Molecular Self-Assembly at Metal-Electrolyte Interfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
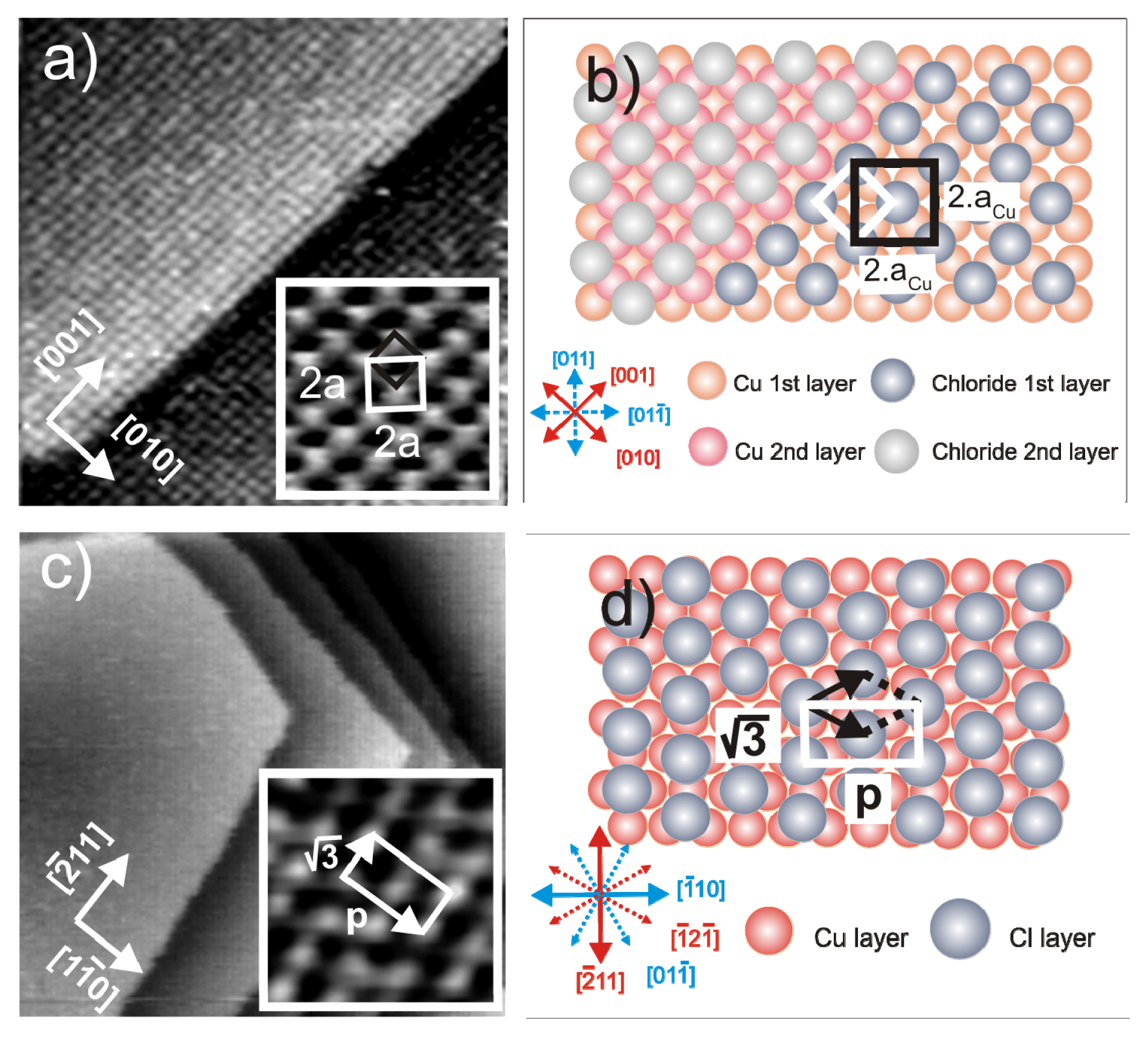
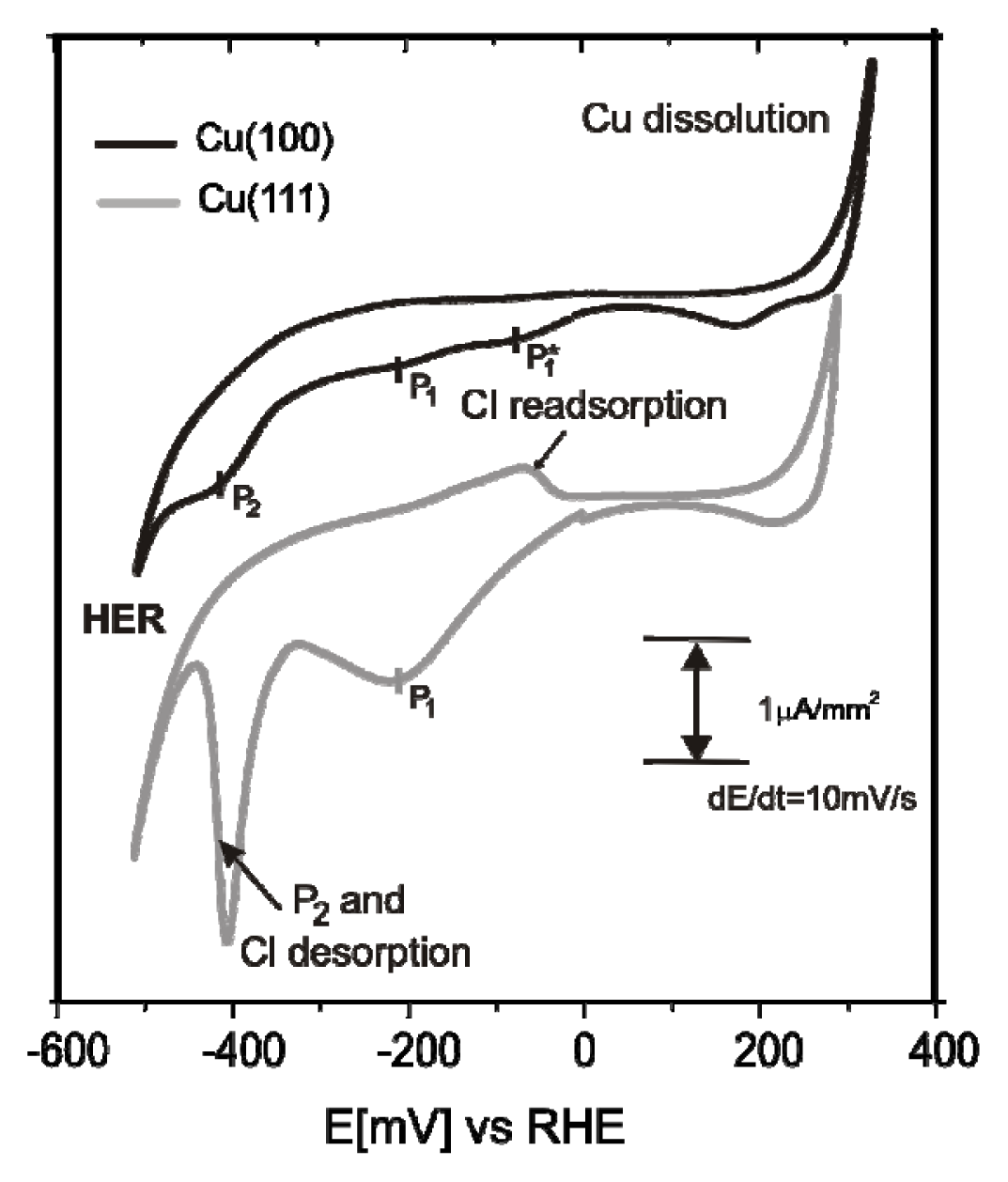
2.1. Structures of the Anion Modified Cu(100) and Cu(111) Substrates
2.2. DBV Adlayers on Chloride Modified Cu(100) and Cu(111)
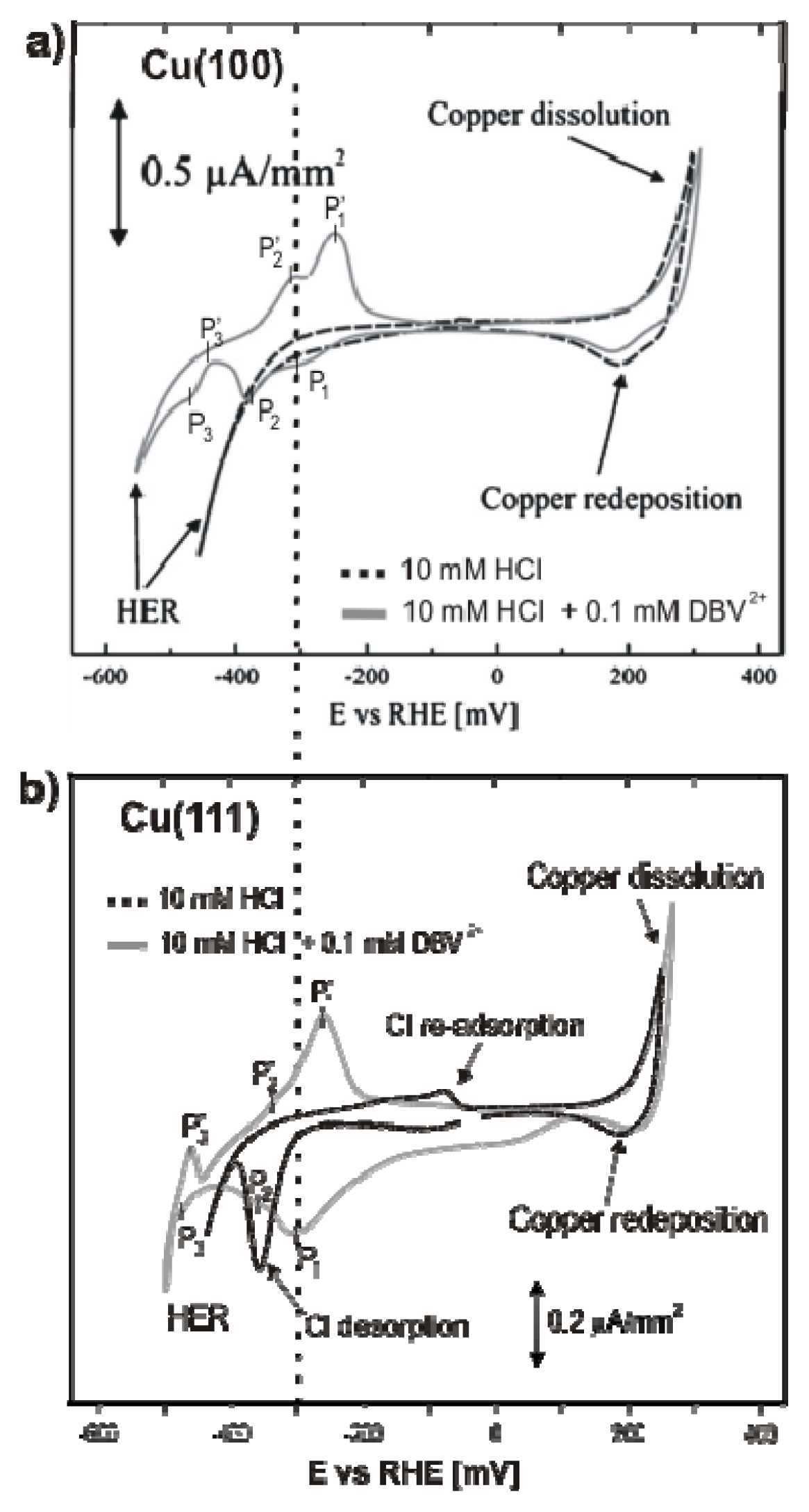
2.2.1. Electrochemistry
2.2.2. Structural Determination
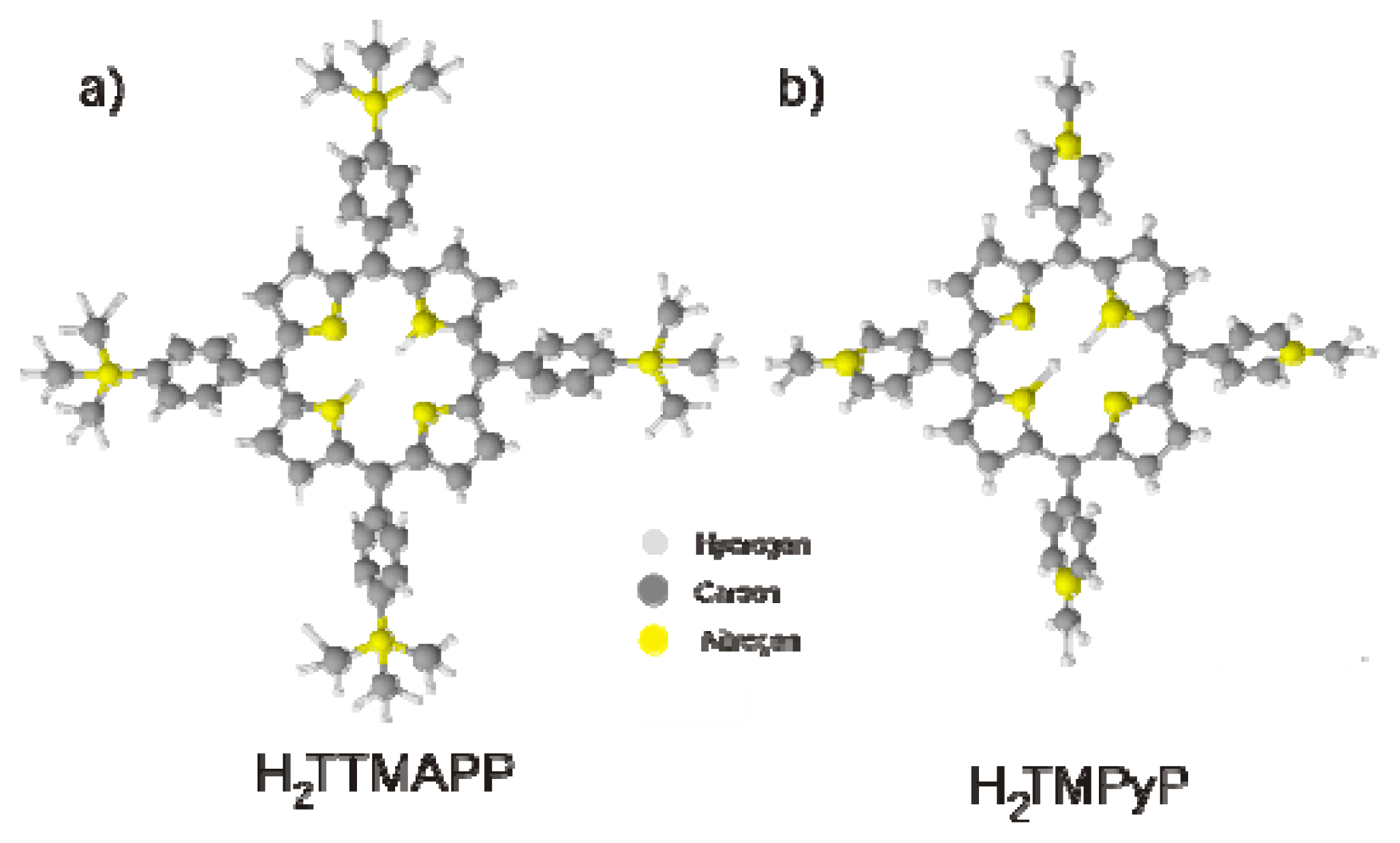
2.3. H2TTMAPP Adlayers on Chloride Modified Cu(100) and Cu(111)
2.3.1. Electrochemical Behavior
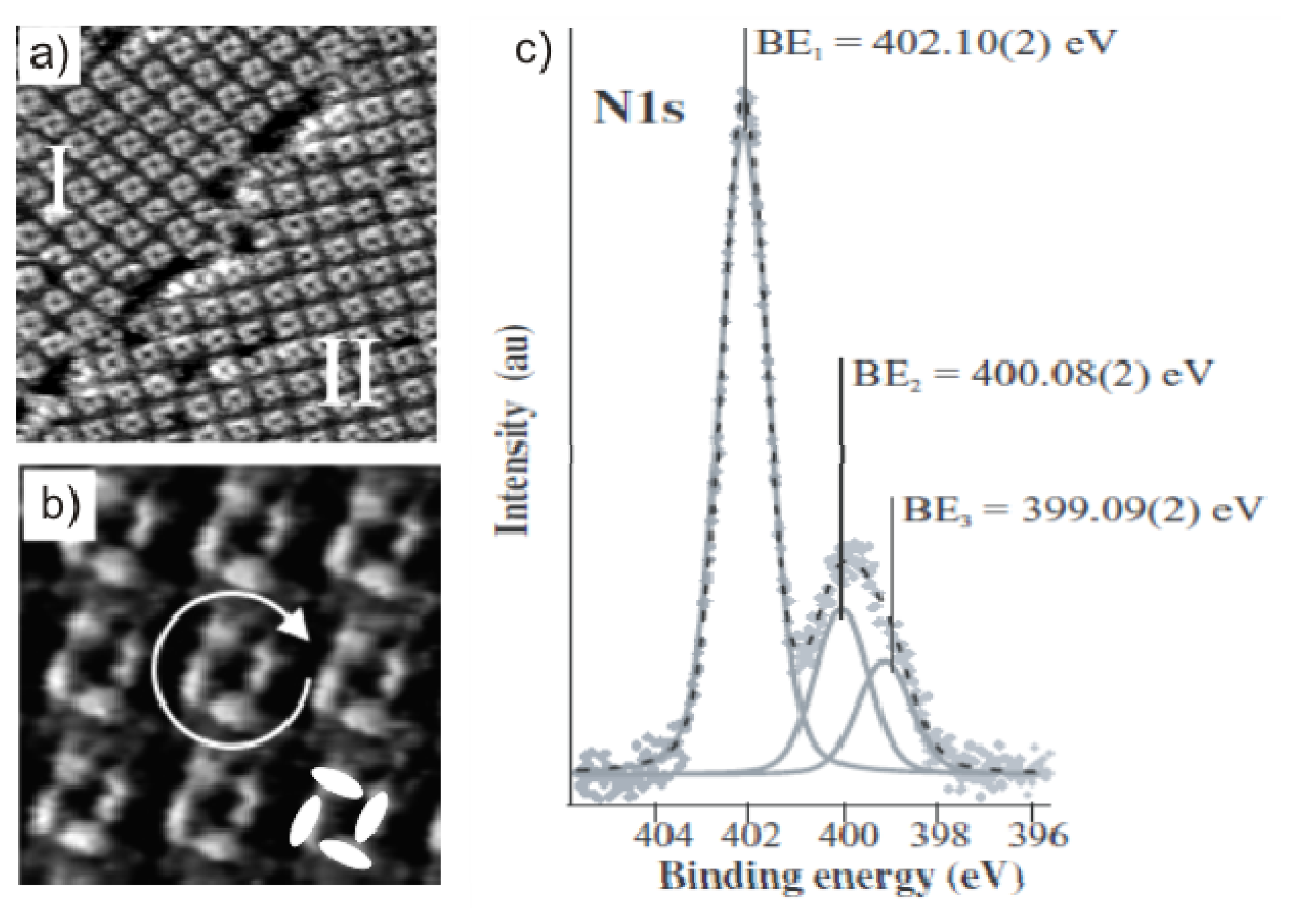
2.3.2. Structural Characterization
2.4. Bimolecular Porphyrin/Viologen Adlayer on Chloride Modified Cu(111) and Cu(100)
2.4.1. Electrochemical Behavior
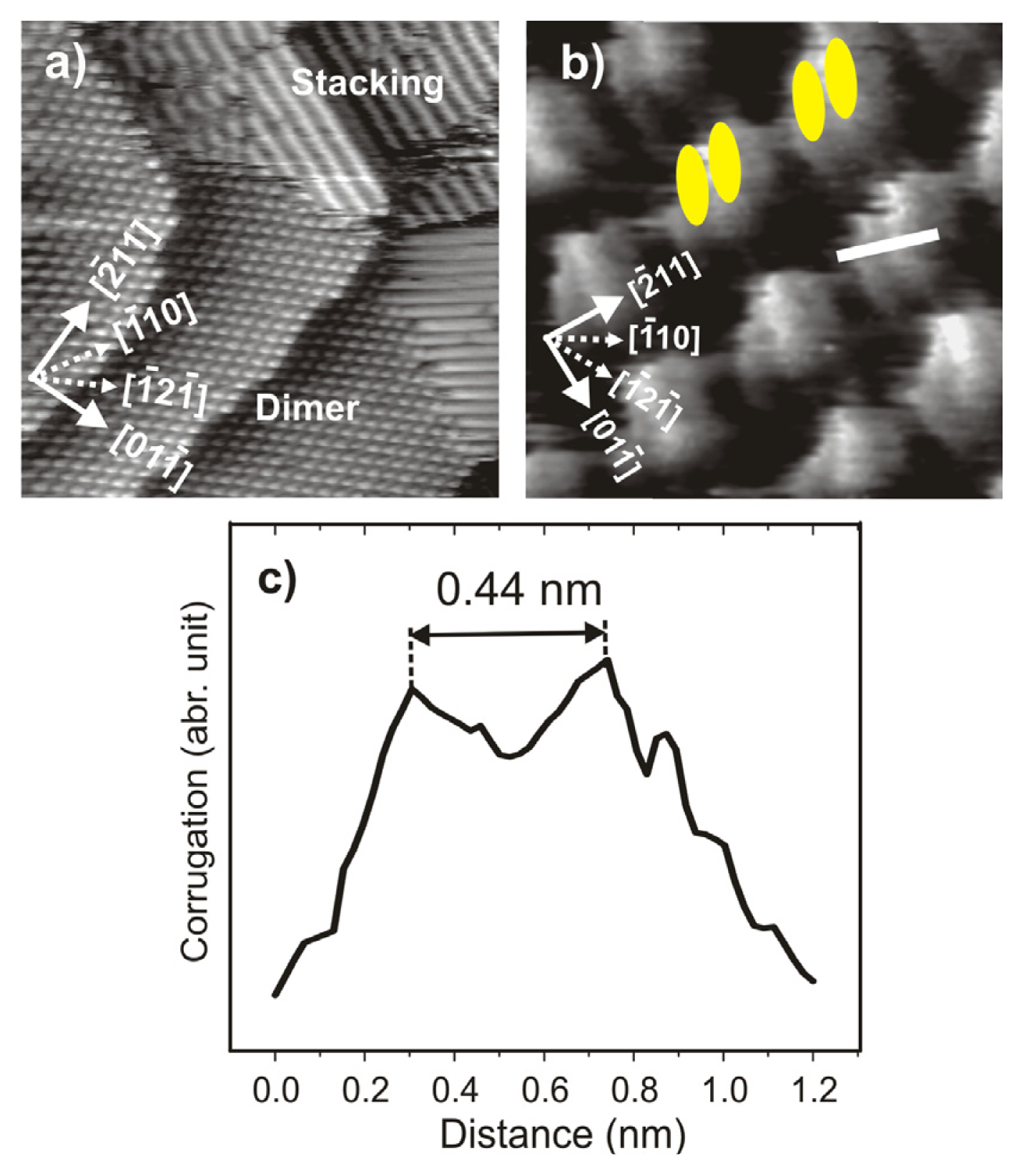
2.4.2. Structural Characterization
2.4.3. Electron transfer Governed Phase Transition
3. Experimental Section
4. Summary
Acknowledgments
References
- Lehn, J.M. Supramolecular Chemistry: Concepts and Perspective, 1st ed; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Lehn, J.M. Supramolecular chemistry—Scope and perspectives molecules, supermolecules, and molecular devices. Angew. Chem. Int. Ed. Engl 1988, 27, 89–112. [Google Scholar]
- Vögtle, F. Supramolecular Chemistry: An Introduction; Wiley & Sons Ltd.: Chichester, UK, 1991. [Google Scholar]
- Kaifer, A.E.; Gomez-Kaifer, M. Supramolecular Electrochemistry; VCH: Weinheim, Germany, 1999. [Google Scholar]
- Guillaud, G.; Simon, J.; Germain, J.P. Metallophthalocyanines: Gas sensors, resistors, and field effect transistors. Coord. Chem. Rev. 1998, 178–180, 1433–1484. [Google Scholar]
- Collman, J.P.; Wagenknecht, P.S.; Hutchison, J.E. Molecular catalysts for multielectron redox reactions of small molecules: The “Cofacial metallodiporphyrin” approach. Angew. Chem. Int. Ed. Engl 1994, 33, 1537–1554. [Google Scholar]
- Balzani, V. Electron Transfer in Chemistry; Wiley-VCH: New York, NY, USA, 2001; Volume 3. [Google Scholar]
- Jasinski, R. Cobalt phthalocyanine as a fuel cell cathode. J. Electrochem. Soc 1965, 112, 526–528. [Google Scholar]
- Alt, H.; Binder, H.; Sandstede, G. Mechanism of the electrocatalytic reduction of oxygen on metal chelates. J. Catal 1973, 28, 8–19. [Google Scholar]
- Savy, M.; Andro, P.; Bernard, C.; Magner, G. Etude de la reduction de l’oxygene sur les phtalocyanines monomeres et polymeres-I. principes fondamentaux, choix de l’ion central. Electrochim. Acta 1973, 18, 191–197. [Google Scholar]
- Otsuki, J.; Nagamine, E.; Kondo, T.; Iwasaki, K.; Asakawa, M.; Miyake, K. Article surface patterning with two-dimensional porphyrin supramolecular arrays. J. Am. Chem. Soc 2005, 127, 10400–10405. [Google Scholar]
- Tao, N.J.; Cardenas, G.; Cunha, F.; Shi, Z. In situ STM and AFM study of protoporphyrin and iron(III) and zinc(II) protoporphyrins adsorbed on graphite in aqueous solutions. Langmuir 1995, 11, 4445–4448. [Google Scholar]
- Tao, N.J. Probing potential-tuned resonant tunneling through redox molecules with scanning tunneling microscopy. Phys. Rev. Lett 1996, 76, 4066–4069. [Google Scholar]
- He, Y.; Ye, T.; Borguet, E. Porphyrin self-assembly at electrochemical interfaces: Role of potential modulated surface mobility. J. Am. Chem. Soc 2002, 124, 11964–11970. [Google Scholar]
- He, Y.; Ye, T.; Borguet, E. Adsorption and electrochemical activity: An in situ electrochemical scanning tunneling microscopy study of electrode reactions and potential-induced adsorption of porphyrins. J. Phys. Chem. B 2006, 110, 6141–6147. [Google Scholar]
- Ogaki, K.; Batina, N.; Kunitake, M.; Itaya, K. In situ scanning tunneling microscopy of ordering processes of adsorbed porphyrin on iodine-modified Ag(111). J. Phys. Chem. B 1996, 100, 7185–7190. [Google Scholar]
- Sashikata, K.; Sutaga, T.; Sugimasa, M.; Itaya, K. In situ scanning tunneling microscopy observation of a porphyrin adlayer on an iodine-modified Pt(100) electrode. Langmuir 1998, 14, 2896–2902. [Google Scholar]
- Kunitake, M.; Batina, N.; Itaya, K. Self-organized porphyrin array on iodine-modified Au(111) in electrolyte solutions: In situ scanning tunneling microscopy study. Langmuir 1995, 11, 2337–2340. [Google Scholar]
- Wan, L.J.; Shundo, S.; Inukai, J.; Itaya, K. Ordered adlayers of organic molecules on sulfur-modified Au(111): In situ scanning tunneling microscopy study. Langmuir 2000, 16, 2164–2168. [Google Scholar]
- Nguyen, T.M.H.; Wandelt, K.; Broekmann, P. Stable anion–cation layers on Cu(111) under reactive conditions. J. Phys. Chem. C 2008, 112, 10176–10186. [Google Scholar]
- Hai, N.T.M.; Gašparović, B.; Wandelt, K.; Broekmann, P. Phase transition in ordered porphyrin layers on iodide modified Cu(111): An EC-STM study. Surf. Sci 2007, 601, 2597–2602. [Google Scholar]
- Xu, B.; Tao, N.J. Measurement of single-molecule resistance by repeated formation of molecular junctions. Science 2003, 301, 1221–2123. [Google Scholar]
- Haiss, W.; Zaling, H.V.; Higgins, S.J.; Bethell, D.; Horbenreich, H.; Schiffrin, D.J.; Nichols, R.J. Redox state dependence of single molecule conductivity. J. Am. Chem. Soc 2003, 125, 15294–15295. [Google Scholar]
- Haiss, W.; Nichols, R.J.; van Zaling, H.; Higgins, S.J.; Bethell, D.; Schiffrin, D. Measurement of single molecule conductivity using the spontaneous formation of molecular wires. J. Phys. Chem. Chem. Phys 2004, 6, 4330–4337. [Google Scholar]
- Li, Z.; Han, B.; Meszaros, G.; Pobelov, I.; Wandlowski, Th.; Blaszczyk, A.; Mayor, M. Two-dimensional assembly and local redox-activity of molecular hybrid htructures in an electrochemical environment. Faraday Discuss. 2006, 131, 121–143. [Google Scholar]
- Imahori, H.; Norieda, H.; Yamada, H.; Nishimura, Y.; Yamazaki, I.; Sakata, Y.; Fukuzumi, S. Light harvesting and photocurrent generation by gold electrodes modified with mixed self-assembled monolayers of boron-dipyrrin and ferrocene-porphyrin-fullerene triad. J. Am. Chem. Soc 2001, 123, 100–110. [Google Scholar]
- Bird, C.L.; Kuhn, A.T. Electrochemistry of the viologens. Chem. Soc. Rev 1981, 10, 49–82. [Google Scholar]
- Monk, P.M.S. The Viologens: Physicochemical Properties, Synthesis and Applications of the Salts of 4,4′-bipyridines, 1st ed; John Wiley and Sons Ltd: Chichester, UK, 1998. [Google Scholar]
- Dretschkow, Th.; Wandlowski, Th. 2,2′-Bipyridine on Au(111)—A novel order/disorder adlayer transition. Electrochim. Acta 1999, 45, 731–740. [Google Scholar]
- Diao, Y.X.; Han, M.J.; Wan, L.J.; Itaya, K.; Uchida, T.; Miyake, H.; Yamakata, A.; Osawa, M. Adsorbed structures of 4,4′-bipyridine on Cu(111) in acid studied by STM and IR. Langmuir 2006, 22, 3640–3646. [Google Scholar]
- Beckers, E.H.A.; Meskers, S.C.J.; Schenning, A.P.H.J.; Chen, Z.; Wurthner, K.; Janssen, R.A.J. Charge separation and recombination in photoexcited oligo(p-phenylene vinylene): Perylene bisimide arrays close to the marcus inverted region. J. Phys. Chem. A 2004, 108, 6933–6937. [Google Scholar]
- Hipps, K.W.; Scudiero, L.; Barlow, D.E.; Cooke, M.P., Jr. A self-organized 2-dimensional bifunctional structure formed by supramolecular design. J. Am. Chem. Soc. 2002, 124, 2126–2127. [Google Scholar]
- Scudiero, L.; Hipps, K.W.; Barlow, D.E. A Self-organized two-dimensional bimolecular structure. J. Phys. Chem. B 2003, 107, 2903–2909. [Google Scholar]
- Yoshimoto, S.; Higa, N.; Itaya, K. Two-dimensional supramolecular organization of copper octaethylporphyrin and cobalt phthalocyanine on Au(111): Molecular assembly control at an electrochemical interface. J. Am. Chem. Soc 2004, 126, 8540–8545. [Google Scholar]
- Suto, K.; Yoshimoto, S.; Itaya, K. Two-dimensional self-organization of phthalocyanine and porphyrin: Dependence on the crystallographic orientation of Au. J. Am. Chem. Soc 2003, 125, 14976–14977. [Google Scholar]
- Kobayashi, K.; Fujisaki, F.; Yoshimina, T.; Nik, K. An analysis of the voltammetric adsorption waves of methyl viologen. Bull. Chem. Soc. Jpn 1986, 59, 3715–3722. [Google Scholar]
- Arihara, K.; Ohsaka, T.; Kitamura, F. Characteristic cyclic voltammograms of alkyl viologens at single crystal gold electrodes. Phys. Chem. Chem. Phys 2002, 4, 1002–2005. [Google Scholar]
- Pham, D.T.; Gentz, K.; Zörlein, C.; Nguyen, T.M.H.; Tsay, S.L.; Kirchner, B.; Kossmann, S.; Wandelt, K.; Broekmann, P. Surface redox chemistry of adsorbed viologens on Cu(100). New J. Chem 2006, 30, 1439–1451. [Google Scholar]
- Pham, D.T. Self-Assembly of Viologen Molecules at Metal/Electrolyte Interfaces under Non-Reactive and Reactive Conditions. Ph.D. Dissertation, Rheinische Friedrich-Wilhelms-University Bonn, Bonn, Germany, 2011. [Google Scholar]
- Suggs, D.W.; Bard, A.J. Scanning tunneling microscopic study with atomic resolution of the dissolution of Cu(100) electrodes in aqueous chloride media. J. Phys. Chem. B 1995, 99, 8349–8355. [Google Scholar]
- Vogt, M.R.; Lachenwitzer, A.; Magnussen, O.M.; Behm, R.J. In situ STM study of the initial stages of corrosion of Cu(100) electrodes in sulfuric and hydrochloric acid solution. Surf. Sci 1998, 399, 49–69. [Google Scholar]
- Magnussen, O.M. Ordered anion adlayers on metal electrode surfaces. Chem. Rev 2002, 102, 679–726. [Google Scholar]
- Ehlers, C.B.; Stickney, J.L. Surface chemistry of electrodes: Cu(111) in aqueous HCl. J. Vac. Sci. Technol 1989, A7, 1801–1805. [Google Scholar]
- Kruft, M.; Wohlmann, B.; Stuhlmann, C.; Wandelt, K. Chloride adsorption on Cu(111) electrodes in dilute HCl solutions. Surf. Sci. 1997, 377–379, 601–604. [Google Scholar]
- Wilms, M.; Broekmann, P.; Kruft, M.; Stuhlmann, C.; Wandelt, K. STM investigation of step orientation and surface dynamics of Cu(111) in hydrochloric acid electrolyte. Appl. Phys 1998, A66, 473–475. [Google Scholar]
- Peljhan, S.; Kokalj, A. Adsorption of Chlorine on Cu(111): A density-functional theory study. J. Phys. Chem. C 2009, 113, 14363–14376. [Google Scholar]
- Inukai, J.; Osawa, Y.; Itaya, K. Adlayer structures of chlorine, bromine, and iodine on Cu(111) electrode in solution: In situ STM and ex situ LEED studies. J. Phys. Chem. B 1998, 102, 10034–10340. [Google Scholar]
- Langhus, D.L.; Wilson, G.S. Analysis of time dependent spectra generated from spectro electrochemical experiments. Anal. Chem 1979, 51, 1134–1139. [Google Scholar]
- Heumann, S.; Hai, N.T.M.; Broekmann, P.; Wandelt, K.; Zajonz, H.; Dosch, H.; Renner, F. X-ray diffraction and STM study of reactive surfaces under electrochemical control: Cl and I on Cu(100). J. Phys. Chem. B 2006, 110, 24955–24963. [Google Scholar]
- Migani, A., III; as, F. A systematic study of the structure and bonding of halogens on low-index transition metal surfaces. J. Phys. Chem. B 2006, 110, 11894–11906. [Google Scholar]
- Tolentino, H.C.N.; De Santis, M.; Gauthier, Y.; Langlais, V. Chlorine chemisorption on Cu(001) by surface X-ray diffraction: Geometry and substrate relaxation. Surf. Sci 2007, 601, 2962–1966. [Google Scholar]
- Batina, N.; Kunitake, M.; Itaya, K. Highly ordered molecular arrays formed on iodine-modified Au(111) in solution: in situ STM imaging. J. Electroal. Chem 1996, 405, 245–250. [Google Scholar]
- Broekmann, P.; Wilms, M.; Kruft, M.; Stuhlmann, C.; Wandelt, K. In situ STM investigation of specific anion adsorption on Cu(111). J. Electroal. Chem 1999, 467, 307–324. [Google Scholar]
- Pham, D.T.; Tsay, S.L.; Gentz, K.; Zorlein, C.; Kossmann, S.; Stay, J.S.; Kirchner, B.; Wandelt, K.; Broekmann, P. Quasi reversible chloride adsorption/desorption through a polycationic organic film on Cu(100). Phys. Chem. C 2007, 111, 16428–16436. [Google Scholar]
- Safarowsky, C.; Wandelt, K.; Broekmann, P. Formation of supramolecular cavitands on copper electrode surfaces. Langmuir 2004, 20, 8261–8269. [Google Scholar]
- Breuer, S.; Pham, D.T.; Huemann, S.; Gentz, K.; Zoerlein, C.; Hunger, R.; Wandelt, K.; Broekmann, P. SXPS studies of porphyrin-adsorption at copper/electrolyte interfaces. New J. Phys 2008, 10, 125033. [Google Scholar]
- Liu, X.; Neoh, K.G.; Zhao, L.P.; Kang, E.T. Surface functionalization of glass and polymeric substrates via graft copolymerization of viologen in an aqueous medium. Langmuir 2002, 18, 2914–2921. [Google Scholar]
- Phan, T.H.; Wandelt, K. Adsorption features and structural transition of dibenzyl viologen adlayer at electrolyte/Cu(111) interface under non-reactive and reactive conditions. 2013. submitted for publication. [Google Scholar]
- Phan, T.H. In situ Characterization of Self-assembled Organic Layers at Anion Modified Metal/Electrolyte Interfaces. Ph.D. Dissertation, Rheinische Friedrich-Wilhelms-University Bonn, Bonn, Germany, 2012. [Google Scholar]
- Weck, M.; Dunn, A.R.; Matsumoto, K.; Coates, G.W.; Lobkoysky, E.B.; Grubbs, R.H. Influence of perfluoroarene–arene interactions on the phase behavior of liquid crystalline and polymeric materials. Angew. Chem. Int. Ed 1999, 38, 2741–2745. [Google Scholar]
- Dretschkow, Th.; Wandlowski, Th. Structural studies of 2,2′-bipyridine on Au(100). J. Electroanal. Chem. 1999, 467, 207–216. [Google Scholar]
- Phan, T.H.; Wandelt, K. Self-assembly of metal free porphyrin layers at copper-electrolyte interfaces: Dependence on substrate symmetry. Surf. Sci 2013, 607, 82–91. [Google Scholar]
- Langhus, D.L.; Wilson, G.S. Spectroelectrochemistry and cyclic voltammetry of the ee mechanism in a porphyrin diacid reduction. Anal. Chem 1979, 51, 1139–1144. [Google Scholar]
- Neri, B.P.; Wilson, G.S. Electrochemical studies of mesotetra(4-N-methylpyridyl)porphine in acid solution. Anal. Chem 1972, 44, 1002–1009. [Google Scholar]
- Phan, T.H.; Wandelt, K. Porphyrin adsorption on a Cu(111) electrode surface: Potential dependent in situ STM studies. Available online: http://ma.ecsdl.org/content/MA2011-02/43/2509.abstract accessed on 16 February 2013.
- Phan, T.H.; Wandelt, K. In situ scanning tunneling microscopy of potential dependent bimolecular component self-assembly consisting of viologen and porphyrin at an electrolyte/electrode interface. 2013. submitted for publication. [Google Scholar]
- Hai, N.T.M. Preparation and Characterization of Copper-Iodide Thin Films and Organic Supramolecular Layers at Copper/Electrolyte Interfaces. Ph.D. Dissertation, Rheinische Friedrich-Wilhelms-University Bonn, Bonn, Germany, 2011. [Google Scholar]
















© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Phan, T.H.; Wandelt, K. Molecular Self-Assembly at Metal-Electrolyte Interfaces. Int. J. Mol. Sci. 2013, 14, 4498-4524. https://doi.org/10.3390/ijms14034498
Phan TH, Wandelt K. Molecular Self-Assembly at Metal-Electrolyte Interfaces. International Journal of Molecular Sciences. 2013; 14(3):4498-4524. https://doi.org/10.3390/ijms14034498
Chicago/Turabian StylePhan, Thanh Hai, and Klaus Wandelt. 2013. "Molecular Self-Assembly at Metal-Electrolyte Interfaces" International Journal of Molecular Sciences 14, no. 3: 4498-4524. https://doi.org/10.3390/ijms14034498
APA StylePhan, T. H., & Wandelt, K. (2013). Molecular Self-Assembly at Metal-Electrolyte Interfaces. International Journal of Molecular Sciences, 14(3), 4498-4524. https://doi.org/10.3390/ijms14034498