3. Experimental Section
3.1. General Procedures and Instrumentation
All moisture-sensitive reactions were carried out under a static atmosphere of nitrogen. All reagents were of commercial quality. Lipase from Pseudomonas cepacia (PS), Amano Pharmaceuticals Co., Japan, 30 units/mg, was employed in this work. TLC: Merk silica gel 60 F254 plates. Column chromatography (CC): silica gel. GC-MS analyses: HP-6890 gas chromatograph equipped with a 5973 mass detector, using a HP-5MS column (30 m × 0.25 mm, 0.25 μm firm thickness; Hewlett Packard). Chiral GC analyses: DANI-HT-86.10 gas chromatograph; enantiomer excesses determined on a CHIRASIL DEX CB-Column. Optical rotations: Jasco-DIP-181 digital polarimeter. 1H and 13C Spectra: CDCl3 solution at room temperature; Bruker-AC-400 spectrometer at 400 and 100 MHz, respectively; chemical shifts in ppm relative to internal SiMe4 (=0 ppm), J values in Hz. IR spectra were recorded on a Perkin-Elmer 2000 FT-IR spectrometer; films; ν in cm−1. Melting points were measured on a Reichert apparatus, equipped with a Reichert microscope, and are uncorrected. Microanalyses were determined on an analyzer 1106 from Carlo Erba.
3.2. Synthesis of (E)-3-(2,6,6-Trimethylcyclohex-2-en-1-yl)prop-2-en-1-ol 2
2 was prepared according to reported literature [
21] as a colourless oil;
1H NMR (400 MHz, CDCl
3) δ 5.75 (d,
J = 16.0 Hz, 1H), 5.64 (dt,
J = 16.0, 6.0 Hz, 1H), 5.45 (d,
J = 7.0 Hz, 1H), 4.35 (d,
J = 14.0 Hz, 2H), 2.65 (d,
J = 6.2 Hz, 1H), 1.75 (s, 3H), 1.65–1.53 (m, 2H), 1.49–1.41 (m, 2H), 1.31 (br s, 1H), 0.98 (s, 6H).
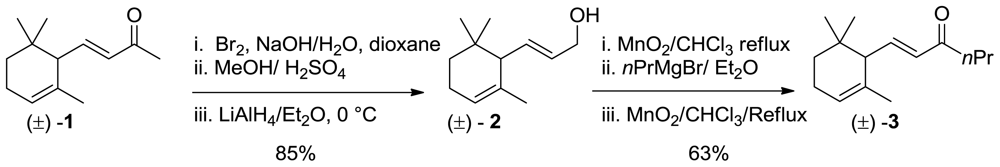
3.3. Synthesis of (E)-1-(2,6,6-Trimethylcyclohex-2-en-1-yl)hex-1-en-3-one (3)
To a mixture of 2 (25 g, 64.4 mmol) in CHCl3 (200 mL), MnO2 (30 g, 345 mmol) was added, and the mixture was stirred at reflux for 6 h. The mixture was then cooled, filtered (using filter paper) and the organic phase was concentrated under reduced pressure to afford an oil (13 g). The latter was dissolved in dry diethyl ether (150 mL) and treated under stirring with an excess of n-propylmagnesium bromide (100 mL of a 1 M solution in ether) keeping the reaction temperature under 5 °C by external cooling (ice bath). The usual work-up afforded crude carbinol that was dissolved in CHCl3 (200 mL) and treated with MnO2 (30 g, 345 mmol) stirring at reflux for 12 h. After filtration and concentration, the crude ketone was purified by chromatography (hexane/Et2O 95:5) and bulb to bulb distillation (oven temperature 105 °C, 0.4 mmHg) to afford pure 3 (8.4 g, 63% yield), 98% chemical purity (GC)) as a colorless oil;1H NMR (400 MHz, CDCl3) δ 6.64 (dd, J = 15.6, 9.6 Hz, 1H), 6.06 (d, J = 15.6 Hz, 1H), 5.48 (bs, 1H), 2.52 (t, J = 7.2 Hz, 2H), 2.27 (d, J = 10 Hz, 1H), 2.04 (m, 1H), 1.67 (ddd, J = 13.4, 6.5, 1.4 Hz, 1H), 1.55 (s, 3H), 1.41 (m, 2H), 1.23 (m, 2H), 0.94 (t, J = 1.7 Hz, 3H), 0.93 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz) δ 212.8, 135.3, 132.6, 121.4, 73.2, 54.5, 40.0, 32.0, 27.9, 27.3, 19.0, 14.3; IR (nujol, cm−1) 1684, 1619, 1554. GC-MS m/z: 221.18 (M+, 1); Anal. Calcd for C15H24O: C, 81.76; H, 10.98.Found: C, 81.75; H, 10.90.
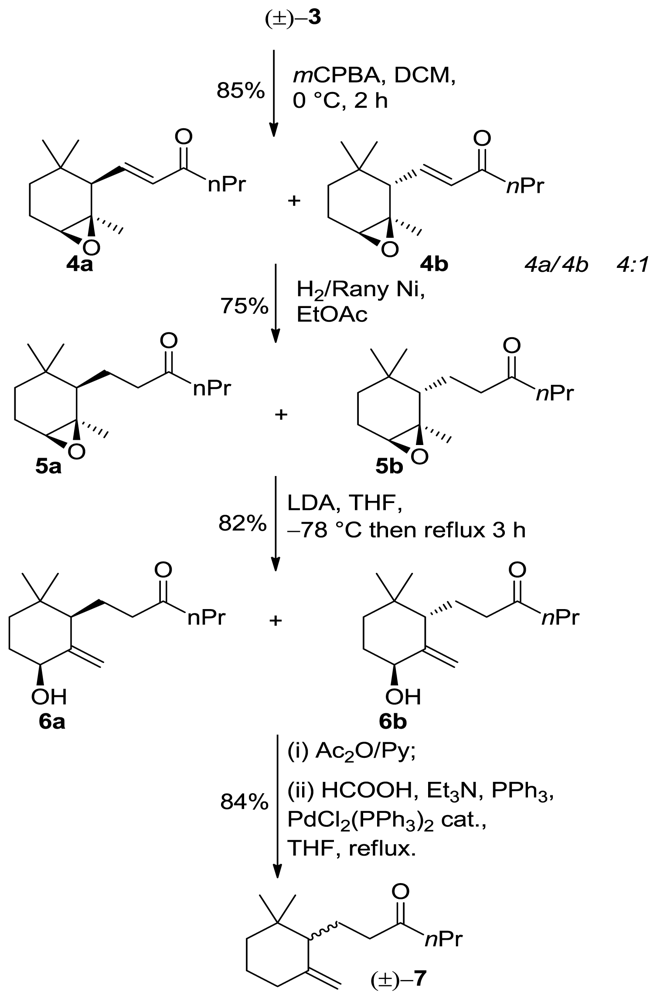
3.4. General Procedure for the Synthesis of Adducts 4a/4b
m-Chloroperbenzoic acid (7.4 g, of 75% wet acid, 32 mmol) was added to a solution of racemic α-ionone isomer 3 (7 g, 30 mmol) in methylene chloride (75 mL) at 0 °C. The reaction mixture was stirred at 0 °C for 2 h and then filtered in order to remove the m-clorobenzoic acid precipitate. The organic phase was washed with saturated Na2SO3 solution (50 mL) and saturated NaHCO3 solution (50 mL), respectively, dried over Na2SO4 and concentrated under reduced pressure. The residue was chromatographed on a silica gel column (hexane/Et2O 9:1) to give the corresponding α-epoxy-derivatives.
3.4.1. (E)-1-[(1R,2R,6S)-1,3,3-Trimethyl-7-oxabicyclo[4.1.0]heptan-2-yl]hex-1-en-3-one (4a)
1H NMR (400 MHz, CDCl3) δ 5.45-5.41 (m, 1H), 5.39 (bs, 1H), 5.48 (bs, 1H), 4.10 (bs, 1H), 2.10 (d, J = 6.4 Hz, 1H), 2.00 (m, 2H), 1.59 (dd, J = 4.4, 1.6 Hz, 1H), 1.54 (m, 2H), 1.40 (m, 2H), 1.37 (s, 3H), 0.94 (td, J = 7.2, 1.6 Hz, 3H), 0.93 (s, 3H), 0.81(s, 3H); 13C NMR (100 MHz) δ 212.8, 135.3, 132.6, 121.4, 73.2, 54.5, 40.0, 32.0, 27.9, 27.3, 19.0, 14.3; IR (nujol, cm−1) 1684; GC-MS m/z: 237.18 (M+, 1); Anal. Calcd for C15H24O2: C, 76.23; H, 10.24. Found: C, 76.34; H, 10.55.
3.4.2. (E)-1-[(1R,2S,6S)-1,3,3-Trimethyl-7-Oxabicyclo[4.1.0]heptan-2-yl]hex-1-en-3-One (4b)
1H NMR (400 MHz, CDCl3) δ 5.47-5.43 (m, 1H), 5.41 (bs, 1H), 5.52 (bs, 1H), 4.13 (bs, 1H), 2.14 (d, J = 6.4 Hz, 1H), 2.00 (m, 2H), 1.61 (dd, J = 4.4, 1.6 Hz, 1H), 1.54 (m, 2H), 1.45 (m, 2H), 1.37 (s, 3H), 0.94 (td, J = 7.2, 1.6 Hz, 3H), 0.93 (s, 3H), 0.81(s, 3H); 13C NMR (100 MHz) δ 212.4, 135.1, 132.3, 121.1, 73.3, 54.1, 40.2, 32.5, 27.9, 27.3, 19.1, 14.1; IR (nujol, cm−1) 1680; GC-MS m/z: 237.18 (M+, 1); Anal. Calcd for C15H24O2: C, 76.23; H, 10.24. Found: C, 76.34; H, 10.55.
3.5. General Procedure for the Synthesis of Adducts 5a/5b
H2 (360 mL, 1 equiv) was adsorbed of a solution of 4a or 4b (4.5 g, 19mmol, 1 equiv) in EtOAc (150 mL) and excess of Rany-Ni for 2 h. The reaction mixture was filtered off and concentrated under reduced pressure. The residue was chromatographed on a silica gel column (hexane/EtOAc 9:1) to give the corresponding product.
3.5.1. 1-[(1R,2R,6S)-1,3,3-Trimethyl-7-Oxabicyclo[4.1.0]heptan-2-yl]hexan-3-One (5a)
(3.4 g, 74% yield, 98% chemical purity (GC)) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 2.85 (bs, 1H), 2.67–2.59 (m, 2H), 2.43–2.31 (m, 2H), 1.86–1.74 (m, 3H), 1.68–1.58 (m, 2H), 1.56–1.43 (m, 2H), 1.34–1.27 (m, 2H), 1.24 (s, 3H), 0.86 (t, J = 14.8 Hz, 3H), 0.81 (s, 3H), 0.76 (s, 3H); 13C NMR (100 MHz) δ 2.11.4, 60.2, 46.5, 45.0, 42.3, 31.8, 28.4, 27.9, 27.5, 27.0, 26.9, 22.3, 21.8, 17.6, 14.1; IR (nujol, cm−1) 1684, 1109, 990. GC-MS m/z: 239.19 (M+, 1); Anal. Calcd for C15H26O2: C, 75.58; H, 10.99. Found: C, 75.55; H, 11.02.
3.5.2. 1-[(1R,2S,6S)-1,3,3-Trimethyl-7-Oxabicyclo[4.1.0]heptan-2-yl]hexan-3-One (5b)
1H NMR (400 MHz, CDCl3) δ 2.83 (bs, 1H), 2.64–2.57 (m, 2H), 2.45–2.33 (m, 2H), 1.83–1.75 (m, 3H), 1.67–1.55 (m, 2H), 1.52–1.45 (m, 2H), 1.32–1.25 (m, 2H), 1.25 (s, 3H), 0.87 (t, J = 14.8 Hz, 3H), 0.82 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz) δ 211.2, 60.3, 46.4, 45.2, 42.2, 31.9, 28.5, 27.9, 27.3, 27.1, 26.9, 22.3, 21.8, 17.6, 14.1; IR (nujol, cm−1) 1683, 1109, 990. GC-MS m/z: 239.19 (M+, 1); 55 (15). Anal. Calcd for C15H26O2: C, 75.58; H, 10.99. Found: C, 75.55; H, 11.02.
3.6. General Procedure for the Synthesis of Adducts 6a/6b
nBuLi (3.0 mL of a 10 M solution in hexane) was added dropwise to a cooled (−78 °C) solution of iPr2NH (5.3 g, 50 mmol) in dry THF (90 mL) under nitrogen. The mixture was stirred at this temperature for 30 min. then a solution of the epoxide 5a or 5b (2.5 g, 10 mmol) in dry THF (20 mL) was added dropwise. The reaction was gradually warmed to r.t (1 h) and then was heated under reflux until no more starting epoxide was detected by TLC analysis (3 h). After cooling to room temperature, the mixture was poured into a mixture of crushed ice and 5% HCl solution. (80 mL) and extracted with Et2O (3 × 200 mL). The organic phase was successively washed with saturated aqueous NH4Cl solution (100 mL), brine, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography (eluting from hexane/AcOEt 9:1 to hexane/AcOEt 1:1) to give allylic alcohol.
3.6.1. 1-((1R,5S)-5-Hydroxy-2,2-Dimethyl-6-Methylenecyclohexyl)hexan-3-One (6a)
6a (1.95 g, 82% yield, 98% chemical purity (GC)) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 5.16 (bs, 1H), 4.64 (bs, 1H), 3.94 (bs, 1H), 2.56–2.48 (m, 2H), 2.34–2.26 (m, 2H), 1.91–1.81 (m, 2H), 1.75–1.67 (m, 2H), 1.64–1.54 (m, 2H), 1.51–1.39 (m, 2H), 1.29–1.24 (m, 2H), 0.97 (s, 3H), 0.89 (t, J = 14.8 Hz, 3H), 0.73 (s, 3H); 13C NMR (100 MHz) δ 2.11.4, 60.2, 46.5, 45.0, 42.3, 31.8, 28.4, 27.9, 27.5, 27.0, 26.9, 22.3, 21.8, 17.6, 14.1; IR (nujol, cm−1) 3334, 1684, 1619. GC-MS m/z: 239.19 (M+, 1); Anal. Calcd for C15H26O2: C, 75.58; H, 10.99. Found: C, 75.65; H, 10.99.
3.6.2. 1-((1S,5S)-5-Hydroxy-2,2-Dimethyl-6-Methylenecyclohexyl)hexan-3-One (6b)
1H NMR (400 MHz, CDCl3) δ 5.18 (bs, 1H), 4.66 (bs, 1H), 3.95 (bs, 1H), 2.58–2.47 (m, 2H), 2.33–2.28 (m, 2H), 1.92–1.83 (m, 2H), 1.75–1.67 (m, 2H), 1.64–1.54 (m, 2H), 1.53–1.38 (m, 2H), 1.29–1.24 (m, 2H), 0.98 (s, 3H), 0.90 (t, J = 14.8 Hz, 3H), 0.75 (s, 3H); 13C NMR (100 MHz) δ 2.11.4, 60.2, 46.5, 45.0, 42.3, 31.8, 28.4, 27.9, 27.5, 27.0, 26.9, 22.3, 21.8, 17.6, 14.1; IR (nujol, cm−1) 3335, 1681, 1620. GC-MS m/z: 239.19 (M+, 1); Anal. Calcd for C15H26O2: C, 75.58; H, 10.99. Found: C, 75.65; H, 10.99.
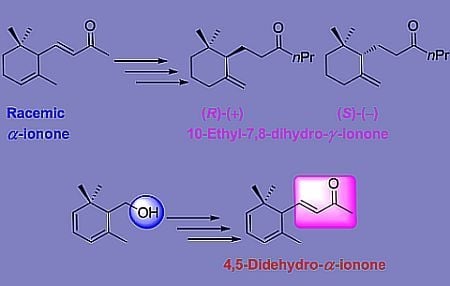
3.7. General Procedure for Reduction of Allylic Alcohol 6a to 10-Ethyl-7,8-dihydro-γ-ionone Isomers 7
A sample of compound 6a (500 mg, 2.0 mmol) was converted into the corresponding acetate by treatment with pyridine (5 mL) and Ac2O (5 mL) at room temperature for 24 h. The crude product was added to a solution of formic acid (180 mg, 4.0 mmol), Et3N (440 mg, 4.4 mmol), (PPh3)2PdCl2 (41 mg, 0.06 mmol) and triphenylphosphine (75 mg, 0.3 mmol) in dry THF (20 mL). The mixture was refluxed under a static nitrogen atmosphere until reduction was complete (2 h, TLC analysis). The reaction was then diluted with ether (30 mL) and washed with water (20 mL), 5% HCl solution (20 mL), saturated aqueous NaHCO3 solution (20 mL), and brine. The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by chromatography (hexane/Et2O 95:5) and bulb-to-bulb distillation to give γ-ionone isomers 7 (84% yield, 97% isomeric purity).
10-Ethyl-7,8-dihydro-γ-ionone (±)-7 (99% chemical purity (GC)) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 4.75 (bs, 1H), 4.50 (d, J = 2.4 Hz,1H), 2.38–2.30 (m, 2H), 2.27–2.18(m, 2H), 2.05–1.94(m, 2H), 1.84–1.76(m, 3H), 1.70–1.55 (m, 2H), 1.54–1.46 (m, 2H), 1.30–1.17 (m, 2H), 0.91 (s, 3H), 0.89 (t, J = 7.2 Hz, 3H), 0.86 (s, 3H); 13C NMR (100 MHz) δ 211.8, 149.5, 109.7, 53.9, 45.3, 41.7, 36.2, 35.2, 32.5, 28.6, 26.8, 23.9, 20.7, 17.7, 14.4; IR (nujol, cm−1) 1684, 1619. GC-MS m/z (rel intensity) 223.20 (M+, 1); Anal. Calcd for C15H26O: C, 81.02; H, 11.79. Found: C, 81.14; H, 11.67.
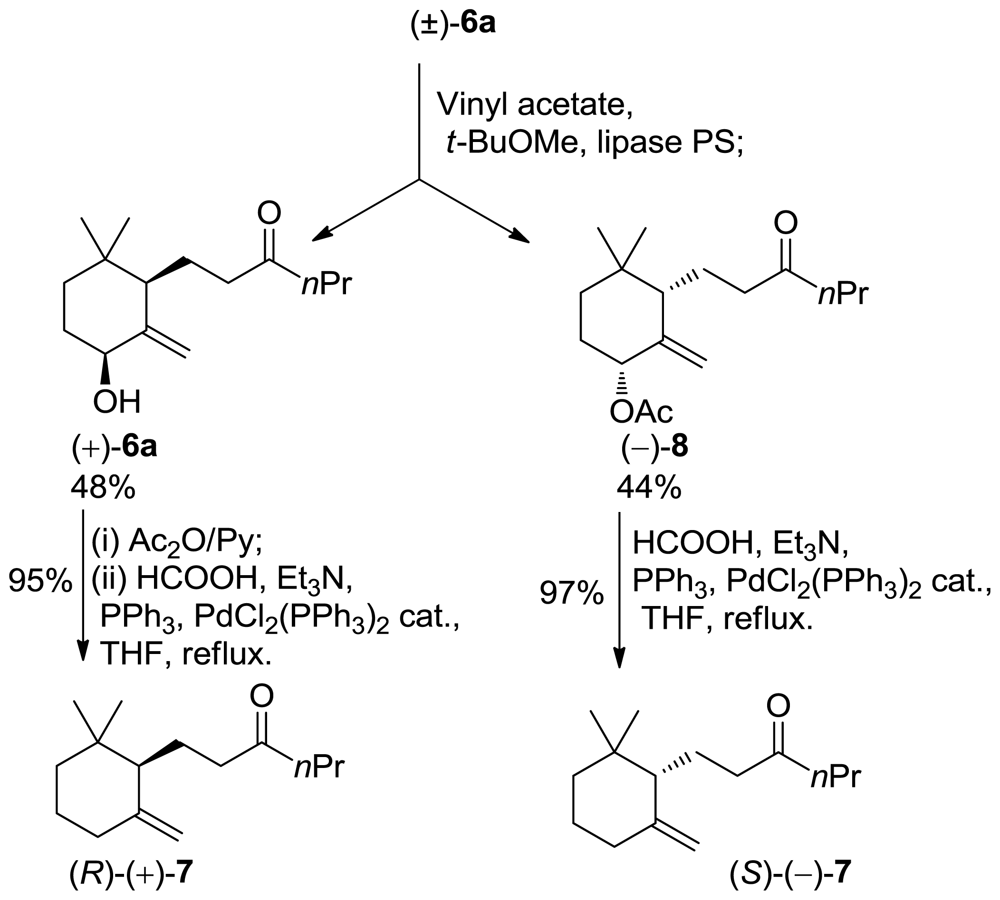
3.9. Synthesis of Enantioenriched 10-Ethyl-7,8-dihydro-γ-ionone Isomers
The above obtained compounds (−)-8, (+)-8 were submitted to the reductive deoxygenation procedure described above in Section 3.7 to afford 10-ethyl-7,8-dihydro-γ-ionone isomers (−)-7, (+)-7 respectively. The latter compounds showed the following analytical data.
3.9.1. (S)-1-(2,2-Dimethyl-6-Methylenecyclohexyl)hexan-3-One (−)-7
(Colorless oil, 99% chemical purity, 97% regioisomeric purity (chiral GC); [α]24D = −20.2° (c = 1.0 g/dL, CHCl3); IR, 1H NMR, MS: in accordance with that of (±)-7.
3.9.2. (R)-1-(2,2-Dimethyl-6-methylenecyclohexyl)hexan-3-one (+)-7
(Colorless oil; 99% chemical purity, 94% regioisomeric purity (chiral GC); [α]24D= +17.3° (c = 1.0 g/dL, CHCl3); IR, 1H NMR, MS: in accordance with that of (±)-7.
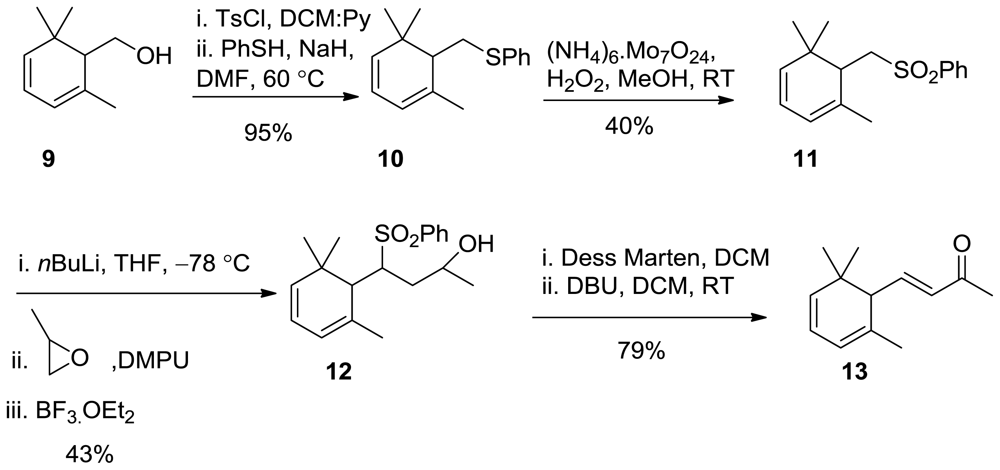
3.10. Synthesis of Phenyl((2,6,6-Trimethylcyclohexa-2,4-Dien-1-yl)methyl)Sulfane 10
In a 100 mL round bottom flask, 9 (3.5 g, 2.3 mmol) was dissolved in a mixture of Py:DCM (1:1, 12 mL). TsCl (5.7 g, 3 mmol, 1.3 equiv) was added into reaction mixture and stirred at room temperature for 1.5 h. Water was added and extracted with Et2O (3 × 100 mL); the organic phase was washed with sat. NaHCO3, then 5% HCL, brine, dried over Na2SO4, and concentrated under reduced pressure, and used for the next step without further purification in quantitative yield.
In a round bottom flask, thiophenol (2.6 g, 23.54mmol, 1.2 equiv) was dissolved in DMF (30 mL), NaH (1.4 g 60%, 3 equiv) was added portion wise. After 15 min, TsCl (6.2 g, 19.62 mmol) was added. The reaction mixture was stirred at room temperature for 1 h, then at 60 °C for 2.5 h, then cooled to room temperature 0.1 N NaOH (30 mL) was added and extracted with Et2O, the organic phase was washed with water, brine, dried over Na2SO4, and concentrated under reduced pressure, and the crude was purified by CC using nhexane:EtOAc 100:2; 4.8 g, 95% yield.
1H NMR (400 MHz, CDCl3) δ 7.41–7.23 (m, 5H), 5.20–5.87 (m, 3H), 3.58–3.95 (m, 2H), 2.8 (t, J = 10.4Hz, 1H), 1.85 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); 13C NMR (100 MHz) δ 155.8, 154.9, 153.5, 128.9, 125.4, 117.6, 114.7, 53.3, 38.7, 33.2, 24.4, 22.3 IR (nujol, cm−1) 1680, 1606, 1454, 1100, 990. GC-MS m/z: 245.13 (M+, 1); Anal. Calcd for C16H20S: C, 78.63; H, 8.25. Found: C, 78.65; H, 8.35.
3.11. Synthesis of (((2,6,6-Trimethylcyclohexa-2,4-dien-1-yl)Methyl)Sulfonyl)Benzene (11)
In a round bottom flask, 10 (4.5 g, 18.44 mmol) was dissolved in MeOH (30 mL) and cooled at 0 °C. H2O2 (4 mL of 30%), and (NH4)2MoO4 (5.4 g, 22.12 mmol, 1.2 equiv) were added. The reaction mixture was warmed to room temperature, and stirred overnight. Na2S2O5 (solid ~ calculated 5 g) was added in order to remove the excess oxidant. Extracted with DCM, dried over Na2SO4, and concentrated under reduced pressure, and the crude was purified by CC using nhexane:EtOAc 100:10; 1.15 g, 40% yield.1H NMR (400 MHz, CDCl3) δ 7.43–7.25 (m, 5H), 5.89–5.21 (m, 3H), 3.98–3.68 (m, 2H), 2.8 (t, J = 10.4Hz, 1H), 1.86 (s, 3H), 1.05 (s, 3H), 0.95 (s, 3H); 13C NMR (100 MHz) δ 155.8, 154.9, 153.5, 128.9, 125.4, 117.6, 114.7, 53.3, 38.7, 33.2, 24.4, 22.3 IR (nujol, cm−1) 1680, 1606, 1454, 1100, 990. GC-MS m/z: 277.12 (M+, 1); Anal. Calcd for C16H20O2S: C, 69.53; H, 11.58. Found: C, 69.54; H, 11.60.
3.12. Synthesis of 4-(Phenylsulfonyl)-4-(2,6,6-Trimethylcyclohexa-2,4-Dien-1-yl)butan-2-ol (12)
In a three neck 100 mL round bottom flask, nBuLi (0.4 mL of a 10 M solution in hexane) was added dropwise to a cooled (−78 °C) solution of 11 (1.15 g, 4.16 mmol) in dry THF (25 mL) under nitrogen, then stirred for 30 min. Subsequently, a mixture of propylene epoxide (1.6 mL) in DMPU [1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone] (5 mL) was added, then BF3OEt2 (3 mL) was added and the reaction mixture was stirred for 5 h at −78 °C. The reaction mixture was allowed to warm up at room temperature and stirred overnight. Water (100 mL) was added and extracted with DCM (3 × 100 mL); the organic phase was washed with brine, dried over Na2SO4, and concentrated under reduced pressure, and the crude was purified by CC using nhexane:EtOAc 10:3 to 5:5 to afford 12. (N.B: recovered starting material 500 mg)
(600 mg, 43% yield, 98% chemical purity (GC)) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.84–7.82 (m, 2h, Ph), 7.58–7.48 (m, 3h, Ph), 5.81 (d, J = 15.8 Hz, 1H), 5.67 (d, J = 15.8 Hz, 1H), 5.35–5.25(m, 1H), 3.77 (m, 1H), 3.59 (bs, 1H), 3.11(m, 1H), 2.54(d, J = 15.8 Hz, 1H), 2.28(t, J = 4.8 Hz, 1H), 2.09(s, 3H), 1.31 (bs, 3H), 1.29 (bs, 3H), 0.95 (d, J = 6 Hz, 1H) 0.89 (s, 3H); 13C NMR (100 MHz) δ 153.9, 144.7, 137.7, 133.8, 129.9, 123.4, 117.9, 64.8, 57.9, 45.1, 36.7, 25.2, 23.5; IR (nujol, cm−1) 3360, 1684, 1619, 1454, 1109, 990. GC-MS m/z: 335.16 (M+, 1); Anal. Calcd for C19H26O3S: C, 68.23; H, 7.84; S, 9.59. Found: C, 68.35; H, 7.85; S, 9.62.
3.13. Synthesis of (E)-4-(2,6,6-Trimethylcyclohexa-2,4-Dien-1-yl)but-3-en-2-One (13)
In a round bottom flask, 12 (0.5 g, 0.15 mmol) was dissolved in DCM (10 mL). Dess Martin (1.3 equiv, 0.82 g, 0.19 mmol) was added portion wise. After the reaction was stirred at room temperature for 2 h, Na2S2O5 (solid~calculated 0.5 g) was added in order to remove the excess oxidant. Extracted with DCM. The organic phase was washed with water, brine, dried over Na2SO4, and concentrated under reduced pressure. The crude was dissolved in DCM (5 mL), and DBU (0.1 mL) was added. The reaction was stirred at room temperature for 30 min, and the reaction mixture was washed with water, brine, dried over Na2SO4, and concentrated under reduced pressure. The crude was purified by CC using EtOAc: nhexane 7:3.
(0.15 g, 79% yield, 98% chemical purity (GC)) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 6.65 (dd, J = 15.6, 9.6 Hz, 1H), 6.06 (d, J = 15.6 Hz, 1H), 5.89–5.21 (m, 3H), 2.8 (t, J = 10.4Hz, 1H), 2.27(s, 3H), 1.86 (s, 3H), 1.05 (s, 3H), 0.95 (s, 3H); 13C NMR (100 MHz) δ 206.9, 156.2, 144.7, 133.7, 119.7, 117.9, 60.5 30.6, 28.3, 26.2. IR (nujol, cm−1) 1684, 1619, 1454, 1109, 990. GC-MS m/z: 191.14 (M+, 1); Anal. Calcd for C13H18O: C, 82.06; H, 9.53. Found: C, 82.11; H, 9.54.



{kind=link}
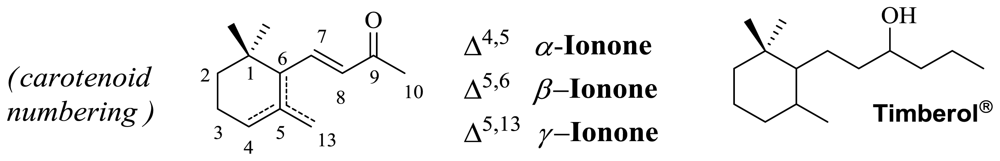{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




