Mass Spectrometry-Based Proteomics for the Analysis of Chromatin Structure and Dynamics


Abstract
:1. Introduction
2. Fundamentals of Mass Spectrometry Technology
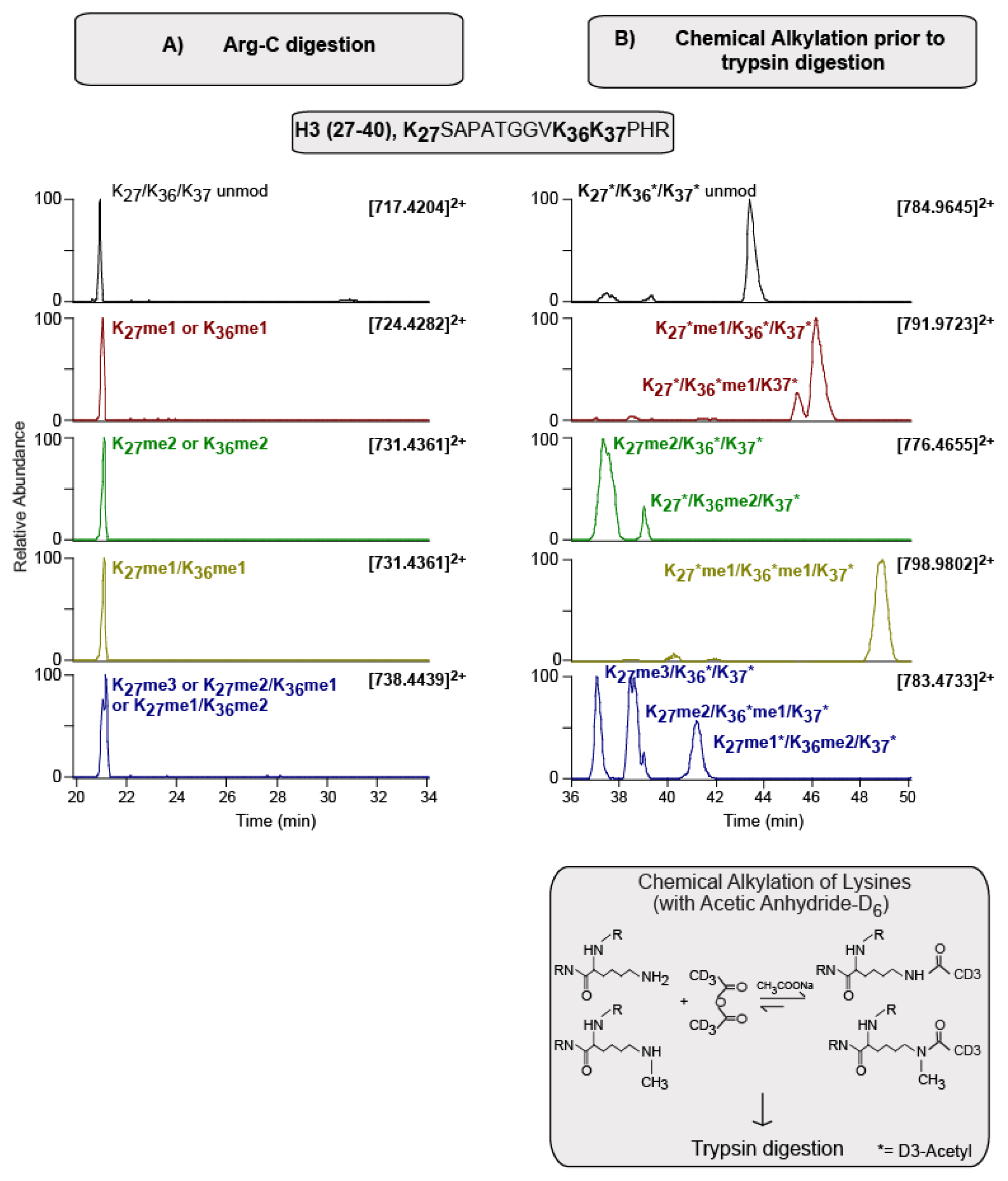
2.1. From “Bottom Up” to “Top Down”, via “Middle Down” MS Approaches in hPTM Research
2.2. Bioinformatics Tools for hPTM Analysis
2.3. Quantitative MS-Based Approaches in Epigenetic Research
3. Mass Spectrometry Analysis of Histone Variants and Their Modifications
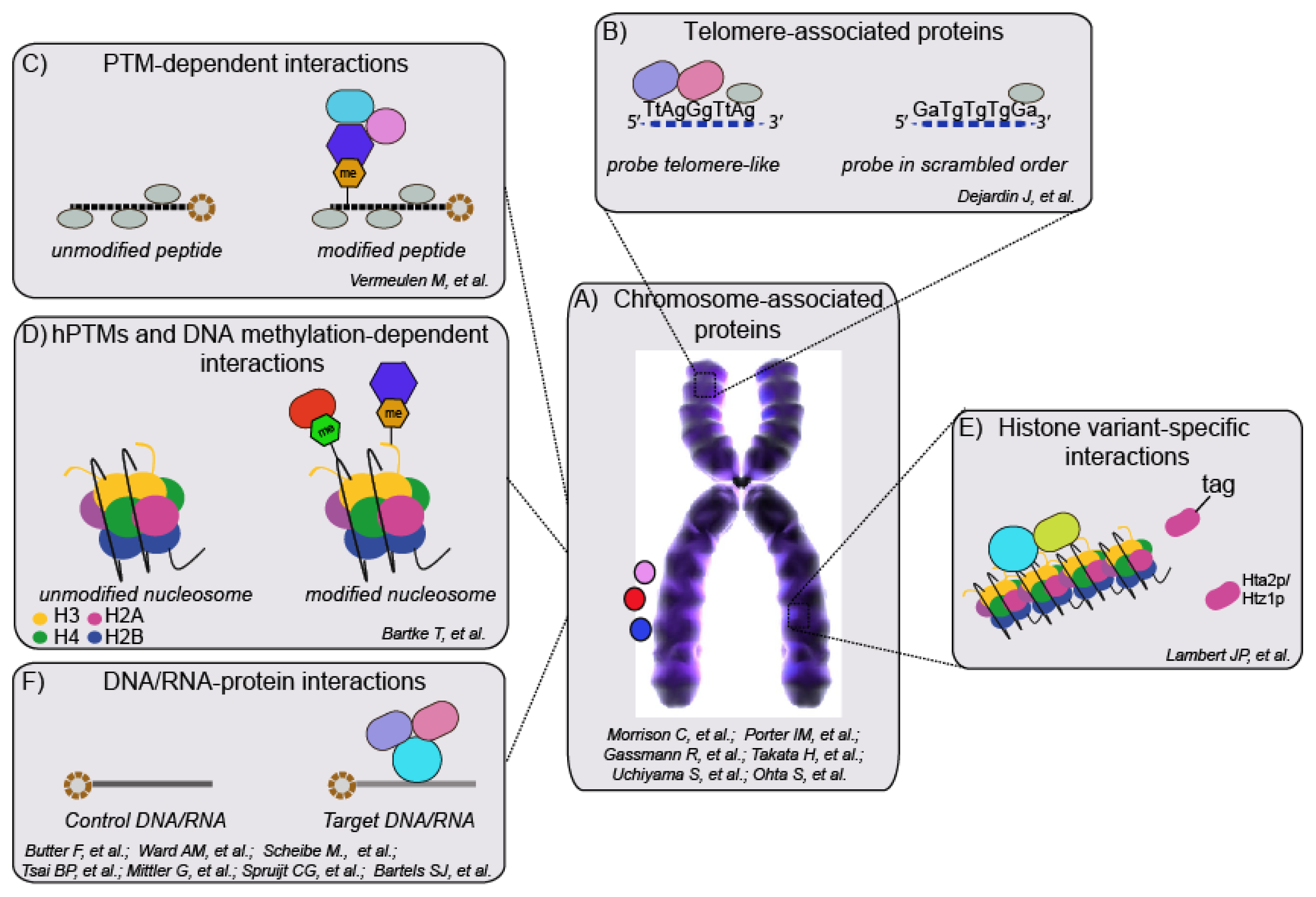
4. Interaction Proteomics to Study Chromatin Architecture
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar]
- Dillon, N. Heterochromatin structure and function. Biol. Cell 2004, 96, 631–637. [Google Scholar]
- Beisel, C.; Paro, R. Silencing chromatin: comparing modes and mechanisms. Nat. Rev. Genet 2011, 12, 123–135. [Google Scholar]
- Grewal, S.I.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet 2007, 8, 35–46. [Google Scholar]
- Probst, A.V.; Dunleavy, E.; Almouzni, G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell Biol 2009, 10, 192–206. [Google Scholar]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol 2011, 41, 10–13. [Google Scholar]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar]
- Nightingale, K.P.; O’Neill, L.P.; Turner, B.M. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev 2006, 16, 125–136. [Google Scholar]
- Turner, B.M. Defining an epigenetic code. Nat. Cell Biol 2007, 9, 2–6. [Google Scholar]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes. Dev 2009, 23, 781–783. [Google Scholar]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci 2006, 31, 89–97. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes. Dev 2002, 16, 6–21. [Google Scholar]
- Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mattick, J.S. The eukaryotic genome as an RNA machine. Science 2008, 319, 1787–1789. [Google Scholar]
- Mattick, J.S.; Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mehler, M.F. RNA regulation of epigenetic processes. Bioessays 2009, 31, 51–59. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res 2011, 21, 381–395. [Google Scholar]
- Campos, E.I.; Reinberg, D. Histones: Annotating chromatin. Annu. Rev. Genet 2009, 43, 559–599. [Google Scholar]
- Spotswood, H.T.; Turner, B.M. An increasingly complex code. J. Clin. Invest 2002, 110, 577–582. [Google Scholar]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar]
- Taverna, S.D.; Li, H.; Ruthenburg, A.J.; Allis, C.D.; Patel, D.J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol 2007, 14, 1025–1040. [Google Scholar]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol 2005, 6, 838–849. [Google Scholar]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet 2010, 11, 285–296. [Google Scholar]
- Park, P.J. Epigenetics meets next-generation sequencing. Epigenetics 2008, 3, 318–321. [Google Scholar]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar]
- Ku, C.S.; Naidoo, N.; Wu, M.; Soong, R. Studying the epigenome using next generation sequencing. J. Med. Genet 2011, 48, 721–730. [Google Scholar]
- Cheung, P.; Tanner, K.G.; Cheung, W.L.; Sassone-Corsi, P.; Denu, J.M.; Allis, C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 2000, 5, 905–915. [Google Scholar]
- Lo, W.S.; Trievel, R.C.; Rojas, J.R.; Duggan, L.; Hsu, J.Y.; Allis, C.D.; Marmorstein, R.; Berger, S.L. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell 2000, 5, 917–926. [Google Scholar]
- Peach, S.E.; Rudomin, E.L.; Udeshi, N.D.; Carr, S.A.; Jaffe, J.D. Quantitative Assessment of Chromatin Immunoprecipitation Grade Antibodies Directed against Histone Modifications Reveals Patterns of Co-occurring Marks on Histone Protein Molecules. Mol. Cell Proteomics 2012, 11, 128–137. [Google Scholar]
- Fuchs, S.M.; Krajewski, K.; Baker, R.W.; Miller, V.L.; Strahl, B.D. Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr. Biol 2011, 21, 53–58. [Google Scholar]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar]
- Sidoli, S.; Cheng, L.; Jensen, O.N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteomics 2012, 75, 3419–3433. [Google Scholar]
- Taverna, S.D.; Ueberheide, B.M.; Liu, Y.; Tackett, A.J.; Diaz, R.L.; Shabanowitz, J.; Chait, B.T.; Hunt, D.F.; Allis, C.D. Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. USA 2007, 104, 2086–2091. [Google Scholar]
- Beck, H.C. Mass spectrometry in epigenetic research. Methods Mol. Biol 2010, 593, 263–282. [Google Scholar]
- Britton, L.M.; Gonzales-Cope, M.; Zee, B.M.; Garcia, B.A. Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 2011, 8, 631–643. [Google Scholar]
- Eberl, H.C.; Mann, M.; Vermeulen, M. Quantitative proteomics for epigenetics. Chembiochem 2011, 12, 224–234. [Google Scholar]
- Garcia, B.A.; Shabanowitz, J.; Hunt, D.F. Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol 2007, 11, 66–73. [Google Scholar]
- Villar-Garea, A.; Imhof, A. The analysis of histone modifications. Biochim. Biophys. Acta 2006, 1764, 1932–1939. [Google Scholar]
- Tipton, J.D.; Tran, J.C.; Catherman, A.D.; Ahlf, D.R.; Durbin, K.R.; Kelleher, N.L. Analysis of intact protein isoforms by mass spectrometry. J. Biol. Chem 2011, 286, 25451–25458. [Google Scholar]
- Plazas-Mayorca, M.D.; Zee, B.M.; Young, N.L.; Fingerman, I.M.; LeRoy, G.; Briggs, S.D.; Garcia, B.A. One-pot shotgun quantitative mass spectrometry characterization of histones. J. Proteome Res 2009, 8, 5367–5374. [Google Scholar]
- Zee, B.M.; Young, N.L.; Garcia, B.A. Quantitative proteomic approaches to studying histone modifications. Curr. Chem. Genomics 2011, 5, 106–114. [Google Scholar]
- Imhof, A.; Bonaldi, T. “Chromatomics” the analysis of the chromatome. Mol. Biosyst 2005, 1, 112–116. [Google Scholar]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng 2009, 11, 49–79. [Google Scholar]
- Walther, T.C.; Mann, M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol 2010, 190, 491–500. [Google Scholar]
- Hillenkamp, F.; Karas, M. Mass spectrometry of peptides and proteins by matrix-assisted ultraviolet laser desorption/ionization. Methods Enzymol 1990, 193, 280–295. [Google Scholar]
- Hillenkamp, F.; Karas, M.; Beavis, R.C.; Chait, B.T. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal. Chem 1991, 63, 1193A–1203A. [Google Scholar]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar]
- Lagarrigue, M.; Lavigne, R.; Guevel, B.; Com, E.; Chaurand, P.; Pineau, C. Matrix-assisted laser desorption/ionization imaging mass spectrometry: A promising technique for reproductive research. Biol. Reprod. 2012, 86. [Google Scholar] [CrossRef]
- Wilm, M.; Mann, M. Analytical properties of the nanoelectrospray ion source. Anal. Chem 1996, 68, 1–8. [Google Scholar]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 1996, 379, 466–469. [Google Scholar]
- Shen, Y.; Zhao, R.; Berger, S.J.; Anderson, G.A.; Rodriguez, N.; Smith, R.D. High-efficiency nanoscale liquid chromatography coupled on-line with mass spectrometry using nanoelectrospray ionization for proteomics. Anal. Chem 2002, 74, 4235–4249. [Google Scholar]
- Shen, Y.; Moore, R.J.; Zhao, R.; Blonder, J.; Auberry, D.L.; Masselon, C.; Pasa-Tolic, L.; Hixson, K.K.; Auberry, K.J.; Smith, R.D. High-efficiency on-line solid-phase extraction coupling to 15–150-microm-i.d. column liquid chromatography for proteomic analysis. Anal. Chem 2003, 75, 3596–3605. [Google Scholar]
- Shukla, A.K.; Futrell, J.H. Tandem mass spectrometry: dissociation of ions by collisional activation. J. Mass Spectrom 2000, 35, 1069–1090. [Google Scholar]
- Kelleher, N.L.; Zubarev, R.A.; Bush, K.; Furie, B.; Furie, B.C.; McLafferty, F.W.; Walsh, C.T. Localization of labile posttranslational modifications by electron capture dissociation: the case of gamma-carboxyglutamic acid. Anal. Chem 1999, 71, 4250–4253. [Google Scholar]
- Mikesh, L.M.; Ueberheide, B.; Chi, A.; Coon, J.J.; Syka, J.E.; Shabanowitz, J.; Hunt, D.F. The utility of ETD mass spectrometry in proteomic analysis. Biochim. Biophys. Acta 2006, 1764, 1811–1822. [Google Scholar]
- Syka, J.E.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar]
- Zubarev, R.A.; Horn, D.M.; Fridriksson, E.K.; Kelleher, N.L.; Kruger, N.A.; Lewis, M.A.; Carpenter, B.K.; McLafferty, F.W. Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem 2000, 72, 563–573. [Google Scholar]
- Olsen, J.V.; Ong, S.E.; Mann, M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell Proteomics 2004, 3, 608–614. [Google Scholar]
- Bonaldi, T.; Imhof, A.; Regula, J.T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics 2004, 4, 1382–1396. [Google Scholar]
- Jufvas, A.; Stralfors, P.; Vener, A.V. Histone variants and their post-translational modifications in primary human fat cells. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Haimberger, Z.W.; Johnson, C.O.; Wolf, A.J.; Gafken, P.R.; Zhang, Z.; Parthun, M.R.; Gottschling, D.E. Heritable chromatin structure: Mapping “memory” in histones H3 and H4. Proc. Natl. Acad. Sci. USA 2002, 99, S16454–S16461. [Google Scholar]
- Garcia, B.A.; Mollah, S.; Ueberheide, B.M.; Busby, S.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc 2007, 2, 933–938. [Google Scholar]
- Bonaldi, T.; Regula, J.T.; Imhof, A. The use of mass spectrometry for the analysis of histone modifications. Methods Enzymol 2004, 377, 111–130. [Google Scholar]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 2006, 1, 2856–2860. [Google Scholar]
- Trelle, M.B.; Salcedo-Amaya, A.M.; Cohen, A.M.; Stunnenberg, H.G.; Jensen, O.N. Global histone analysis by mass spectrometry reveals a high content of acetylated lysine residues in the malaria parasite Plasmodium falciparum. J. Proteome Res 2009, 8, 3439–3450. [Google Scholar]
- Loyola, A.; Bonaldi, T.; Roche, D.; Imhof, A.; Almouzni, G. PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 2006, 24, 309–316. [Google Scholar]
- Trelle, M.B.; Jensen, O.N. Functional proteomics in histone research and epigenetics. Expert Rev. Proteomics 2007, 4, 491–503. [Google Scholar]
- Kelleher, N.L. Top-down proteomics. Anal. Chem 2004, 76, 197A–203A. [Google Scholar]
- Boyne, M.T., 2nd; Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L. Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J. Proteome Res. 2006, 5, 248–253. [Google Scholar]
- Breuker, K.; Oh, H.; Lin, C.; Carpenter, B.K.; McLafferty, F.W. Nonergodic and conformational control of the electron capture dissociation of protein cations. Proc. Natl. Acad. Sci. USA 2004, 101, 14011–14016. [Google Scholar]
- Thomas, C.E.; Kelleher, N.L.; Mizzen, C.A. Mass spectrometric characterization of human histone H3: a bird’s eye view. J. Proteome Res 2006, 5, 240–247. [Google Scholar]
- Eliuk, S.M.; Maltby, D.; Panning, B.; Burlingame, A.L. High resolution electron transfer dissociation studies of unfractionated intact histones from murine embryonic stem cells using on-line capillary LC separation: determination of abundant histone isoforms and post-translational modifications. Mol. Cell Proteomics 2010, 9, 824–837. [Google Scholar]
- Tian, Z.; Zhao, R.; Tolic, N.; Moore, R.J.; Stenoien, D.L.; Robinson, E.W.; Smith, R.D.; Pasa-Tolic, L. Two-dimensional liquid chromatography system for online top-down mass spectrometry. Proteomics 2010, 10, 3610–3620. [Google Scholar]
- Garcia, B.A. What does the future hold for Top Down mass spectrometry? J. Am. Soc. Mass Spectrom 2010, 21, 193–202. [Google Scholar]
- Young, N.L.; DiMaggio, P.A.; Plazas-Mayorca, M.D.; Baliban, R.C.; Floudas, C.A.; Garcia, B.A. High throughput characterization of combinatorial histone codes. Mol. Cell Proteomics 2009, 8, 2266–2284. [Google Scholar]
- DiMaggio, P.A., Jr; Young, N.L.; Baliban, R.C.; Garcia, B.A.; Floudas, C.A. A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol. Cell Proteomics 2009, 8, 2527–2543. [Google Scholar]
- Frank, A.M.; Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L.; Pevzner, P.A. Interpreting top-down mass spectra using spectral alignment. Anal. Chem 2008, 80, 2499–2505. [Google Scholar]
- Guan, S.; Burlingame, A.L. Data processing algorithms for analysis of high-resolution MSMS spectra of peptides with complex patterns of posttranslational modifications. Mol. Cell Proteomics 2010, 9, 804–810. [Google Scholar]
- Matthiesen, R.; Trelle, M.B.; Hojrup, P.; Bunkenborg, J.; Jensen, O.N. VEMS 3.0: Algorithms and computational tools for tandem mass spectrometry based identification of post-translational modifications in proteins. J. Proteome Res 2005, 4, 2338–2347. [Google Scholar]
- Wilkins, M.R.; Gasteiger, E.; Gooley, A.A.; Herbert, B.R.; Molloy, M.P.; Binz, P.A.; Ou, K.; Sanchez, J.C.; Bairoch, A.; Williams, K.L.; et al. High-throughput mass spectrometric discovery of protein post-translational modifications. J. Mol. Biol 1999, 289, 645–657. [Google Scholar]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 2008, 26, 1367–1372. [Google Scholar]
- Savitski, M.M.; Nielsen, M.L.; Zubarev, R.A. ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteomics 2006, 5, 935–948. [Google Scholar]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open mass spectrometry search algorithm. J. Proteome Res 2004, 3, 958–964. [Google Scholar]
- Colinge, J.; Masselot, A.; Cusin, I.; Mahe, E.; Niknejad, A.; Argoud-Puy, G.; Reffas, S.; Bederr, N.; Gleizes, A.; Rey, P.A.; et al. High-performance peptide identification by tandem mass spectrometry allows reliable automatic data processing in proteomics. Proteomics 2004, 4, 1977–1984. [Google Scholar]
- Baliban, R.C.; DiMaggio, P.A.; Plazas-Mayorca, M.D.; Young, N.L.; Garcia, B.A.; Floudas, C.A. A novel approach for untargeted post-translational modification identification using integer linear optimization and tandem mass spectrometry. Mol. Cell. Proteomics 2010, 9, 764–779. [Google Scholar]
- Zamdborg, L.; LeDuc, R.D.; Glowacz, K.J.; Kim, Y.B.; Viswanathan, V.; Spaulding, I.T.; Early, B.P.; Bluhm, E.J.; Babai, S.; Kelleher, N. ProSight PTM 2.0: Improved protein identification and characterization for top down mass spectrometry. Nucleic Acids Res 2007, 35, W701–W706. [Google Scholar]
- Chalkley, R.J.; Baker, P.R.; Huang, L.; Hansen, K.C.; Allen, N.P.; Rexach, M.; Burlingame, A.L. Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry dataset acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large datasets. Mol. Cell. Proteomics 2005, 4, 1194–1204. [Google Scholar]
- Ahrne, E.; Ohta, Y.; Nikitin, F.; Scherl, A.; Lisacek, F.; Muller, M. An improved method for the construction of decoy peptide MS/MS spectra suitable for the accurate estimation of false discovery rates. Proteomics 2011, 11, 4085–4095. [Google Scholar]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 1994, 5, 976–989. [Google Scholar]
- Liu, J.; Erassov, A.; Halina, P.; Canete, M.; Nguyen, D.V.; Chung, C.; Cagney, G.; Ignatchenko, A.; Fong, V.; Emili, A. Sequential interval motif search: Unrestricted database surveys of global MS/MS data sets for detection of putative post-translational modifications. Anal. Chem 2008, 80, 7846–7854. [Google Scholar]
- Craig, R.; Beavis, R.C. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom 2003, 17, 2310–2316. [Google Scholar]
- Marshall, A.G.; Hendrickson, C.L. High-resolution mass spectrometers. Annu. Rev. Anal. Chem 2008, 1, 579–599. [Google Scholar]
- Nielsen, M.L.; Vermeulen, M.; Bonaldi, T.; Cox, J.; Moroder, L.; Mann, M. Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 2008, 5, 459–460. [Google Scholar]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. BioAnal. Chem 2007, 389, 1017–1031. [Google Scholar]
- Nikolov, M.; Schmidt, C.; Urlaub, H. Quantitative mass spectrometry-based proteomics: An overview. Methods Mol. Biol 2012, 893, 85–100. [Google Scholar]
- Voigt, P.; LeRoy, G.; Drury, W.J., 3rd; Zee, B.M.; Son, J.; Beck, D.B.; Young, N.L.; Garcia, B.A.; Reinberg, D. Asymmetrically modified nucleosomes. Cell 2012, 151, 181–193. [Google Scholar]
- Montes de Oca, R.; Shoemaker, C.J.; Gucek, M.; Cole, R.N.; Wilson, K.L. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Yuan, X.; Gu, X.; Crabb, J.S.; Yue, X.; Shadrach, K.; Hollyfield, J.G.; Crabb, J.W. Quantitative proteomics: Comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol. Cell Proteomics 2010, 9, 1031–1046. [Google Scholar]
- Salim, K.; Kehoe, L.; Minkoff, M.S.; Bilsland, J.G.; Munoz-Sanjuan, I.; Guest, P.C. Identification of differentiating neural progenitor cell markers using shotgun isobaric tagging mass spectrometry. Stem Cells Dev 2006, 15, 461–470. [Google Scholar]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell Proteomics 2002, 1, 376–386. [Google Scholar]
- Stunnenberg, H.G.; Vermeulen, M. Towards cracking the epigenetic code using a combination of high-throughput epigenomics and quantitative mass spectrometry-based proteomics. Bioessays 2011, 33, 547–551. [Google Scholar]
- Ong, S.E.; Mann, M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol 2007, 359, 37–52. [Google Scholar]
- Pimienta, G.; Chaerkady, R.; Pandey, A. SILAC for global phosphoproteomic analysis. Methods Mol. Biol 2009, 527, 107–116. [Google Scholar]
- Bonenfant, D.; Towbin, H.; Coulot, M.; Schindler, P.; Mueller, D.R.; van Oostrum, J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol. Cell Proteomics 2007, 6, 1917–1932. [Google Scholar]
- Pesavento, J.J.; Yang, H.; Kelleher, N.L.; Mizzen, C.A. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell Biol 2008, 28, 468–486. [Google Scholar]
- Scharf, A.N.; Meier, K.; Seitz, V.; Kremmer, E.; Brehm, A.; Imhof, A. Monomethylation of lysine 20 on histone H4 facilitates chromatin maturation. Mol. Cell Biol 2009, 29, 57–67. [Google Scholar]
- Jung, H.R.; Pasini, D.; Helin, K.; Jensen, O.N. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell Proteomics 2010, 9, 838–850. [Google Scholar]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell Proteomics 2009, 8, 2796–2808. [Google Scholar]
- Cuomo, A.; Moretti, S.; Minucci, S.; Bonaldi, T. SILAC-based proteomic analysis to dissect the “histone modification signature” of human breast cancer cells. Amino Acids 2011, 41, 387–399. [Google Scholar]
- Geiger, T.; Cox, J.; Ostasiewicz, P.; Wisniewski, J.R.; Mann, M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 2010, 7, 383–385. [Google Scholar]
- Geiger, T.; Wisniewski, J.R.; Cox, J.; Zanivan, S.; Kruger, M.; Ishihama, Y.; Mann, M. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat. Protoc 2011, 6, 147–157. [Google Scholar]
- Zee, B.M.; Levin, R.S.; Dimaggio, P.A.; Garcia, B.A. Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 2010, 3. [Google Scholar] [CrossRef]
- Ong, S.E.; Mittler, G.; Mann, M. Identifying and quantifying in vivo methylation sites by heavy methyl SILAC. Nat. Methods 2004, 1, 119–126. [Google Scholar]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes. Dev 2006, 20, 1557–1562. [Google Scholar]
- Zee, B.M.; Levin, R.S.; Xu, B.; LeRoy, G.; Wingreen, N.S.; Garcia, B.A. In vivo residue-specific histone methylation dynamics. J. Biol. Chem 2010, 285, 3341–3350. [Google Scholar]
- Sweet, S.M.; Li, M.; Thomas, P.M.; Durbin, K.R.; Kelleher, N.L. Kinetics of re-establishing H3K79 methylation marks in global human chromatin. J. Biol. Chem 2010, 285, 32778–32786. [Google Scholar]
- Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L. Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal. Chem 2006, 78, 4271–4280. [Google Scholar]
- Garcia, B.A.; Pesavento, J.J.; Mizzen, C.A.; Kelleher, N.L. Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 2007, 4, 487–489. [Google Scholar]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar]
- Darwanto, A.; Curtis, M.P.; Schrag, M.; Kirsch, W.; Liu, P.; Xu, G.; Neidigh, J.W.; Zhang, K. A modified “cross-talk” between histone H2B Lys-120 ubiquitination and H3 Lys-79 methylation. J. Biol. Chem 2010, 285, 21868–21876. [Google Scholar]
- Yuan, G.; Zhu, B. Histone variants and epigenetic inheritance. Biochim. Biophys. Acta 2012, 1819, 222–229. [Google Scholar]
- Hake, S.B.; Allis, C.D. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc. Natl. Acad. Sci. USA 2006, 103, 6428–6435. [Google Scholar]
- Hake, S.B.; Garcia, B.A.; Duncan, E.M.; Kauer, M.; Dellaire, G.; Shabanowitz, J.; Bazett-Jones, D.P.; Allis, C.D.; Hunt, D.F. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem 2006, 281, 559–568. [Google Scholar]
- McKittrick, E.; Gafken, P.R.; Ahmad, K.; Henikoff, S. Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. USA 2004, 101, 1525–1530. [Google Scholar]
- Johnson, L.; Mollah, S.; Garcia, B.A.; Muratore, T.L.; Shabanowitz, J.; Hunt, D.F.; Jacobsen, S.E. Mass spectrometry analysis of Arabidopsis histone H3 reveals distinct combinations of post-translational modifications. Nucleic Acids Res 2004, 32, 6511–6518. [Google Scholar]
- Garcia, B.A.; Siuti, N.; Thomas, C.E.; Mizzen, C.A.; Kelleher, N.L. Characterization of neurohistone variants and post-translational modifications by electron capture dissociation mass spectrometry. Int. J. Mass Spectrom 2007, 259, 184–196. [Google Scholar]
- Garcia, B.A.; Thomas, C.E.; Kelleher, N.L.; Mizzen, C.A. Tissue-specific expression and post-translational modification of histone H3 variants. J. Proteome Res 2008, 7, 4225–4236. [Google Scholar]
- Bonenfant, D.; Coulot, M.; Towbin, H.; Schindler, P.; van Oostrum, J. Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell Proteomics 2006, 5, 541–552. [Google Scholar]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar]
- Ikura, T.; Tashiro, S.; Kakino, A.; Shima, H.; Jacob, N.; Amunugama, R.; Yoder, K.; Izumi, S.; Kuraoka, I.; Tanaka, K.; et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol. Cell Biol 2007, 27, 7028–7040. [Google Scholar]
- Rangasamy, D.; Greaves, I.; Tremethick, D.J. RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat. Struct. Mol. Biol 2004, 11, 650–655. [Google Scholar]
- Chu, F.; Nusinow, D.A.; Chalkley, R.J.; Plath, K.; Panning, B.; Burlingame, A.L. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol. Cell Proteomics 2006, 5, 194–203. [Google Scholar]
- Bernstein, E.; Muratore-Schroeder, T.L.; Diaz, R.L.; Chow, J.C.; Changolkar, L.N.; Shabanowitz, J.; Heard, E.; Pehrson, J.R.; Hunt, D.F.; Allis, C.D. A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis. Proc. Natl. Acad. Sci. USA 2008, 105, 1533–1538. [Google Scholar]
- Siuti, N.; Roth, M.J.; Mizzen, C.A.; Kelleher, N.L.; Pesavento, J.J. Gene-specific characterization of human histone H2B by electron capture dissociation. J. Proteome Res 2006, 5, 233–239. [Google Scholar]
- Zhang, L.; Eugeni, E.E.; Parthun, M.R.; Freitas, M.A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003, 112, 77–86. [Google Scholar]
- Lu, S.; Xie, Y.M.; Li, X.; Luo, J.; Shi, X.Q.; Hong, X.; Pan, Y.H.; Ma, X. Mass spectrometry analysis of dynamic post-translational modifications of TH2B during spermatogenesis. Mol. Hum. Reprod 2009, 15, 373–378. [Google Scholar]
- Medzihradszky, K.F.; Zhang, X.; Chalkley, R.J.; Guan, S.; McFarland, M.A.; Chalmers, M.J.; Marshall, A.G.; Diaz, R.L.; Allis, C.D.; Burlingame, A.L. Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS). Mol. Cell Proteomics 2004, 3, 872–886. [Google Scholar]
- Villar-Garea, A.; Forne, I.; Vetter, I.; Kremmer, E.; Thomae, A.; Imhof, A. Developmental regulation of N-terminal H2B methylation in Drosophila melanogaster. Nucleic Acids Res 2012, 40, 1536–1549. [Google Scholar]
- Talasz, H.; Sarg, B.; Lindner, H.H. Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma 2009, 118, 693–709. [Google Scholar]
- Wisniewski, J.R.; Zougman, A.; Kruger, S.; Mann, M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell Proteomics 2007, 6, 72–87. [Google Scholar]
- Garcia, B.A.; Busby, S.A.; Barber, C.M.; Shabanowitz, J.; Allis, C.D.; Hunt, D.F. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J. Proteome Res 2004, 3, 1219–1227. [Google Scholar]
- Weiss, T.; Hergeth, S.; Zeissler, U.; Izzo, A.; Tropberger, P.; Zee, B.M.; Dundr, M.; Garcia, B.A.; Daujat, S.; Schneider, R. Histone H1 variant-specific lysine methylation by G9a/KMT1C and Glp1/KMT1D. Epigenetics Chromatin 2010, 3. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Imhof, A. Fine mapping of posttranslational modifications of the linker histone H1 from Drosophila melanogaster. PLoS One 2008, 3. [Google Scholar] [CrossRef] [Green Version]
- Daujat, S.; Zeissler, U.; Waldmann, T.; Happel, N.; Schneider, R. HP1 binds specifically to Lys26-methylated histone H1.4, whereas simultaneous Ser27 phosphorylation blocks HP1 binding. J. Biol. Chem 2005, 280, 38090–38095. [Google Scholar]
- Zheng, Y.; John, S.; Pesavento, J.J.; Schultz-Norton, J.R.; Schiltz, R.L.; Baek, S.; Nardulli, A.M.; Hager, G.L.; Kelleher, N.L.; Mizzen, C.A. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II. J. Cell Biol 2010, 189, 407–415. [Google Scholar]
- Shiio, Y.; Eisenman, R.N.; Yi, E.C.; Donohoe, S.; Goodlett, D.R.; Aebersold, R. Quantitative proteomic analysis of chromatin-associated factors. J. Am. Soc. Mass Spectrom 2003, 14, 696–703. [Google Scholar]
- Porter, I.M.; McClelland, S.E.; Khoudoli, G.A.; Hunter, C.J.; Andersen, J.S.; McAinsh, A.D.; Blow, J.J.; Swedlow, J.R. Bod1, a novel kinetochore protein required for chromosome biorientation. J. Cell Biol 2007, 179, 187–197. [Google Scholar] [Green Version]
- Gassmann, R.; Henzing, A.J.; Earnshaw, W.C. Novel components of human mitotic chromosomes identified by proteomic analysis of the chromosome scaffold fraction. Chromosoma 2005, 113, 385–397. [Google Scholar]
- Uchiyama, S.; Kobayashi, S.; Takata, H.; Ishihara, T.; Hori, N.; Higashi, T.; Hayashihara, K.; Sone, T.; Higo, D.; Nirasawa, T.; et al. Proteome analysis of human metaphase chromosomes. J. Biol. Chem 2005, 280, 16994–17004. [Google Scholar]
- Takata, H.; Uchiyama, S.; Nakamura, N.; Nakashima, S.; Kobayashi, S.; Sone, T.; Kimura, S.; Lahmers, S.; Granzier, H.; Labeit, S.; et al. A comparative proteome analysis of human metaphase chromosomes isolated from two different cell lines reveals a set of conserved chromosome-associated proteins. Genes. Cells 2007, 12, 269–284. [Google Scholar]
- Morrison, C.; Henzing, A.J.; Jensen, O.N.; Osheroff, N.; Dodson, H.; Kandels-Lewis, S.E.; Adams, R.R.; Earnshaw, W.C. Proteomic analysis of human metaphase chromosomes reveals topoisomerase II alpha as an Aurora B substrate. Nucleic Acids Res 2002, 30, 5318–5327. [Google Scholar]
- Khoudoli, G.A.; Gillespie, P.J.; Stewart, G.; Andersen, J.S.; Swedlow, J.R.; Blow, J.J. Temporal profiling of the chromatin proteome reveals system-wide responses to replication inhibition. Curr. Biol 2008, 18, 838–843. [Google Scholar]
- Ohta, S.; Bukowski-Wills, J.C.; Sanchez-Pulido, L.; de Alves, F.L.; Wood, L.; Chen, Z.A.; Platani, M.; Fischer, L.; Hudson, D.F.; Ponting, C.P.; et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 2010, 142, 810–821. [Google Scholar]
- Dejardin, J.; Kingston, R.E. Purification of proteins associated with specific genomic Loci. Cell 2009, 136, 175–186. [Google Scholar]
- Vermeulen, M.; Mulder, K.W.; Denissov, S.; Pijnappel, W.W.; van Schaik, F.M.; Varier, R.A.; Baltissen, M.P.; Stunnenberg, H.G.; Mann, M.; Timmers, H.T. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 2007, 131, 58–69. [Google Scholar]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; Mann, M. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010, 143, 470–484. [Google Scholar]
- Nikolov, M.; Stutzer, A.; Mosch, K.; Krasauskas, A.; Soeroes, S.; Stark, H.; Urlaub, H.; Fischle, W. Chromatin affinity purification and quantitative mass spectrometry defining the interactome of histone modification patterns. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef]
- Li, X.; Foley, E.A.; Molloy, K.R.; Li, Y.; Chait, B.T.; Kapoor, T.M. Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions. J. Am. Chem. Soc 2012, 134, 1982–1985. [Google Scholar]
- Liu, H.; Galka, M.; Iberg, A.; Wang, Z.; Li, L.; Voss, C.; Jiang, X.; Lajoie, G.; Huang, Z.; Bedford, M.T.; Li, S.S. Systematic identification of methyllysine-driven interactions for histone and nonhistone targets. J. Proteome Res 2010, 9, 5827–5836. [Google Scholar]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar]
- Torrente, M.P.; Zee, B.M.; Young, N.L.; Baliban, R.C.; LeRoy, G.; Floudas, C.A.; Hake, S.B.; Garcia, B.A. Proteomic interrogation of human chromatin. PLoS One 2011, 6, e24747. [Google Scholar]
- Lambert, J.P.; Mitchell, L.; Rudner, A.; Baetz, K.; Figeys, D. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol. Cell Proteomics 2009, 8, 870–882. [Google Scholar]
- Byrum, S.D.; Raman, A.; Taverna, S.D.; Tackett, A.J. ChAP-MS: A method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep 2012, 2, 198–205. [Google Scholar]
- Soldi, M.; Bonaldi, T. The proteomic investigation of chromatin functional domains reveals novel synergisms among distinct heterochromatin components. Mol. Cell Proteomics 2013, in press. http://www.mcponline.org/content/early/2013/01/14/mcp.M112.024307.full.pdf. [Google Scholar]
- Mittler, G.; Butter, F.; Mann, M. A SILAC-based DNA protein interaction screen that identifies candidate binding proteins to functional DNA elements. Genome. Res 2009, 19, 284–293. [Google Scholar]
- Spruijt, C.G.; Bartels, S.J.; Brinkman, A.B.; Tjeertes, J.V.; Poser, I.; Stunnenberg, H.G.; Vermeulen, M. CDK2AP1/DOC-1 is a bona fide subunit of the Mi-2/NuRD complex. Mol. Biosyst 2010, 6, 1700–1706. [Google Scholar]
- Bartels, S.J.; Spruijt, C.G.; Brinkman, A.B.; Jansen, P.W.; Vermeulen, M.; Stunnenberg, H.G. A SILAC-based screen for Methyl-CpG binding proteins identifies RBP-J as a DNA methylation and sequence-specific binding protein. PLoS One 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Butter, F.; Kappei, D.; Buchholz, F.; Vermeulen, M.; Mann, M. A domesticated transposon mediates the effects of a single-nucleotide polymorphism responsible for enhanced muscle growth. EMBO Rep 2010, 11, 305–311. [Google Scholar]
- Markljung, E.; Jiang, L.; Jaffe, J.D.; Mikkelsen, T.S.; Wallerman, O.; Larhammar, M.; Zhang, X.; Wang, L.; Saenz-Vash, V.; Gnirke, A.; et al. ZBED6, a novel transcription factor derived from a domesticated DNA transposon regulates IGF2 expression and muscle growth. PLoS Biol. 2009, 7. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Kanhere, A.; Viiri, K.; Araujo, C.C.; Rasaiyaah, J.; Bouwman, R.D.; Whyte, W.A.; Pereira, C.F.; Brookes, E.; Walker, K.; Bell, G.W.; et al. Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol. Cell 2010, 38, 675–688. [Google Scholar]
- Butter, F.; Scheibe, M.; Morl, M.; Mann, M. Unbiased RNA-protein interaction screen by quantitative proteomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10626–10631. [Google Scholar]
- Scheibe, M.; Butter, F.; Hafner, M.; Tuschl, T.; Mann, M. Quantitative mass spectrometry and PAR-CLIP to identify RNA-protein interactions. Nucleic Acids Res 2012, 40, 9897–9902. [Google Scholar]
- Urlaub, H.; Kuhn-Holsken, E.; Luhrmann, R. Analyzing RNA-protein crosslinking sites in unlabeled ribonucleoprotein complexes by mass spectrometry. Methods Mol. Biol 2008, 488, 221–245. [Google Scholar]
- Ward, A.M.; Bidet, K.; Yinglin, A.; Ler, S.G.; Hogue, K.; Blackstock, W.; Gunaratne, J.; Garcia-Blanco, M.A. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol 2011, 8, 1173–1186. [Google Scholar]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell Proteomics 2011, 10. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Software | Freely available | Unbiased PTM search | Reference |
|---|---|---|---|
| FindMod | + | − | [81] |
| Mascot | − | − | [82] |
| MaxQuant | + | − | [83] |
| Modificomb | + | + | [84] |
| OMSSA | + | − | [85] |
| Phenyx | − | − | [86] |
| PILOT PTM | + | + | [87] |
| ProSightPTM 2.0 | + | + | [88] |
| Protein Prospector | + | + | [89] |
| QuickMod | + | + | [90] |
| SEQUEST | − | − | [91] |
| SIMS | + | + | [92] |
| VEMS 3.0 | + | − | [80] |
| X!Tandem | + | − | [93] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Soldi, M.; Cuomo, A.; Bremang, M.; Bonaldi, T. Mass Spectrometry-Based Proteomics for the Analysis of Chromatin Structure and Dynamics. Int. J. Mol. Sci. 2013, 14, 5402-5431. https://doi.org/10.3390/ijms14035402
Soldi M, Cuomo A, Bremang M, Bonaldi T. Mass Spectrometry-Based Proteomics for the Analysis of Chromatin Structure and Dynamics. International Journal of Molecular Sciences. 2013; 14(3):5402-5431. https://doi.org/10.3390/ijms14035402
Chicago/Turabian StyleSoldi, Monica, Alessandro Cuomo, Michael Bremang, and Tiziana Bonaldi. 2013. "Mass Spectrometry-Based Proteomics for the Analysis of Chromatin Structure and Dynamics" International Journal of Molecular Sciences 14, no. 3: 5402-5431. https://doi.org/10.3390/ijms14035402
APA StyleSoldi, M., Cuomo, A., Bremang, M., & Bonaldi, T. (2013). Mass Spectrometry-Based Proteomics for the Analysis of Chromatin Structure and Dynamics. International Journal of Molecular Sciences, 14(3), 5402-5431. https://doi.org/10.3390/ijms14035402