Oxidative Stress Mediates the Disruption of Airway Epithelial Tight Junctions through a TRPM2-PLCγ1-PKCα Signaling Pathway

Abstract
:1. Introduction
2. Results and Discussion
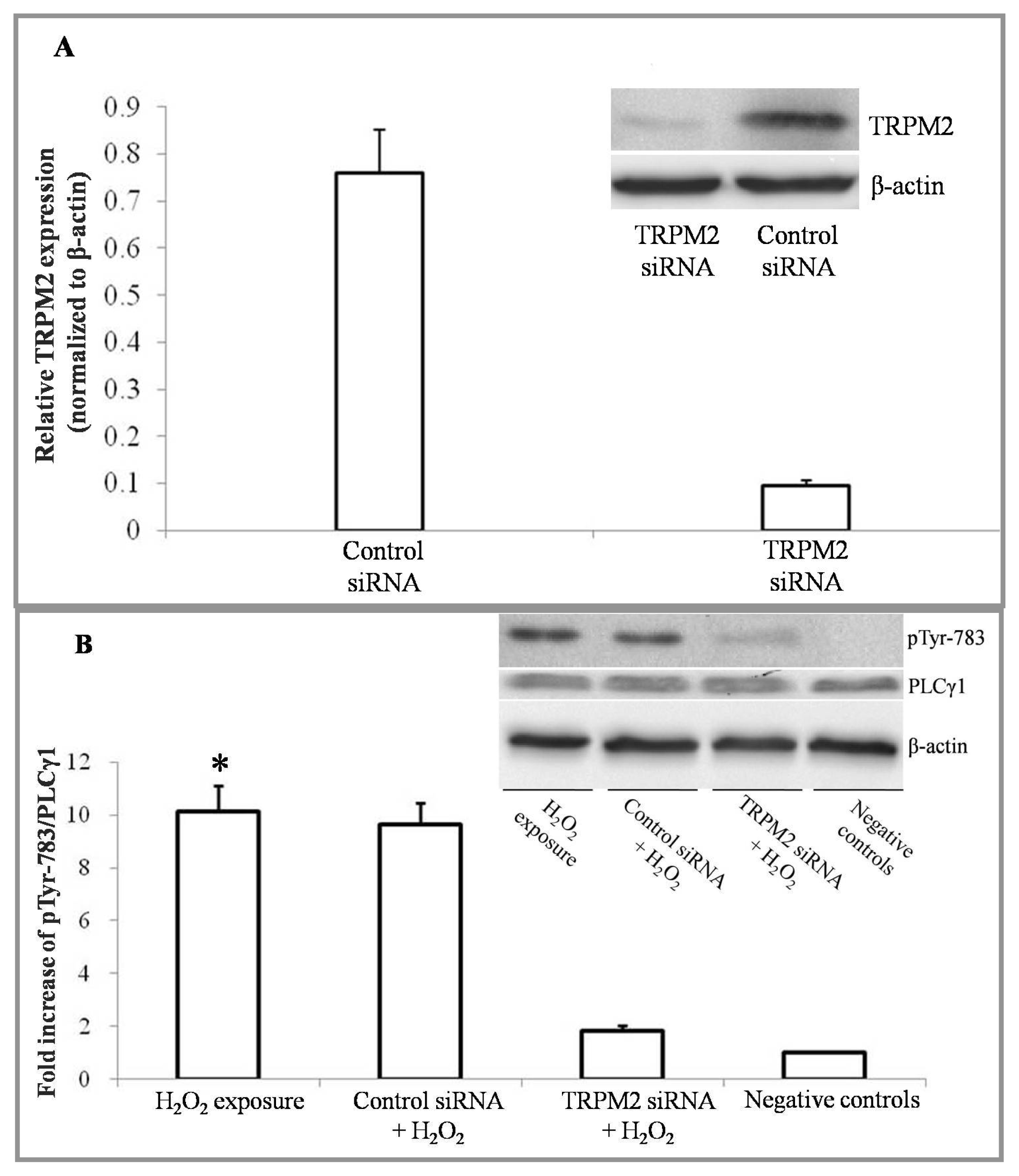
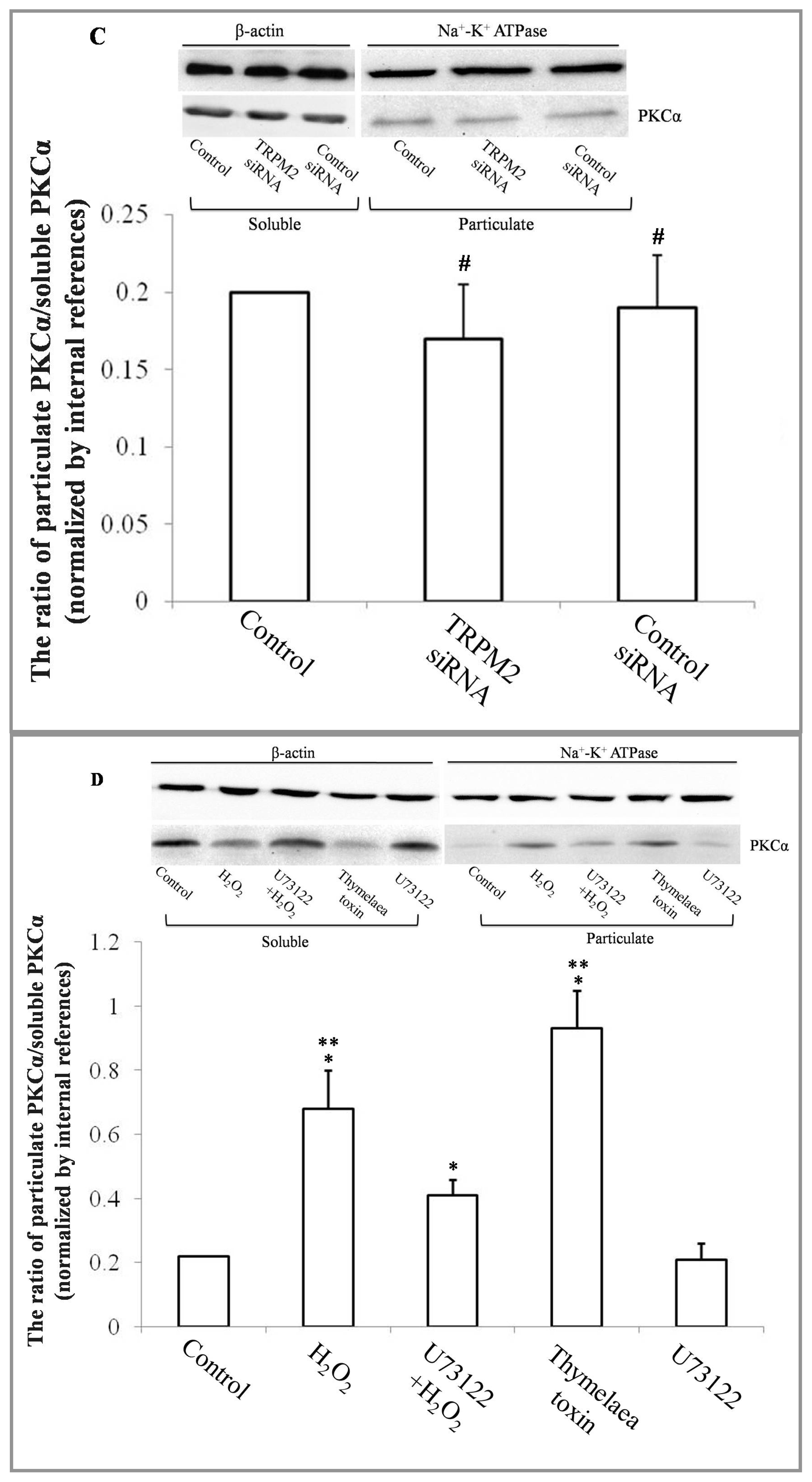
2.1. H2O2 Exposure Activates PLCγ1 and Subsequently, PKCα in a TRPM2 Dependent Manner
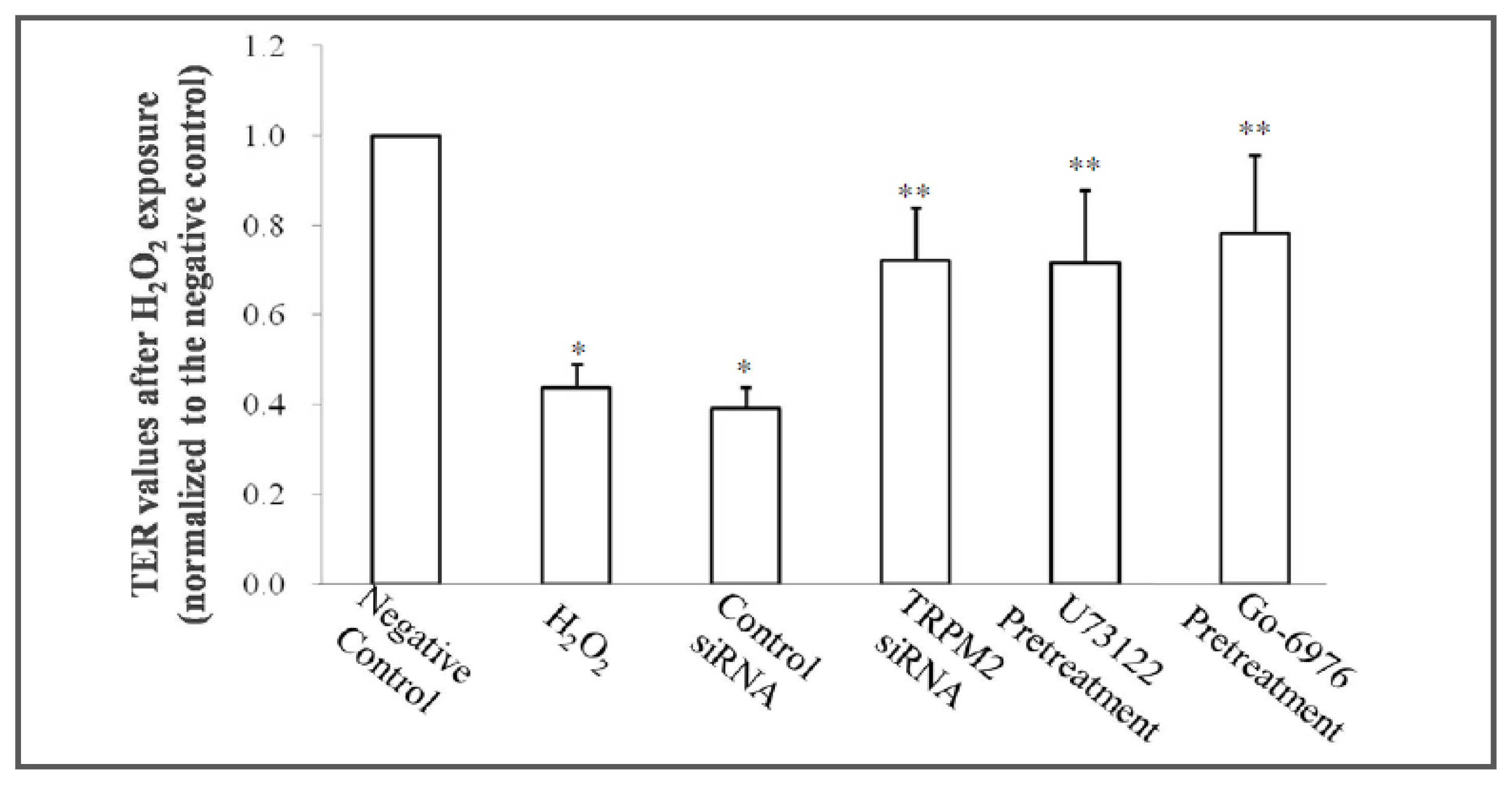
2.2. TRPM2 siRNA and Pretreatment with a PLCγ- or a PKCα-Specific Inhibitor Attenuate the Hyperpermeability Induced by an Oxidative Reaction
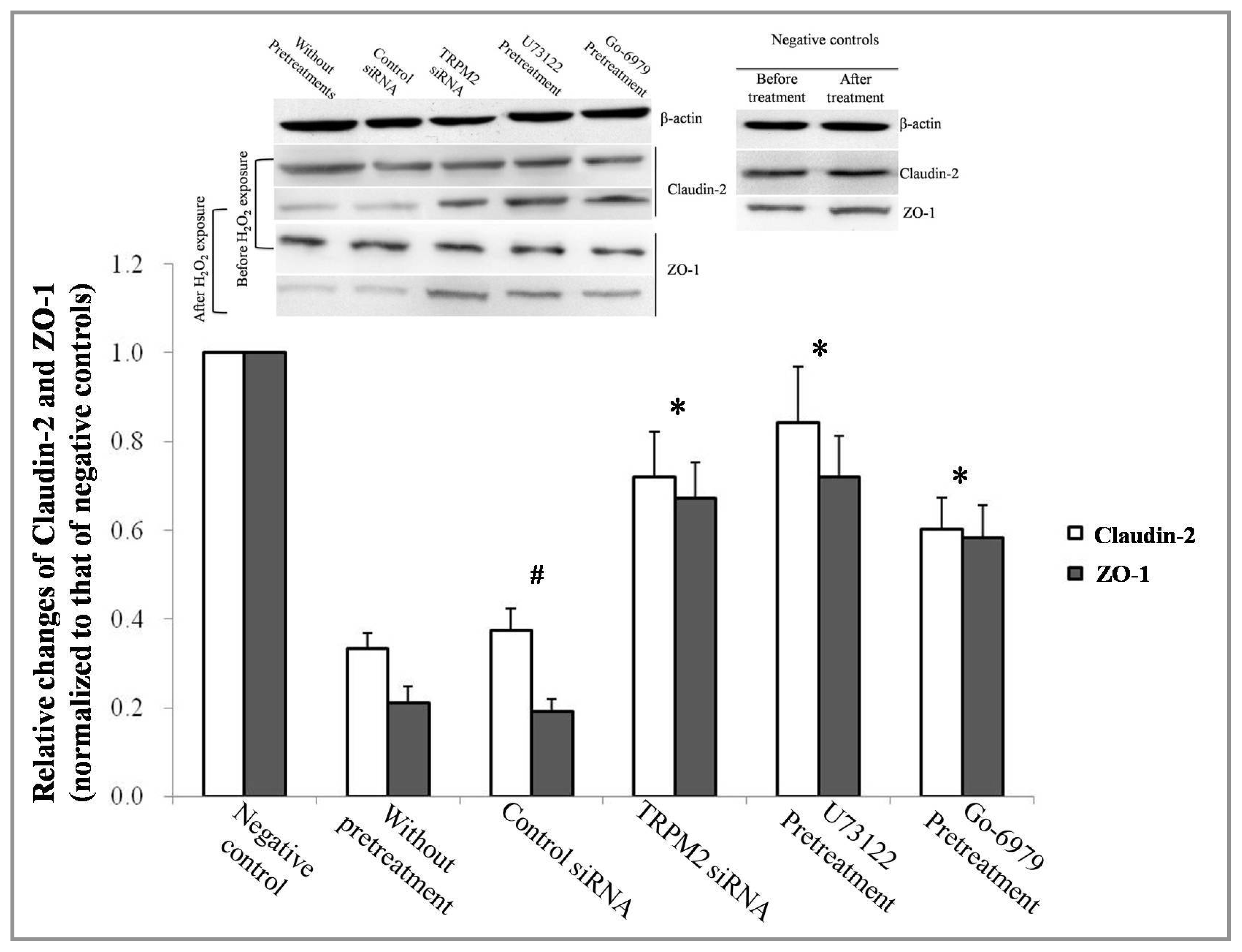
2.3. Effect of TRPM2-PLCγ1-PKCα on ZO-1 and Claudin-2 Expression
3. Experimental Section
3.1. Materials
3.2. Cell Culture and Treatment
3.3. Small Interfering RNA Transfection
3.4. H2O2 Exposure
3.5. Epithelial Barrier Function of 16HBE Cells
3.6. Western Blot
3.7. PLCγ1 and PKCα Activity Assay
3.8. Statistical Analysis
4. Conclusions
Supplemental Information
ijms-14-09475-s001.pdfAcknowledgments
Conflict of Interest
References
- Tomita, K.; Barnes, P.J.; Adcock, I.M. The effect of oxidative stress on histone acetylation and IL-8 release. Biochem. Biophys. Res. Commun 2003, 301, 572–577. [Google Scholar]
- Van Itallie, C.M.; Anderson, J.M. The molecular physiology of tight junction pores. Physiology 2004, 19, 331–338. [Google Scholar]
- Balda, M.S.; Matter, K. Tight junctions and the regulation of gene expression. Biochim. Biophys. Acta 2009, 1788, 761–767. [Google Scholar]
- Wang, F.; Daugherty, B.; Keise, L.L.; Wei, Z.; Foley, J.P.; Savani, R.C.; Koval, M. Heterogeneity of claudin expression by alveolar epithelial cells. Am. J. Respir. Cell Mol. Biol 2003, 29, 62–70. [Google Scholar]
- Koval, M. Tight junctions, but not too tight: Fine control of lung permeability by claudins. Am. J. Physiol. Lung Cell Mol. Physiol 2009, 297, L217–L218. [Google Scholar]
- Peter, Y.; Comellas, A.; Levantini, E.; Ingenito, E.P.; Shapiro, S.D. Epidermal growth factor receptor and claudin-2 participate in A549 permeability and remodeling: Implications for non-small cell lung cancer tumor colonization. Mol. Carcinog 2009, 48, 488–497. [Google Scholar]
- Sun, Y.; Minshall, R.D.; Hu, G. Role of claudins in oxidant-induced alveolar epithelial barrier dysfunction. Methods Mol. Biol 2011, 762, 291–301. [Google Scholar]
- Denker, B.M.; Nigam, S.K. Molecular structure and assembly of the tight junction. Am. J. Physiol 1998, 274, F1–F9. [Google Scholar]
- Chen, W.; Hu, J.; Zhang, Z.; Chen, L.; Xie, H.; Dong, N.; Chen, Y.; Liu, Z. Localization and expression of zonula occludins-1 in the rabbit corneal epithelium following exposure to benzalkonium chloride. PLoS One 2012, 7, e40893. [Google Scholar]
- Han, X.; Fink, M.P.; Yang, R.; Delude, R.L. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock 2004, 21, 261–270. [Google Scholar]
- Watson, P.M.; Anderson, J.M.; Vanltallie, C.M.; Doctrow, S.R. The tight-junction-specific protein ZO-1 is a component of the human and rat blood-brain barriers. Neurosci. Lett 1991, 129, 6–10. [Google Scholar]
- Relova, A.J.; Shahana, S.; Makeeva, N.; Roomans, G.M. Effect of cytokines on ICAM-1 and ZO-1 expression on human airway epithelial cells. Cell Biol. Int 2005, 29, 768–777. [Google Scholar]
- Clarke, H.; Ginanni, N.; Laughlin, K.V.; Smith, J.B.; Pettit, G.R.; Mullin, J.M. The transient increase of tight junction permeability induced by bryostatin 1 correlates with rapid downregulation of protein kinase C-alpha. Exp. Cell Res 2000, 261, 239–249. [Google Scholar]
- Song, J.C.; Hanson, C.M.; Tsai, V.; Farokhzad, O.C.; Lotz, M.; Matthews, J.B. Regulation of epithelial transport and barrier function by distinct protein kinase C isoforms. Am. J. Physiol. Cell Physiol 2001, 281, C649–C661. [Google Scholar]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res 2008, 102, 347–355. [Google Scholar]
- Kraft, R.; Grimm, C.; Grosse, K.; Hoffmann, A.; Sauerbruch, S.; Kettenmann, H.; Schultz, G.; Harteneck, C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am. J. Physiol. Cell Physiol 2004, 286, C129–C137. [Google Scholar]
- Son, H.; Lim, Y.; Kim, J.; Park, H.; Choi, S.; Han, I.; Kim, W.S.; Park, S.; Bae, Y.; Oh, E.S. Protein kinase Calpha can undergo membrane localization via an alternative phosphatidylinositol 4,5-bisphosphate-dependent pathway. Arch. Biochem. Biophys 2006, 454, 1–6. [Google Scholar]
- Gonzalez-Pacheco, F.R.; Caramelo, C.; Castilla, M.A.; Deudero, J.J.; Arias, J.; Yague, S.; Jimenez, S.; Bragado, R.; Alvarez-Arroyo, M.V. Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: Role of phospholipase C gamma. Nephrol. Dial. Transplant 2002, 17, 392–398. [Google Scholar]
- Hong, J.H.; Moon, S.J.; Byun, H.M.; Kim, M.S.; Jo, H.; Bae, Y.S.; Lee, S.I.; Bootman, M.D.; Roderick, H.L.; Shin, D.M.; et al. Critical role of phospholipase Cgamma1 in the generation of H2O2-evoked [Ca2+]i oscillations in cultured rat cortical astrocytes. J. Biol. Chem 2006, 281, 13057–13067. [Google Scholar]
- Simet, S.M.; Wyatt, T.A.; DeVasure, J.; Yanov, D.; Allen-Gipson, D.; Sisson, J.H. Alcohol increases the permeability of airway epithelial tight junctions in Beas-2B and NHBE cells. Alcohol. Clin. Exp. Res 2012, 36, 432–442. [Google Scholar]
- Marin-Vicente, C.; Gomez-Fernandez, J.C.; Corbalan-Garcia, S. The ATP-dependent membrane localization of protein kinase Calpha is regulated by Ca2+ influx and phosphatidylinositol 4,5-bisphosphate in differentiated PC12 cells. Mol. Biol. Cell 2005, 16, 2848–2861. [Google Scholar]
- Kim, H.K.; Kim, J.W.; Zilberstein, A.; Margolis, B.; Kim, J.G.; Schlessinger, J.; Rhee, S.G. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell 1991, 65, 435–441. [Google Scholar]
- Wang, Z.; Gluck, S.; Zhang, L.; Moran, M.F. Requirement for phospholipase C-gamma1 enzymatic activity in growth factor-induced mitogenesis. Mol. Cell Biol 1998, 18, 590–597. [Google Scholar]
- Tomas, N.M.; Masur, K.; Piecha, J.C.; Niggemann, B.; Zanker, K.S. Akt and phospholipase Cgamma are involved in the regulation of growth and migration of MDA-MB-468 breast cancer and SW480 colon cancer cells when cultured with diabetogenic levels of glucose and insulin. BMC Res. Notes 2012, 5, 214. [Google Scholar]
- Casalino-Matsuda, S.M.; Monzon, M.E.; Forteza, R.M. Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am. J. Respi. Cell Mol. Biol 2006, 34, 581–591. [Google Scholar]
- Boldogh, I.; Bacsi, A.; Choudhury, B.K.; Dharajiya, N.; Alam, R.; Hazra, T.K.; Mitra, S.; Goldblum, R.M.; Sur, S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J. Clin. Invest 2005, 115, 2169–2179. [Google Scholar]
- Fukui, A.; Naito, Y.; Handa, O.; Kugai, M.; Tsuji, T.; Yoriki, H.; Qin, Y.; Adachi, S.; Higashimura, Y.; Mizushima, K.; et al. Acetyl salicylic acid induces damage to intestinal epithelial cells by oxidation-related modifications of ZO-1. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 303, G927–G936. [Google Scholar]
- Eisfeld, J.; Luckhoff, A. Trpm2. Handb. Exp. Pharmacol 2007, 179, 237–252. [Google Scholar]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem 2005, 280, 6138–6148. [Google Scholar]
- Martin, E.; Rosenthal, R.E.; Fiskum, G. Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res 2005, 79, 240–247. [Google Scholar]
- Yadav, V.R.; Song, T.; Joseph, L.; Mei, L.; Zheng, Y.M.; Wang, Y.X. Important role of PLCgamma1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2012. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TER values before treatments (Ω·cm2) | TER values after treatments (Ω·cm2) | |
|---|---|---|
| Negative control | 351.41 ± 30.91 | 379.39 ± 41.11 * |
| H2O2 exposure | 360.83 ± 48.94 # | 166.04 ± 34.31 |
| Control siRNA | 345.41 ± 26.92 # | 148.78 ± 29.19 |
| TRPM2 siRNA | 376.67 ± 56.54 # | 273.40 ± 44.80 * |
| U73122 pretreatment | 349.05 ± 49.71 # | 271.83 ± 38.50 * |
| Go-6976 pretreatment | 364.42 ± 56.74 # | 296.44 ± 29.99 * |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, R.; Li, Q.; Zhou, X.-D.; Perelman, J.M.; Kolosov, V.P. Oxidative Stress Mediates the Disruption of Airway Epithelial Tight Junctions through a TRPM2-PLCγ1-PKCα Signaling Pathway. Int. J. Mol. Sci. 2013, 14, 9475-9486. https://doi.org/10.3390/ijms14059475
Xu R, Li Q, Zhou X-D, Perelman JM, Kolosov VP. Oxidative Stress Mediates the Disruption of Airway Epithelial Tight Junctions through a TRPM2-PLCγ1-PKCα Signaling Pathway. International Journal of Molecular Sciences. 2013; 14(5):9475-9486. https://doi.org/10.3390/ijms14059475
Chicago/Turabian StyleXu, Rui, Qi Li, Xiang-Dong Zhou, Juliy M. Perelman, and Victor P. Kolosov. 2013. "Oxidative Stress Mediates the Disruption of Airway Epithelial Tight Junctions through a TRPM2-PLCγ1-PKCα Signaling Pathway" International Journal of Molecular Sciences 14, no. 5: 9475-9486. https://doi.org/10.3390/ijms14059475
APA StyleXu, R., Li, Q., Zhou, X. -D., Perelman, J. M., & Kolosov, V. P. (2013). Oxidative Stress Mediates the Disruption of Airway Epithelial Tight Junctions through a TRPM2-PLCγ1-PKCα Signaling Pathway. International Journal of Molecular Sciences, 14(5), 9475-9486. https://doi.org/10.3390/ijms14059475