Nec-1 Enhances Shikonin-Induced Apoptosis in Leukemia Cells by Inhibition of RIP-1 and ERK1/2

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
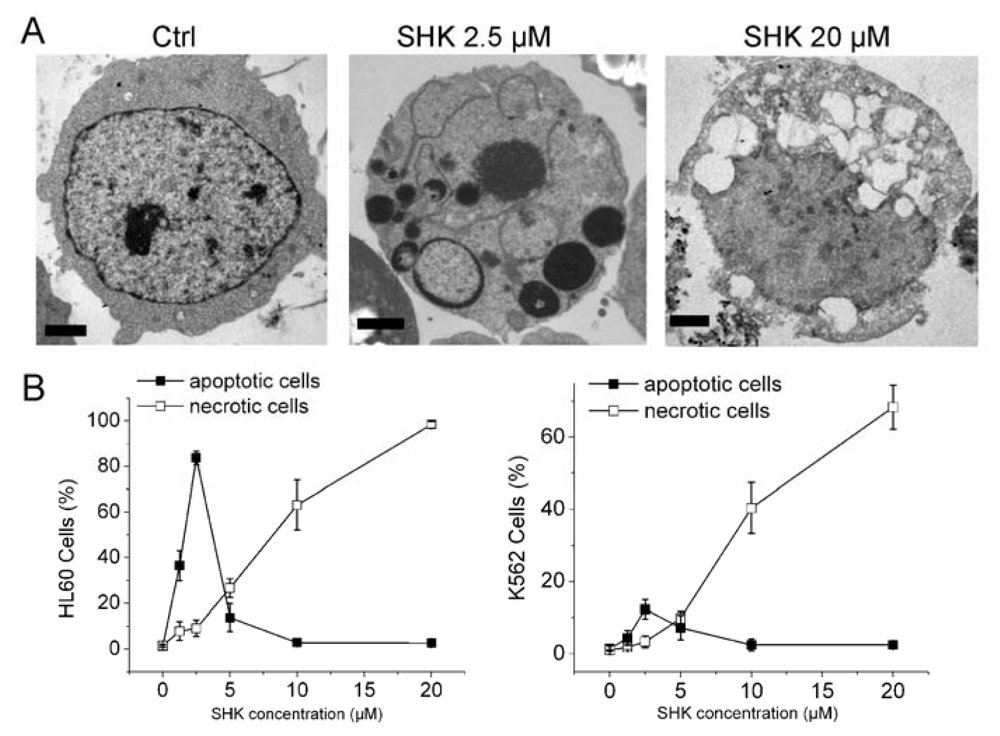
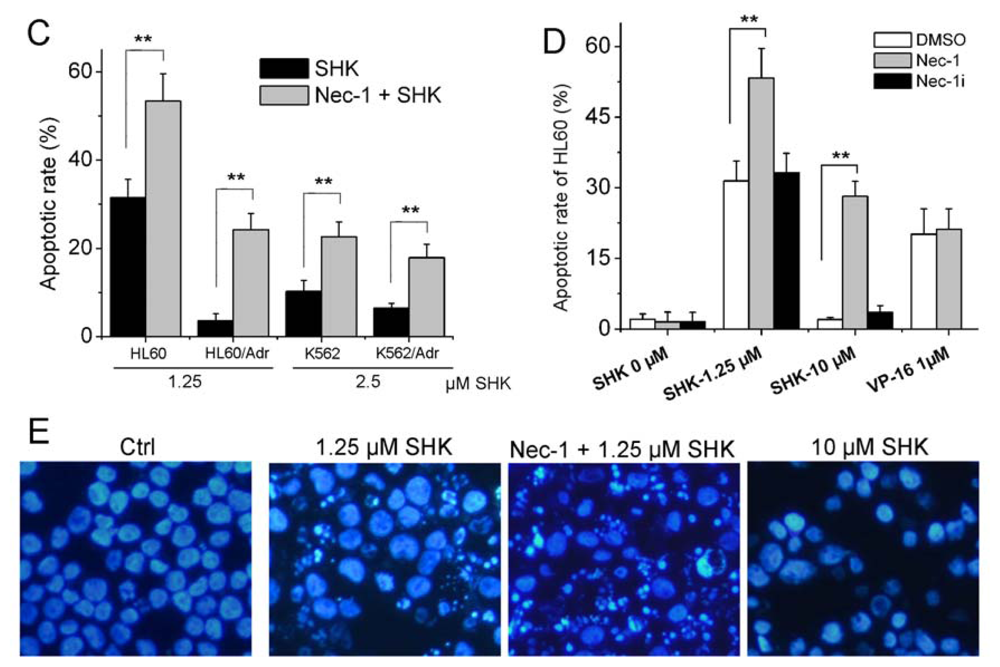
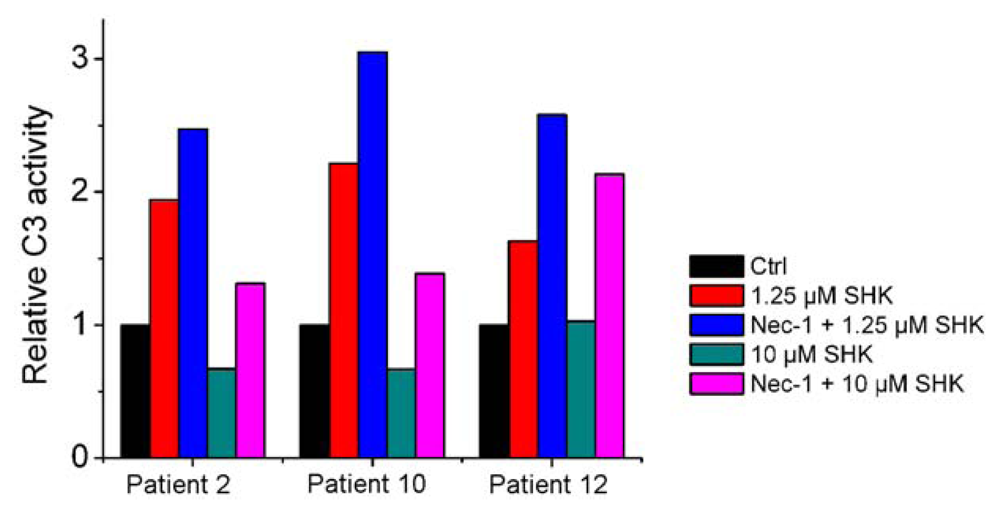
2.1.1. Nec-1 Enhances Shikonin-Induced Apoptosis in both Leukemia Cell Lines and Primary Leukemia Cells
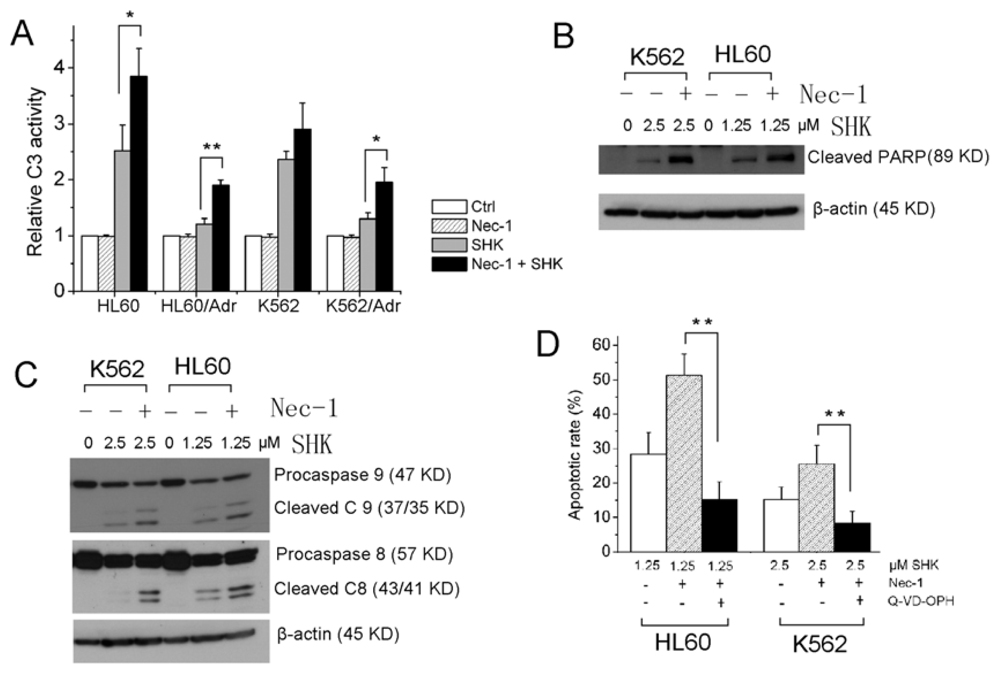
2.1.2. Nec-1 Enhances Shikonin-Induced Apoptosis through Increasing Caspases Activation in Leukemia Cells
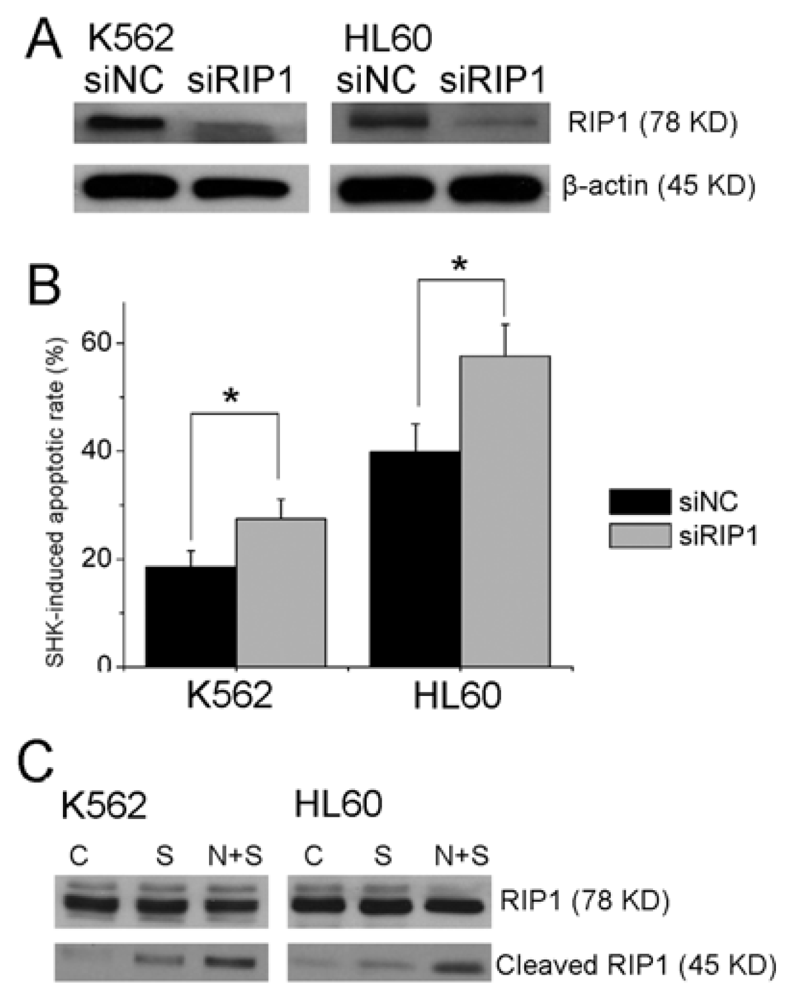
2.1.3. Knockdown of RIP1 Sensitizes Shikonin Induced Apoptosis
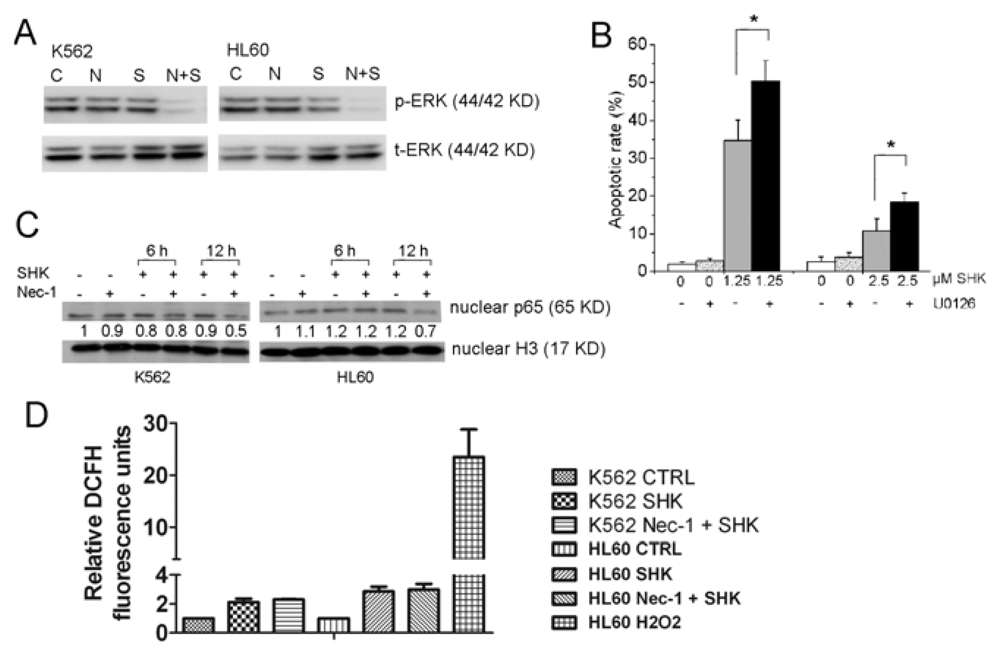
2.1.4. ERK1/2, but Not NF-κB and ROS Are Involved in the Nec-1 Enhancement of Apoptosis
2.2. Discussion
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Cell Cultures
3.3. Primary Human Leukemia Cells
3.4. Treatment of Cells with Shikonin in the Presence or Absence of Nec-1
3.5. Electron Microscopy
3.6. Vital Dye Exclusion Assay and Hoechst-Staining
3.7. Measurement of Caspase-3 Activity
3.8. Western Blot Analysis
3.9. Small Interfering RNA Knockdown of RIP1
3.10. Reactive Oxygen Species Detection
3.11. Statistical Analyses
4. Conclusions
Acknowledgments
- Author ContributionsConceived and designed the experiments: W.H., H.P. Performed the experiments: W.H., J.X., Y.F., Z.W. Analyzed the data: W.H., H.P. Wrote the paper: W.H.
- Financial DisclosureThis work was supported by the National Natural Science Foundation of China (grant number 30901740, http://www.nsfc.gov.cn), China National Ministry of Education Grant (grant number 20090101120124, http://www.moe.edu.cn), Zhejiang Natural Sciences Foundation Grant (grant number Y2090166, http://www.zjnsf.gov.cn), and Research Projects of Science Technology Department of Zhejiang Province (grant number 2010c33168, http://www.zjinfo.gov.cn) to W. Han. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol 2010, 11, 700–714. [Google Scholar]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 2005, 1, 112–119. [Google Scholar]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol 2008, 4, 313–321. [Google Scholar]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar]
- Smith, C.C.; Davidson, S.M.; Lim, S.Y.; Simpkin, J.C.; Hothersall, J.S.; Yellon, D.M. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc. Drugs Ther 2007, 21, 227–233. [Google Scholar]
- Li, Y.; Yang, X.; Ma, C.; Qiao, J.; Zhang, C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat’s cultured cortical neurons. Neurosc. Lett 2008, 447, 120–123. [Google Scholar]
- Han, W.; Li, L.; Qiu, S.; Lu, Q.; Pan, Q.; Gu, Y.; Luo, J.; Hu, X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer Ther 2007, 6, 1641–1649. [Google Scholar]
- Ahn, B.Z.; Baik, K.U.; Kweon, G.R.; Lim, K.; Hwang, B.D. Acylshikonin analogues: Synthesis and inhibition of DNA topoisomerase-I. J. Med. Chem 1995, 38, 1044–1047. [Google Scholar]
- Bailly, C. Topoisomerase I poisons and suppressors as anticancer drugs. Curr. Med. Chem 2000, 7, 39–58. [Google Scholar]
- Kim, S.H.; Kang, I.C.; Yoon, T.J.; Park, Y.M.; Kang, K.S.; Song, G.Y.; Ahn, B.Z. Antitumor activities of a newly synthesized shikonin derivative, 2-hyim-DMNQ-S-33. Cancer Lett 2001, 172, 171–175. [Google Scholar]
- Nakaya, K.; Miyasaka, T. A shikonin derivative, beta-hydroxyisovalerylshikonin, is an ATP-non-competitive inhibitor of protein tyrosine kinases. Anticancer Drugs 2003, 14, 683–693. [Google Scholar]
- Xuan, Y.; Hu, X. Naturally-occurring shikonin analogues—A class of necroptotic inducers that circumvent cancer drug resistance. Cancer Lett 2009, 274, 233–242. [Google Scholar]
- Yang, H.; Zhou, P.; Huang, H.; Chen, D.; Ma, N.; Cui, Q.C.; Shen, S.; Dong, W.; Zhang, X.; Lian, W.; et al. Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int. J. Cancer 2009, 124, 2450–2459. [Google Scholar]
- Chang, I.C.; Huang, Y.J.; Chiang, T.I.; Yeh, C.W.; Hsu, L.S. Shikonin induces apoptosis through reactive oxygen species/extracellular signal-regulated kinase pathway in osteosarcoma cells. Biol. Pharm. Bull 2010, 33, 816–824. [Google Scholar]
- Han, W.; Xie, J.; Li, L.; Liu, Z.; Hu, X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis 2009, 14, 674–686. [Google Scholar]
- Festjens, N.; Vanden Berghe, T.; Cornelis, S.; Vandenabeele, P. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ 2007, 14, 400–410. [Google Scholar]
- Cho, Y.; McQuade, T.; Zhang, H.; Zhang, J.; Chan, F.K. RIP1-Dependent and independent effects of necrostatin-1 in necrosis and T cell activation. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 1999, 13, 2514–2526. [Google Scholar]
- Vandenabeele, P.; Declercq, W.; van Herreweghe, F.; Vanden Berghe, T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal 2010, 3. [Google Scholar] [CrossRef]
- Galluzzi, L.; Aaronson, S.A.; Abrams, J.; Alnemri, E.S.; Andrews, D.W.; Baehrecke, E.H.; Bazan, N.G.; Blagosklonny, M.V.; Blomgren, K.; Borner, C.; et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 2009, 16, 1093–1107. [Google Scholar]
- Balmanno, K.; Cook, S.J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ 2009, 16, 368–377. [Google Scholar]
- Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic B-RafV600E inhibits apoptosis and promotes ERK-dependent inactivation of Bad and Bim. J. Biol. Chem 2008, 283, 22128–22135. [Google Scholar]
- Ewings, K.E.; Wiggins, C.M.; Cook, S.J. Bim and the pro-survival Bcl-2 proteins: Opposites attract, ERK repels. Cell Cycle 2007, 6, 2236–2240. [Google Scholar]
- Vanlangenakker, N.; Vanden Berghe, T.; Bogaert, P.; Laukens, B.; Zobel, K.; Deshayes, K.; Vucic, D.; Fulda, S.; Vandenabeele, P.; Bertrand, M.J. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 2010, 18, 656–665. [Google Scholar]
- Bertrand, M.J.; Vandenabeele, P. RIP1’s function in NF-kappaB activation: From master actor to onlooker. Cell Death Differ 2010, 17, 379–380. [Google Scholar]
- Ting, A.T.; Pimentel-Muinos, F.X.; Seed, B. RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J 1996, 15, 6189–6196. [Google Scholar]
- Rebe, C.; Cathelin, S.; Launay, S.; Filomenko, R.; Prevotat, L.; L’Ollivier, C.; Gyan, E.; Micheau, O.; Grant, S.; Dubart-Kupperschmitt, A.; et al. Caspase-8 prevents sustained activation of NF-κB in monocytes undergoing macrophagic differentiation. Blood 2007, 109, 1442–1450. [Google Scholar]
- Vande Walle, L.; Wirawan, E.; Lamkanfi, M.; Festjens, N.; Verspurten, J.; Saelens, X.; Vanden Berghe, T.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi cleaves RIP1 during apoptosis of Ba/F3 cells induced by growth factor withdrawal. Cell Res 2010, 20, 421–433. [Google Scholar]
- Biton, S.; Ashkenazi, A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-α feedforward signaling. Cell 2011, 145, 92–103. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sex | Age | Diagnosis | SHKconc. (μM) | Apoptotic rate (SHK) | Apoptotic rate (SHK + Nec-1) | p value (SHK vs. SHK + Nec-1) |
|---|---|---|---|---|---|---|---|
| 1 | M | 74 | AML | 2.5 | 15.9 ± 2.5 | 24.3 ± 4.8 | 0.05669 |
| 2 | F | 49 | AML | 1.25 | 6.7 ± 3.7 | 20.6 ± 1.7 | 0.00398 ** |
| 3 | M | 24 | AML | 2.5 | 10.1 ± 4.5 | 18.8 ± 3.0 | 0.05162 |
| 4 | M | 28 | CML | 2.5 | 9.3 ± 2.8 | 15.2 ± 3.7 | 0.09042 |
| 5 | F | 54 | CML | 1.25 | 7.5 ± 3.2 | 15.4 ± 2.1 | 0.02372 * |
| 6 | M | 33 | CML | 1.25 | 10.5 ± 2.4 | 19.9 ± 3.2 | 0.01594 * |
| 7 | F | 35 | CML | 2.5 | 11.5 ± 1.9 | 18.5 ± 3.2 | 0.03051 * |
| 8 | M | 43 | CML | 2.5 | 13.1 ± 4.6 | 18.6 ± 3.7 | 0.18326 |
| 9 | M | 53 | AML | 1.25 | 10.1 ± 1.0 | 13.3 ± 2.8 | 0.13721 |
| 10 | F | 18 | AML | 1.25 | 11.0 ± 3.6 | 24.8 ± 2.8 | 0.0065 ** |
| 11 | M | 55 | CML | 2.5 | 15.1 ±3.0 | 26.2 ± 4.0 | 0.01778 * |
| 12 | M | 66 | CML | 2.5 | 13.8 ± 1.8 | 23.1 ± 2.8 | 0.00849 ** |
| 13 | M | 6 | AML | 1.25 | 23.3 ± 5.7 | 36.8 ± 7.2 | 0.0629 |
| 14 | F | 62 | CML | 1.25 | 35.8 ± 6.6 | 78.1 ± 7.7 | 0.00192 ** |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Han, W.; Xie, J.; Fang, Y.; Wang, Z.; Pan, H. Nec-1 Enhances Shikonin-Induced Apoptosis in Leukemia Cells by Inhibition of RIP-1 and ERK1/2. Int. J. Mol. Sci. 2012, 13, 7212-7225. https://doi.org/10.3390/ijms13067212
Han W, Xie J, Fang Y, Wang Z, Pan H. Nec-1 Enhances Shikonin-Induced Apoptosis in Leukemia Cells by Inhibition of RIP-1 and ERK1/2. International Journal of Molecular Sciences. 2012; 13(6):7212-7225. https://doi.org/10.3390/ijms13067212
Chicago/Turabian StyleHan, Weidong, Jiansheng Xie, Yong Fang, Zhanggui Wang, and Hongming Pan. 2012. "Nec-1 Enhances Shikonin-Induced Apoptosis in Leukemia Cells by Inhibition of RIP-1 and ERK1/2" International Journal of Molecular Sciences 13, no. 6: 7212-7225. https://doi.org/10.3390/ijms13067212
APA StyleHan, W., Xie, J., Fang, Y., Wang, Z., & Pan, H. (2012). Nec-1 Enhances Shikonin-Induced Apoptosis in Leukemia Cells by Inhibition of RIP-1 and ERK1/2. International Journal of Molecular Sciences, 13(6), 7212-7225. https://doi.org/10.3390/ijms13067212