonin. Int J Pharm 2005, 296, 122–132. [Google Scholar]
- Yang, S; Zhu, J; Lu, Y; Liang, B; Yang, C. Body distribution of camptothecin solid lipid nanoparticles after oral administration. Pharm Res 1999, 16, 751–757. [Google Scholar]
- He, W; Horn, SW; Hussain, MD. Improved bioavailability of orally administered mifepristone from PLGA nanoparticles. Int J Pharm 2007, 334, 173–178. [Google Scholar]
- Zhang, Q; Shen, Z; Nagai, T. Prolonged hypoglycemic effect of insulin-loaded polybutylcyanoacrylate nanoparticles after pulmonary administration to normal rats. Int J Pharm 2001, 218, 75–80. [Google Scholar]
- De, TK; Hoffman, AS. A reverse microemulsion polymerization method for preparation of bioadhesive polyacrylic acid nanoparticles for mucosal drug delivery: Loading and release of timolol maleate. Artif Cells, Blood Substit, Biotechnol 2001, 29, 31–46. [Google Scholar]
- Desgouilles, S; Vauthier, C; Bazile, D; Vacus, J; Grossiord, J; Veillard, M; Couvreur, P. The design of nanoparticles obtained by solvent evaporation: A comprehensive study. Langmuir 2003, 19, 9504–9510. [Google Scholar]
- Budhian, A; Siegel, SJ; Winey, KI. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int J Pharm 2007, 336, 367–375. [Google Scholar]
- Galindo-Rodríguez, SA; Puel, F; Briançon, S; Allémann, E; Doelker, E; Fessi, H. Comparative scale-up of three methods for producing ibuprofen-loaded nanoparticles. Eur J Pharm Sci 2005, 25, 357–367. [Google Scholar]
- Zweers, MLT; Grijpma, DW; Engbers, GHM; Feijen, J. The preparation of monodisperse biodegradable polyester nanoparticles with a controlled size. J Biomed Mater Res Part B 2003, 66B, 559–566. [Google Scholar]
- Byrappa, K; Ohara, S; Adschiri, T. Nanoparticles synthesis using supercritical fluid technology – towards biomedical applications. Adv Drug Deliv Rev 2008, 60, 299–327. [Google Scholar]
- Varshosaz, J; Hassanzadeh, F; Mahmoudzadeh, M; Sadeghi, A. Preparation of cefuroxime axetil nanoparticles by rapid expansion of supercritical fluid technology. Powder Technol 2009, 189, 97–102. [Google Scholar]
- Wang, G; Uludag, H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv 2008, 5, 499–515. [Google Scholar]
- Krauland, AH; Alonso, MJ. Chitosan/cyclodextrin nanoparticles as macromolecular drug delivery system. Int J Pharm 2007, 340, 134–142. [Google Scholar]
- Gan, Q; Wang, T. Chitosan nanoparticle as protein delivery carrier—systematic examination of fabrication conditions for efficient loading and release. Colloids Surf B 2007, 59, 24–34. [Google Scholar]
- Grant, J; Blicker, M; Piquette-Miller, M; Allen, C. Hybrid films from blends of chitosan and egg phosphatidylcholine for localized delivery of paclitaxel. J Pharm Sci 2005, 94, 1512–1527. [Google Scholar]
- Hafner, A; Lovrić, J; Voinovich, D; Filipović-Grčić, J. Melatonin-loaded lecithin/chitosan nanoparticles: Physicochemical characterisation and permeability through caco-2 cell monolayers. Int J Pharm 2009, 381, 205–213. [Google Scholar]
- Ho, EA; Vassileva, V; Allen, C; Piquette-Miller, M. In vitro and in vivo characterization of a novel biocompatible polymer–lipid implant system for the sustained delivery of paclitaxel. J Control Release 2005, 104, 181–191. [Google Scholar]
- Lim Soo, P; Cho, J; Grant, J; Ho, E; Piquette-Miller, M; Allen, C. Drug release mechanism of paclitaxel from a chitosan–lipid implant system: Effect of swelling, degradation and morphology. Eur J Pharm Biopharm 2008, 69, 149–157. [Google Scholar]
- Sonvico, F; Cagnani, A; Rossi, A; Motta, S; Di Bari, MT; Cavatorta, F; Alonso, MJ; Deriu, A; Colombo, P. Formation of self-organized nanoparticles by lecithin/chitosan ionic interaction. Int J Pharm 2006, 324, 67–73. [Google Scholar]
- Zahedi, P; De Souza, R; Piquette-Miller, M; Allen, C. Chitosan–phospholipid blend for sustained and localized delivery of docetaxel to the peritoneal cavity. Int J Pharm 2009, 377, 76–84. [Google Scholar]
- Beun, S; Glorieux, T; Devaux, J; Vreven, J; Leloup, G. Characterization of nanofilled compared to universal and microfilled composites. Dental Mater 2007, 23, 51–59. [Google Scholar]
- Gojny, FH; Wichmann, MHG; Fiedler, B; Schulte, K. Influence of different carbon nanotubes on the mechanical properties of epoxy matrix composites – a comparative study. Compos Sci Technol 2005, 65, 2300–2313. [Google Scholar]
- Gomoll, AH; Fitz, W; Scott, RD; Thornhill, TS; Bellare, A. Nanoparticulate fillers improve the mechanical strength of bone cement. Acta Orthop 2008, 79, 421–427. [Google Scholar]
- Park, JH; Jana, SC. The relationship between nano- and micro-structures and mechanical properties in pmma–epoxy–nanoclay composites. Polymer 2003, 44, 2091–2100. [Google Scholar]
- Rapoport, L; Nepomnyashchy, O; Verdyan, A; Popovitz-Biro, R; Volovik, Y; Ittah, B; Tenne, R. Polymer nanocomposites with fullerene-like solid lubricant. Adv Eng Mater 2004, 6, 44–48. [Google Scholar]
- Saha, MC; Kabir, ME; Jeelani, S. Enhancement in thermal and mechanical properties of polyurethane foam infused with nanoparticles. Mater Sci Eng A 2008, 479, 213–222. [Google Scholar]
- Zhang, MQ; Rong, MZ; Zhang, HB; Friedrich, K. Mechanical properties of low nano-silica filled high density polyethylene composites. Polym Eng Sci 2003, 43, 490–500. [Google Scholar]
- Tajiri, T; Morita, S; Sakamoto, R; Suzuki, M; Yamanashi, S; Ozaki, Y; Kitamura, S. Release mechanisms of acetaminophen from polyethylene oxide/polyethylene glycol matrix tablets utilizing magnetic resonance imaging. Int J Pharm 2010, 395, 147–153. [Google Scholar]
- Warhurst, D; Craig, J; Adagu, I; Meyer, D; Lee, S. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar J 2003, 2, 26. [Google Scholar]
- Pearlman, DA; Case, DA; Caldwell, JW; Ross, WS; Cheatham, TE; DeBolt, S; Ferguson, D; Seibel, G; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun 1995, 91, 1–41. [Google Scholar]
- Golovnya, RV; Zhuravleva, IL; Zenin, SV; Polyakov, VA; Sergeev, GB. Determination of thermodynamic characteristics of complexing of amines with alkyl and aryl phosphates by the NMR method. Russ Chem Bull 1973, 22, 2528–2530. [Google Scholar]
- Kumar, P; Pillay, V; Choonara, YE; Modi, G; Naidoo, D; du Toit, LC. In silico theoretical molecular modeling for alzheimer’s disease: The nicotine-curcumin paradigm in neuroprotection and neurotherapy. Int J Mol Sci 2011, 12, 694–724. [Google Scholar]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Polymer Solution | Addition of L-dopa | Polymer + L-Dopa + Lecithin | Polymer + L-dopa + Lecithin + TPP |
|---|---|---|---|---|
| A22 | 1.17 | 1.31 | 1.36 | 3.15 |
| B3 | 1.17 | 1.34 | 1.40 | 1.73 |
| B6 | 1.18 | 1.36 | 1.41 | 1.78 |
| B9 | 1.13 | 1.28 | 1.28 | 1.68 |
| B12 | 1.14 | 1.19 | 1.23 | 1.78 |
| B13 | 1.14 | 1.19 | 1.23 | 1.78 |
| B18 | 1.13 | 1.28 | 1.28 | 2.37 |
| B19 | 1.18 | 1.36 | 1.41 | 2.82 |
| Polymer Composition | Polymer Solution | Addition of Lecithin | Addition of TPP |
|---|---|---|---|
| EE100 | 0.0135 | 0.5681 | 0.4876 |
| Chitosan | 0.1382 | 3.3501 | 3.5597 |
| EE100 + Chitosan | 0.0589 | 2.7885 | 3.1930 |
| S/N | Formulation Code | Eudragit (mg) | Chitosan (mg) | L-dopa (mg) | Lecithin (mL) | TPP (mg) |
|---|---|---|---|---|---|---|
| 1 | A22 | 150 | 150 | 100 | 1.00 | 250 |
| 2 | B3 | 150 | 50 | 100 | 1.00 | 50 |
| 3 | B6 | 100 | 100 | 100 | 1.00 | 50 |
| 4 | B9 | 200 | – | 100 | 1.00 | 50 |
| 5 | B12 | 50 | 50 | 100 | 1.00 | 100 |
| 6 | B18 | 200 | – | 100 | 1.00 | 150 |
| 7 | B19 | 100 | 100 | 100 | 1.00 | 150 |
| 8 | C0 | – | 200 | 100 | 1.00 | 150 |
| 9 | C1 | – | 200 | 100 | 1.00 | – |
| 10 | B180 | 200 | – | 100 | 1.00 | – |
| 11 | B190 | 100 | 100 | 100 | 1.00 | – |
| S. No. | Parameter | Value |
|---|---|---|
| 1. | Imaging protocol | FSHEF |
| 2. | Requested gain (%) | 1.90 |
| 3. | Signal strength | 71.62 |
| 4. | Average | 2 |
| 5. | Matrix size | 128 |
| 6. | Repetition time (ms) | 1000.00 |
| 7. | Spin Echo Tau (ms) | 6.00 |
| 8. | Image acquired after | 60 min |
| 9. | Total scans | 64 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ngwuluka, N.C.; Pillay, V.; Choonara, Y.E.; Modi, G.; Naidoo, D.; Toit, L.C.d.; Kumar, P.; Ndesendo, V.M.K.; Khan, R.A. Fabrication, Modeling and Characterization of Multi-Crosslinked Methacrylate Copolymeric Nanoparticles for Oral Drug Delivery. Int. J. Mol. Sci. 2011, 12, 6194-6225. https://doi.org/10.3390/ijms12096194
Ngwuluka NC, Pillay V, Choonara YE, Modi G, Naidoo D, Toit LCd, Kumar P, Ndesendo VMK, Khan RA. Fabrication, Modeling and Characterization of Multi-Crosslinked Methacrylate Copolymeric Nanoparticles for Oral Drug Delivery. International Journal of Molecular Sciences. 2011; 12(9):6194-6225. https://doi.org/10.3390/ijms12096194
Chicago/Turabian StyleNgwuluka, Ndidi C., Viness Pillay, Yahya E. Choonara, Girish Modi, Dinesh Naidoo, Lisa C. du Toit, Pradeep Kumar, Valence M.K. Ndesendo, and Riaz A. Khan. 2011. "Fabrication, Modeling and Characterization of Multi-Crosslinked Methacrylate Copolymeric Nanoparticles for Oral Drug Delivery" International Journal of Molecular Sciences 12, no. 9: 6194-6225. https://doi.org/10.3390/ijms12096194
APA StyleNgwuluka, N. C., Pillay, V., Choonara, Y. E., Modi, G., Naidoo, D., Toit, L. C. d., Kumar, P., Ndesendo, V. M. K., & Khan, R. A. (2011). Fabrication, Modeling and Characterization of Multi-Crosslinked Methacrylate Copolymeric Nanoparticles for Oral Drug Delivery. International Journal of Molecular Sciences, 12(9), 6194-6225. https://doi.org/10.3390/ijms12096194