1. Introduction
Adult stem cells have a much lower capacity than embryonic stem cells to self-renew and differentiate along multiple lineage pathways. However, adult stem cells are immunocompatible, and their use is not restricted by the ethical concerns associated with embryo-derived cells. Mesenchymal stem cells can be easily isolated from many adult tissues such as bone marrow, periosteum, trabecular bone, adipose tissue, synovium, skeletal muscle, and deciduous teeth [
1,
2], and are therefore commonly used to study stem cell multipotency. Skeletal muscle-derived stem cells (MDSCs) have long been considered to be unipotent stem cells capable of differentiating only into myogenic lineage cells [
3]. However, recent evidence supports the existence of multipotent MDSCs that can participate in the healing and regeneration of several tissues [
4–
12]. These reports demonstrate that MDSCs can differentiate into various cell lineages using defined culture conditions, and that large quantities of differentiated cells could be obtained
in vitro. Therefore, MDSCs could be an excellent source of differentiated cell types for tissue engineering and cell therapies.
Despite the remarkable achievements of medical research, the treatment of central nervous system diseases remains challenging. Neural stem cells (NSCs) are capable of self-renewal and differentiation into neurons, astrocytes, and oligodendrocytes. NSC transplantation has shown great potential for the therapy of neurological disorders; however, NSCs commonly cultured
in vitro as neurospheres do not behave as stem cells when transplanted back into the brain [
13,
14]. NSCs proliferate slowly throughout life and only small numbers persist in the adult brain. Moreover, these cells are difficult to isolate from human brain biopsy or autopsy samples. Thus, the ease of isolation and differentiation potential of MDSCs makes them an attractive possible source of functional neuronal or glial cells that could be used in cell therapy of neurological disorders. Indeed, several studies have demonstrated that MDSCs can undergo neuroectodermal differentiation to neurons and astrocytes [
5–
12].
In this study, we investigated the neural differentiation potential of rat MDSCs (rMDSCs) treated with basic fibroblast growth factor (bFGF) and ethosuximide. Several studies have demonstrated that bFGF can induce neural differentiation of stem cells derived from distinct mesenchymal tissues, including endometrial [
15] and adipose [
16] tissues. Ethosuximide is a small heterocyclic ring compound of the succinimide class and is used clinically to treat absence seizures in humans [
17]. The compound primarily acts by blocking T-type voltage-gated calcium channels in thalamic neurons [
18]. To the best of our knowledge, this is the first evidence that ethosuximide induces neurogenesis of rMDSCs
in vitro, suggesting that ethosuximide might be useful in inducing neural differentiation of human MDSCs.
2. Results and Discussion
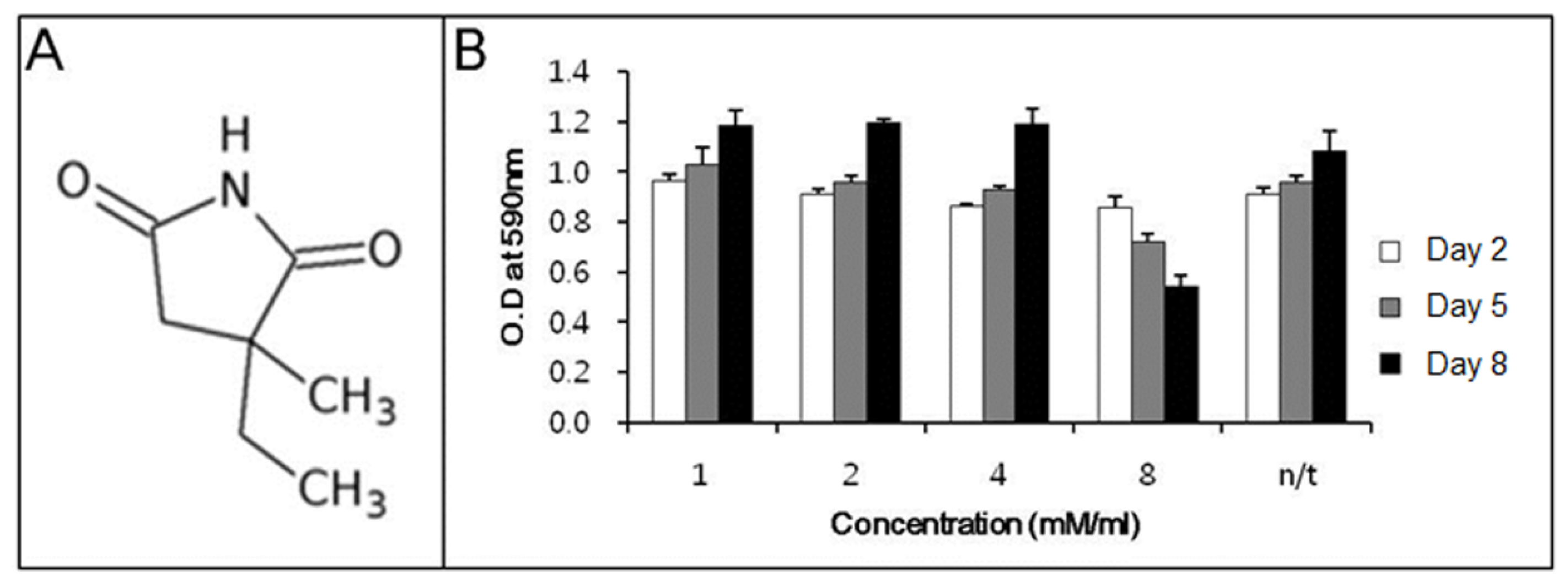
Before determining the ability of ethosuximide as a neurogenic agent, we evaluated its potential cytotoxicity to rMDSCs. There have been no previous reports on the cytotoxic or genotoxic effects of ethosuximide on rMDSCs. Ethosuximide is a succinimide derivative (
Figure 1A) and is highly soluble in aqueous solution. rMDSCs were incubated in the presence or absence of ethosuximide (1, 2, 4, and 8 mM), and the MTT assay was performed at 2, 5, or 8 days after the culture initiation of culture. As shown in
Figure 1B, ethosuximide was toxic to rMDSCs at 8 mM but not at lower concentrations. Cells treated with 8 mM ethosuximide did not proliferate and underwent necrosis after 2 days. However, cells treated with lower concentrations of ethosuximide proliferated well and showed no evidence of cell death over the 8-day incubation period.
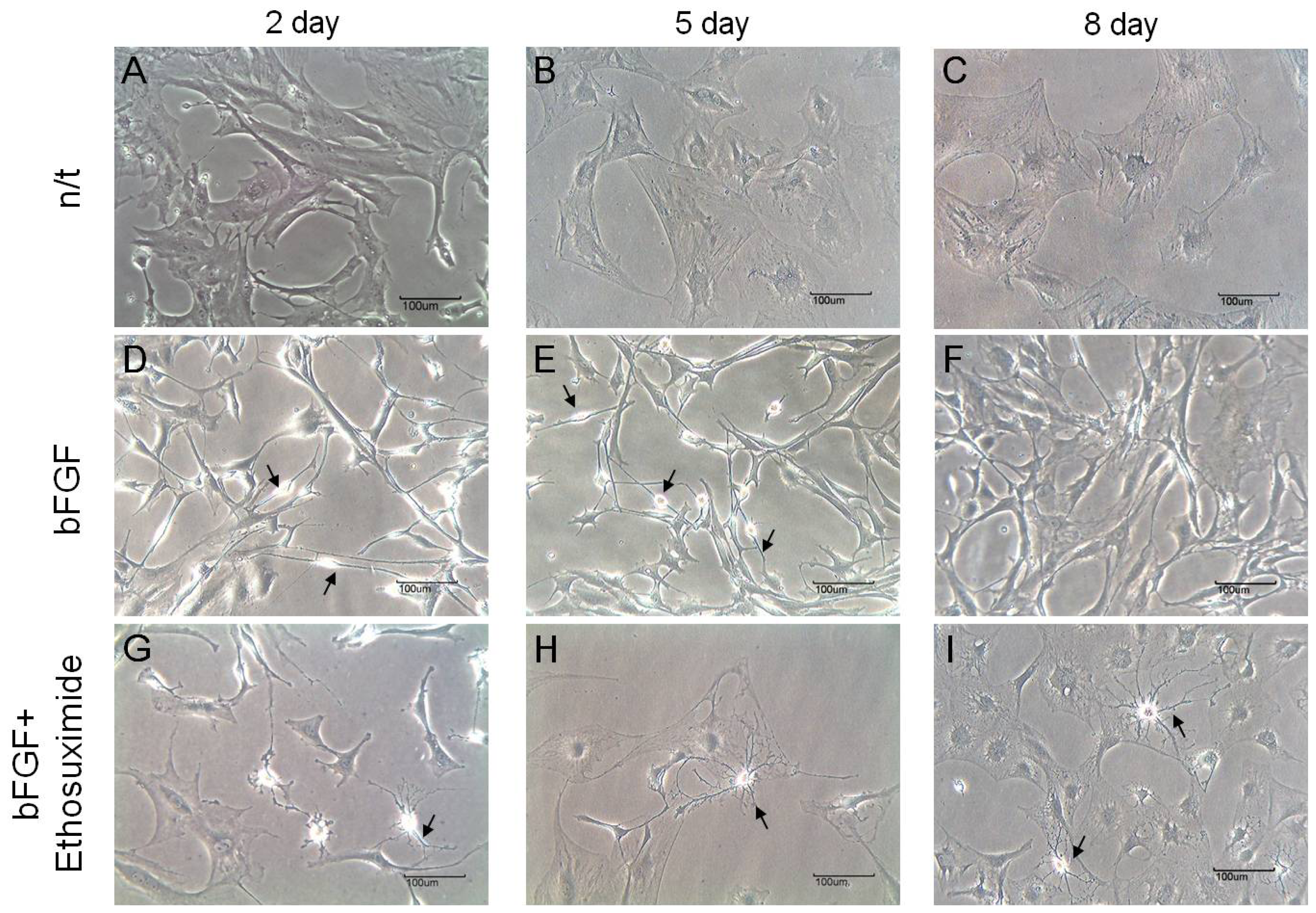
The morphological changes observed in the cultures of rMDSCs treated with bFGF alone or with both bFGF and ethosuximide are shown in
Figure 2. Cells stimulated with bFGF for 24 h and then transferred to medium appeared similar to γ-aminobutyric acid (GABA) neurons, which secrete GABA as their primary neurotransmitter (arrows in
Figure 2D,E). On the other hand, the morphology of rMDSCs treated with both bFGF and ethosuximide was more reminiscent of oligodendrocytes (arrows in
Figure 2G–I). Oligodendrocytes are a variety of neuroglia that play a supporting role for neurons.
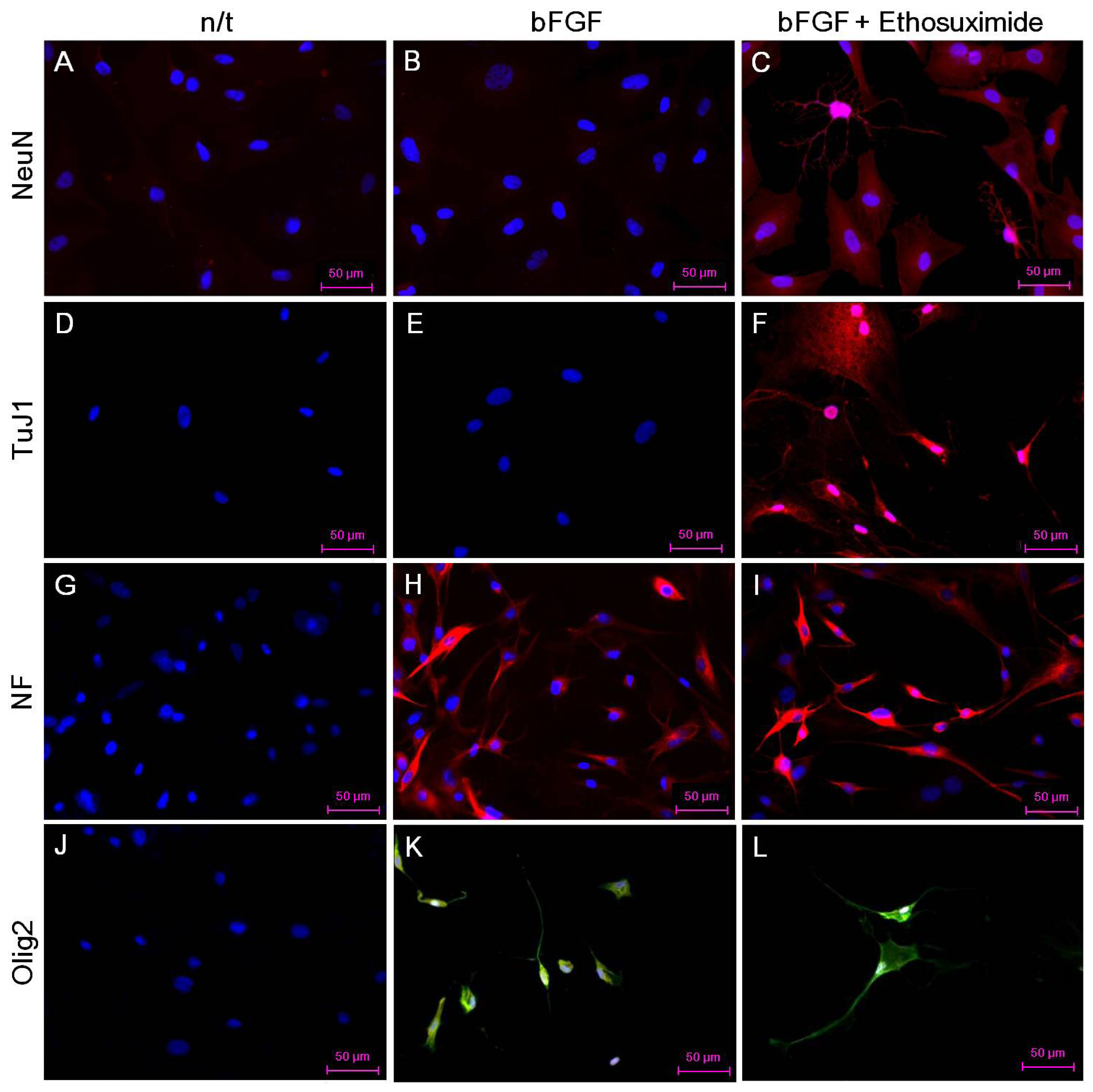
The effect of ethosuximide treatment on lineage-specific differentiation of rMDSCs in culture was monitored by immunocytochemical detection of the neuronal markers NeuN and TuJ1, the oligodendrocyte marker Olig2, and the neurofilament marker NF-MH. These neural markers were not detected in rMDSCs cultured in the absence of bFGF and ethosuximide (
Figure 3A,D,G,J). The rMDSCs stimulated with bFGF alone contained cells expressing NF and Olig2 but not NeuN or TuJ1 (
Figure 3B,E,H,K). However, treatment with both bFGF and ethosuximide induced rMDSCs to differentiate into cells expressing NeuN, TuJ1, and NF (
Figure 3C,F,I) and Olig2 (
Figure 3L), indicating the presence of neuronal lineage and oligodendrocyte lineage cells, respectively. Notably, a larger proportion of cells in these cultures expressed NeuN and TuJ1 than Olig2. These results therefore suggest that rMDSCs treated with ethosuximide differentiated mainly into cells with neural-like characteristics, implying that ethosuximide may stimulate neural differentiation.
To confirm the effects of ethosuximide on the neuronal differentiation of rMDSCs, we performed western blotting analyses of the neuron-specific markers NeuN, TuJ1, NF-M, and NF-H. The results of these experiments were consistent with the immunocytochemical analyses of marker expression shown in
Figure 3. Western blotting of NeuN, TuJ1, and NF confirmed their expression in the rMDSCs (
Figure 4), and the ability of ethosuximide to induce neuronal differentiation was verified by the high levels of NeuN and TuJ1 expression in treated cells (
Figure 4A,B). At day 8, the expression of NeuN was significantly higher in bFGF plus ethosuximide-treated cells than in either untreated cells or cells treated with bFGF alone (
Figure 4A,
p < 0.001). Similarly, TuJ1 expression significantly increased by treatment of bFGF-induced rMDSCs with ethosuximide (
Figure 4B,
p < 0.05). These results therefore suggest that ethosuximide treatment induced differentiation of rMDSCs into cells having a neuronal phenotype.
Although the results of this study do not identify the exact mechanism by which ethosuximide induces the neuronal differentiation of rMDSCs, we suggest that ethosuximide may affect cell signaling. As shown in
Figures 3H,K and
4C,D, rMDSCs treated with bFGF alone expressed Olig2 and high levels of NF, indicating that bFGF alone induced neural differentiation. However, expression of the neuron-specific proteins NeuN and TuJ1 was only observed in cultures treated with both bFGF and ethosuximide. Thus, we hypothesize that rMDSCs are directed towards the neural lineage by bFGF, and ethosuximide acts synergistically to induce further neuronal differentiation. Ethosuximide has been shown to affect the activity of multiple ion channels, including T-type calcium channels, in vertebrate cultured cells [
19,
20]. Therefore, it is possible that ethosuximide may induce signaling for neuronal differentiation by binding to such ion channels in rMDSCs.
3. Experimental Section
3.1. Isolation of Rat MDSCs
All the animals were treated in accordance with the policies and regulations for the care and use of laboratory animals approved by the Institutional Animal Experiment Committee at the College of Medicine, Ajou University. Primary muscle-derived cells were isolated from 4-week-old female Fischer rats by a method described previously [
5,
6]. In brief, the major hind-limb muscles were removed and enzymatically dissociated with 0.2% collagenase type XI (Sigma, Munich, Germany), dispase (Sigma, Munich, Germany), and trypsin (Gibco BRL, Grand Island, NY, USA). Cells were separated from muscle fiber fragments and tissue debris by differential centrifugation and then resuspended in Dulbecco’s modified Eagle’s medium-high glucose (DMEM; Gibco BRL, Grand Island, NY, USA) containing 10% (
v/
v) fetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin (both Invitrogen, Carlsbad, CA, USA). Cells were seeded into flasks for 1 h, and unattached cells were then transferred into new collagen-coated flasks. The subsequent pre-plates were performed in the next 2 h, 3 h, 1 day, 2 days and 3 days. Finally, the rMDSCs were used at passage 5. The identity of the MDSCs was verified by analyzing CD44 and CD45 expression using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) (data not shown).
3.2. Neuronal Differentiation of rMDSCs
rMDSCs were cultured in 6-well plates at a density of 5 × 104 cells/well in DMEM containing 5% (v/v) FBS, 5% (v/v) horse serum (Gibco BRL, Grand Island, NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were incubated for 24 h at 37 °C in a 5% CO2 atmosphere. For pre-induction of neurogenesis, the cells were cultured for 24 h in amplification medium (DMEM containing 20% (v/v) FBS supplemented with 25 ng/mL bFGF (Sigma, St. Louis, MO, USA)). The pre-induction medium was then replaced with DMEM alone or DMEM containing 4 mM ethosuximide (Sigma, St. Louis, MO, USA), and neuronal differentiation was monitored for a further 7 days. rMDSCs incubated for 8 days in the medium alone served as negative controls.
3.3. Analysis of Cytotoxicity
The effect of ethosuximide on cell proliferation and cell death was measured by performing the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) assay. The rMDSCs were cultured in 96-well microplates at a density of 5 × 103 cells/well, according to the neuronal differentiation method described above, except that ethosuximide was present at concentration of 1 mM to 8 mM. On days 2, 5, and 8, MTT was added to each well at a final concentration of 1.0 mg/mL. Four hours later, the supernatant was removed and the insoluble formazan crystals were dissolved in 200 μL of dimethylsulfoxide (Sigma, St. Louis, MO, USA). Absorbance was measured at a wavelength of 590 nm. The relative cell proliferation (%) was related to a 100% confluence per well in the A590 test/A590 100% confluence well.
3.4. Immunocytochemical Analysis
rMDSCs were cultured in 4-well chamber slides (BD Biocoat, Heidelberg, Germany), according to the neuronal differentiation schedule described above. After 5 days of differentiation, the cells were fixed in 4% paraformaldehyde (Medilab, Seoul, Korea) for 20 min at room temperature. For intracellular staining of neuronal nuclear antigen (NeuN), the cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. The primary antibodies were mouse anti-rat NF-MH antibody (recognizing neurofilament 160 kDa and 200 kDa; Abcam, Cambridge, UK), mouse anti-mouse NeuN antibody (Millipore, Billerica, MA, USA), rabbit anti-rat Olig2 antibody (oligodendrocyte transcription factor 2; Millipore, Billerica, MA, USA), and rabbit anti-rat TuJ1 antibody (neuron-specific class III beta-tubulin; Abcam, Cambridge, UK). The cells were then washed and incubated with secondary Alexa Fluor® 488-conjugated goat anti-rabbit antibody (Invitrogen, Carlsbad, CA, USA) or Alexa Fluor® 594-conjugated donkey anti-mouse IgG antibody (Invitrogen, Carlsbad, CA, USA). Nuclei were counterstained with DAPI nucleic acid stain (4′,6-diamino-2-phenylindol dihydrochloride; Invitrogen, Carlsbad, CA, USA). The cells were examined using a fluorescence microscope AxioObserver Z1 equipped with an ApoTome for optical sectioning (Zeiss, Oberkochen, Germany). To detect specific antibody staining, digital images were captured from each fluorescence channel and superimposed using Axiovision™ software (Zeiss).
3.5. Western Blotting Analysis
The cells were incubated for 15 min at 4 °C in RIPA lysis buffer (Cell Signaling Technology, Beverly, MA, USA) and then centrifuged at 12,000 × g for 5 min at 4 °C to remove the insoluble material. The supernatants were collected and protein concentrations were determined using a Nanodrop Spectrophotometer (ND-1000; Thermo Scientific, DE, USA). For western blot analysis, lysates (15 μg protein per sample) were resolved by electrophoresis in NuPAGE® Novex® 4%–12% Bis-Tris gels (Invitrogen, Carlsbad, CA, USA), and proteins were transferred to nitrocellulose membranes (iBlot® Transfer Stack, Invitrogen, Carlsbad, CA, USA). The membranes were probed using antibodies to NF-M, NF-H (both Abcam, UK), NeuN (Millipore, Billerica, MA, USA), TuJ1 (Abcam, Cambridge, UK), and rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, CA, USA) Horseradish peroxidase-conjugated sheep anti-rabbit antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or horseradish peroxidase-conjugated goat anti-mouse IgG (Novus Biologicals, Littleton, CO, USA) were used as secondary antibodies. The reaction products were visualized by chemiluminescence using an ECL western blotting detection kit (GE Healthcare, Milwaukee, WI, USA). Quantification of band densities was performed with an ImageQuant LAS 4000 mini luminescent image analyzer (GE Healthcare, Milwaukee, WI, USA).
3.6. Statistical Analysis
The results of the cytotoxicity assays and western blotting analyses are given as the mean and standard deviation (SD) of 3 independent experiments. The results were analyzed for statistical differences by one-way ANOVA with Bonferroni’s correction for multiple comparisons, using the Prism 3.0 software package (GraphPad Software Inc., San Diego, CA, USA). A p value of 0.05 was considered to be statistically significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}



