Immune-Pineal Axis: Nuclear Factor κB (NF-kB) Mediates the Shift in the Melatonin Source from Pinealocytes to Immune Competent Cells

{kind=link}
Abstract
:1. Introduction
2. NF-κB Signaling
3. Melatonin and the Regulation of NF-κB Activation
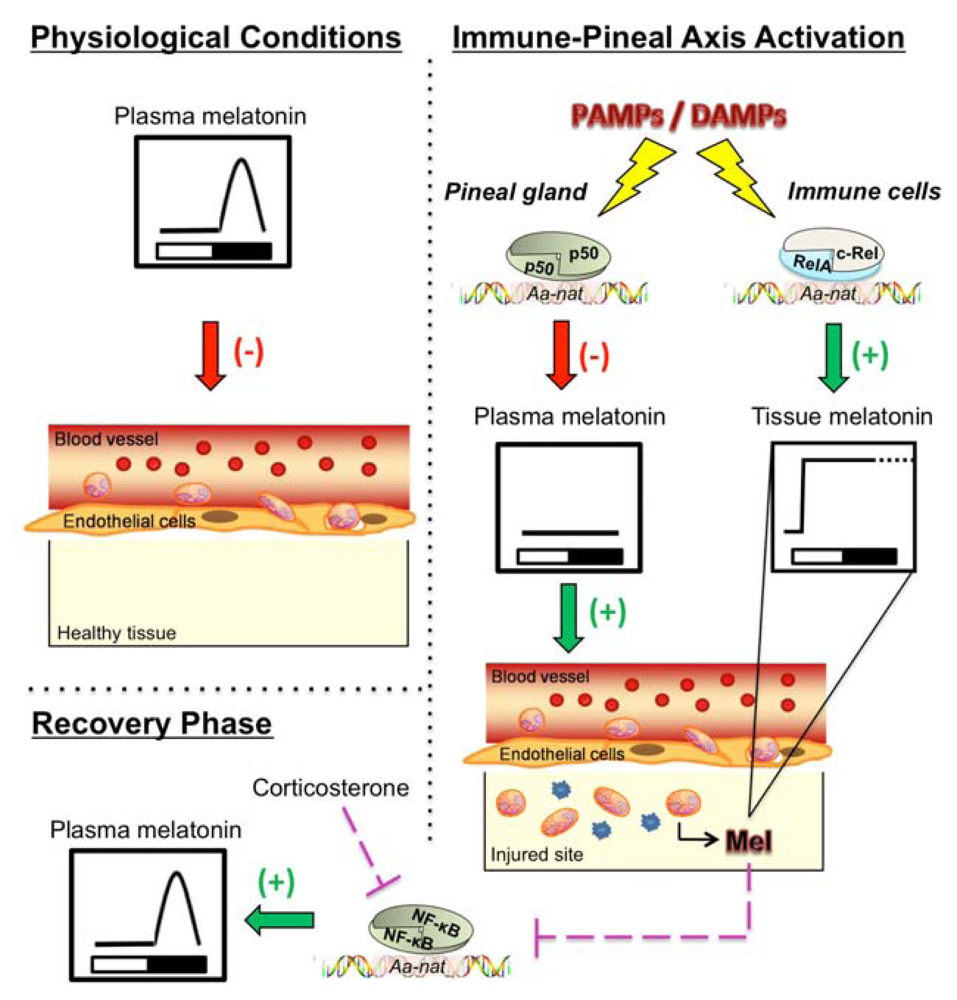
4. NF-κB and the Pineal Gland
5. NF-κB and the Regulation of Melatonin Synthesis in Extra-Pineal Tissues
6. The Immune-Pineal Axis: The NF-κB Pathway Coordinates Pineal and Extra-Pineal Melatonin Synthesis
7. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Reiter, R.J. The melatonin rhythm: Both a clock and a calendar. Experientia 1993, 49, 654–664. [Google Scholar]
- Tan, D.X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res 2007, 42, 28–42. [Google Scholar]
- Guerrero, J.M.; Reiter, R.J. Melatonin-immune system relationships. Curr. Top. Med. Chem 2002, 2, 167–179. [Google Scholar]
- Skwarlo-Sonta, K. Melatonin in immunity: Comparative aspects. Neuro Endocrinol. Lett 2002, 23, 61–66. [Google Scholar]
- Carrillo-Vico, A.; Guerrero, J.M.; Lardone, P.J.; Reiter, R.J. A review of the multiple actions of melatonin on the immune system. Endocrine 2005, 27, 189–200. [Google Scholar]
- Carrillo-Vico, A.; Lardone, P.J.; Alvarez-Sanchez, N.; Rodriguez-Rodriguez, A.; Guerrero, J.M. Melatonin: Buffering the immune system. Int. J. Mol. Sci 2013, 14, 8638–8683. [Google Scholar]
- Radogna, F.; Diederich, M.; Ghibelli, L. Melatonin: A pleiotropic molecule regulating inflammation. Biochem. Pharmacol 2010, 80, 1844–1852. [Google Scholar]
- Haus, E.; Lakatua, D.J.; Swoyer, J.; Sackett-Lundeen, L. Chronobiology in hematology and immunology. Am. J. Anat 1983, 168, 467–517. [Google Scholar]
- Petrovsky, N.; Harrison, L.C. The chronobiology of human cytokine production. Int. Rev. Immunol 1998, 16, 635–649. [Google Scholar]
- Trufakin, V.A.; Shurlygina, A.V.; Michurina, S.V.; Verbitskaja, L.V.; Litvinenko, G.I.; Kovshik, I.G.; Panteleeva, N.G.; Melnikova, E.V.; Bitchaeva, M.V. The influence of experimental desynchronosis on the morphofunctional characteristics of mouse immune system. Alaska Med 2007, 49, 169–176. [Google Scholar]
- Mazzoccoli, G.; A.D.E.C.; Carughi, S.; Greco, A.; Inglese, M.; Perfetto, F.; Tarquini, R. A possible mechanism for altered immune response in the elderly. In Vivo 2010, 24, 471–487. [Google Scholar]
- Haldar, C.; Ahmad, R. Photoimmunomodulation and melatonin. J. Photochem. Photobiol. B 2010, 98, 107–117. [Google Scholar]
- Lotufo, C.M.; Lopes, C.; Dubocovich, M.L.; Farsky, S.H.; Markus, R.P. Melatonin and N-acetylserotonin inhibit leukocyte rolling and adhesion to rat microcirculation. Eur. J. Pharmacol 2001, 430, 351–357. [Google Scholar]
- Bertuglia, S.; Colantuoni, A.; Intaglietta, M. Effect of leukocyte adhesion and microvascular permeability on capillary perfusion during ischemia-reperfusion injury in hamster cheek pouch. Int. J. Microcirc. Clin. Exp 1993, 13, 13–26. [Google Scholar]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar]
- Panayi, G.S.; Corrigall, V.M.; Henderson, B. Stress cytokines: Pivotal proteins in immune regulatory networks: Opinion. Curr. Opin. Immunol 2004, 16, 531–534. [Google Scholar]
- Fernandes, P.A.; Cecon, E.; Markus, R.P.; Ferreira, Z.S. Effect of TNF-alpha on the melatonin synthetic pathway in the rat pineal gland: Basis for a “feedback” of the immune response on circadian timing. J. Pineal Res 2006, 41, 344–350. [Google Scholar]
- da Silveira Cruz-Machado, S.; Carvalho-Sousa, C.E.; Tamura, E.K.; Pinato, L.; Cecon, E.; Fernandes, P.A.; de Avellar, M.C.; Ferreira, Z.S.; Markus, R.P. TLR4 and CD14 receptors expressed in rat pineal gland trigger NFKB pathway. J. Pineal Res 2010, 49, 183–192. [Google Scholar]
- Pontes, G.N.; Cardoso, E.C.; Carneiro-Sampaio, M.M.; Markus, R.P. Injury switches melatonin production source from endocrine (pineal) to paracrine (phagocytes)—melatonin in human colostrum and colostrum phagocytes. J. Pineal Res 2006, 41, 136–141. [Google Scholar]
- Muxel, S.M.; Pires-Lapa, M.A.; Monteiro, A.W.; Cecon, E.; Tamura, E.K.; Floeter-Winter, L.M.; Markus, R.P. NF-kappaB drives the synthesis of melatonin in RAW 264.7 macrophages by inducing the transcription of the arylalkylamine-N-acetyltransferase (AA-NAT) gene. PLoS One 2012, 7, e52010. [Google Scholar]
- Markus, R.P.; Ferreira, Z.S.; Fernandes, P.A.; Cecon, E. The immune-pineal axis: A shuttle between endocrine and paracrine melatonin sources. Neuroimmunomodulation 2007, 14, 126–133. [Google Scholar]
- Markus, R.P.; Ferreira, Z.S. The Immune-pineal axis: The role of pineal and extra-pineal melatonin in modulating inflammation. Adv. Neuroimmune Biol 2011, 1, 95–104. [Google Scholar]
- Gilmore, T.D.; Wolenski, F.S. NF-kappaB: Where did it come from and why? Immunol. Rev 2012, 246, 14–35. [Google Scholar]
- Nabel, G.J.; Verma, I.M. Proposed NF-kappa B/I kappa B family nomenclature. Genes Dev 1993, 7, 2063. [Google Scholar]
- Zhang, Y.L.; Dong, C. MAP kinases in immune responses. Cell Mol. Immunol 2005, 2, 20–27. [Google Scholar]
- Li, J.; Tang, Y.; Cai, D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol 2012, 14, 999–1012. [Google Scholar]
- Colombo, B.M.; Canevali, P.; Magnani, O.; Rossi, E.; Puppo, F.; Zocchi, M.R.; Poggi, A. Defective expression and function of the leukocyte associated Ig-like receptor 1 in B lymphocytes from systemic lupus erythematosus patients. PLoS One 2012, 7, e31903. [Google Scholar]
- Gaudio, E.; Spizzo, R.; Paduano, F.; Luo, Z.; Efanov, A.; Palamarchuk, A.; Leber, A.S.; Kaou, M.; Zanesi, N.; Bottoni, A.; et al. Tcl1 interacts with Atm and enhances NF-kappaB activation in hematologic malignancies. Blood 2012, 119, 180–187. [Google Scholar]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell 2007, 128, 369–381. [Google Scholar]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect Biol 2009, 1, a000141. [Google Scholar]
- Razani, B.; Zarnegar, B.; Ytterberg, A.J.; Shiba, T.; Dempsey, P.W.; Ware, C.F.; Loo, J.A.; Cheng, G. Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal 2010, 3, ra41. [Google Scholar]
- Le Negrate, G. Subversion of innate immune responses by bacterial hindrance of NF-kappaB pathway. Cell Microbiol 2012, 14, 155–167. [Google Scholar]
- Courtine, E.; Pene, F.; Cagnard, N.; Toubiana, J.; Fitting, C.; Brocheton, J.; Rousseau, C.; Gerondakis, S.; Chiche, J.D.; Ouaaz, F.; et al. Critical role of cRel subunit of NF-kappaB in sepsis survival. Infect. Immun 2011, 79, 1848–1854. [Google Scholar]
- Pizzi, M.; Sarnico, I.; Lanzillotta, A.; Battistin, L.; Spano, P. Post-ischemic brain damage: NF-kappaB dimer heterogeneity as a molecular determinant of neuron vulnerability. FEBS J 2009, 276, 27–35. [Google Scholar]
- Sarnico, I.; Lanzillotta, A.; Boroni, F.; Benarese, M.; Alghisi, M.; Schwaninger, M.; Inta, I.; Battistin, L.; Spano, P.; Pizzi, M. NF-kappaB p50/RelA and c-Rel-containing dimers: Opposite regulators of neuron vulnerability to ischaemia. J. Neurochem 2009, 108, 475–485. [Google Scholar]
- Bhakar, A.L.; Tannis, L.L.; Zeindler, C.; Russo, M.P.; Jobin, C.; Park, D.S.; MacPherson, S.; Barker, P.A. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J. Neurosci 2002, 22, 8466–8475. [Google Scholar]
- Pereira, S.G.; Oakley, F. Nuclear factor-kappaB1: Regulation and function. Int. J. Biochem. Cell Biol 2008, 40, 1425–1430. [Google Scholar]
- Saccani, S.; Pantano, S.; Natoli, G. Two waves of nuclear factor kappa B recruitment to target promoters. J. Exp. Med 2001, 193, 1351–1359. [Google Scholar]
- Nelson, D.E.; Ihekwaba, A.E.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar]
- Sanjabi, S.; Williams, K.J.; Saccani, S.; Zhou, L.; Hoffmann, A.; Ghosh, G.; Gerondakis, S.; Natoli, G.; Smale, S.T. A c-Rel subdomain responsible for enhanced DNA-binding affinity and selective gene activation. Genes Dev 2005, 19, 2138–2151. [Google Scholar]
- Mohan, N.; Sadeghi, K.; Reiter, R.J.; Meltz, M.L. The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induced NF-kappa B. Biochem. Mol. Biol. Int 1995, 37, 1063–1070. [Google Scholar]
- Gilad, E.; Wong, H.R.; Zingarelli, B.; Virag, L.; O’Connor, M.; Salzman, A.L.; Szabo, C. Melatonin inhibits expression of the inducible isoform of nitric oxide synthase in murine macrophages: Role of inhibition of NFkappaB activation. FASEB J 1998, 12, 685–693. [Google Scholar]
- Maldonado, M.D.; Mora-Santos, M.; Naji, L.; Carrascosa-Salmoral, M.P.; Naranjo, M.C.; Calvo, J.R. Evidence of melatonin synthesis and release by mast cells. Possible modulatory role on inflammation. Pharmacol. Res 2010, 62, 282–287. [Google Scholar]
- Pedrosa, A.M.; Weinlich, R.; Mognol, G.P.; Robbs, B.K.; Viola, J.P.; Campa, A.; Amarante-Mendes, G.P. Melatonin protects CD4+ T cells from activation-induced cell death by blocking NFAT-mediated CD95 ligand upregulation. J. Immunol 2010, 184, 3487–3494. [Google Scholar]
- Choi, E.Y.; Jin, J.Y.; Lee, J.Y.; Choi, J.I.; Choi, I.S.; Kim, S.J. Melatonin inhibits Prevotella intermedia lipopolysaccharide-induced production of nitric oxide and interleukin-6 in murine macrophages by suppressing NF-kappaB and STAT1 activity. J. Pineal Res 2011, 50, 197–206. [Google Scholar]
- Murakami, Y.; Yuhara, K.; Takada, N.; Arai, T.; Tsuda, S.; Takamatsu, S.; Machino, M.; Fujisawa, S. Effect of melatonin on cyclooxygenase-2 expression and nuclear factor-kappa B activation in RAW264.7 macrophage-like cells stimulated with fimbriae of Porphyromonas gingivalis. In Vivo 2011, 25, 641–647. [Google Scholar]
- Chang, C.C.; Tien, C.H.; Lee, E.J.; Juan, W.S.; Chen, Y.H.; Hung, Y.C.; Chen, T.Y.; Chen, H.Y.; Wu, T.S. Melatonin inhibits matrix metalloproteinase-9 (MMP-9) activation in the lipopolysaccharide (LPS)-stimulated RAW 264.7 and BV2 cells and a mouse model of meningitis. J. Pineal Res 2012, 53, 188–197. [Google Scholar]
- Post, A.; Holsboer, F.; Behl, C. Induction of NF-kappaB activity during haloperidol-induced oxidative toxicity in clonal hippocampal cells: Suppression of NF-kappaB and neuroprotection by antioxidants. J. Neurosci 1998, 18, 8236–8246. [Google Scholar]
- Fang, Q.; Chen, G.; Zhu, W.; Dong, W.; Wang, Z. Influence of melatonin on cerebrovascular proinflammatory mediators expression and oxidative stress following subarachnoid hemorrhage in rabbits. Mediators Inflamm 2009, 2009, 426346. [Google Scholar]
- Bruck, R.; Aeed, H.; Avni, Y.; Shirin, H.; Matas, Z.; Shahmurov, M.; Avinoach, I.; Zozulya, G.; Weizman, N.; Hochman, A. Melatonin inhibits nuclear factor kappa B activation and oxidative stress and protects against thioacetamide induced liver damage in rats. J. Hepatol 2004, 40, 86–93. [Google Scholar]
- Quiroz, Y.; Ferrebuz, A.; Romero, F.; Vaziri, N.D.; Rodriguez-Iturbe, B. Melatonin ameliorates oxidative stress, inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction. Am. J. Physiol. Renal Physiol 2008, 294, F336–F344. [Google Scholar]
- Ozbek, E.; Ilbey, Y.O.; Ozbek, M.; Simsek, A.; Cekmen, M.; Somay, A. Melatonin attenuates unilateral ureteral obstruction-induced renal injury by reducing oxidative stress, iNOS, MAPK, and NF-kB expression. J. Endourol 2009, 23, 1165–1173. [Google Scholar]
- Shang, Y.; Xu, S.P.; Wu, Y.; Jiang, Y.X.; Wu, Z.Y.; Yuan, S.Y.; Yao, S.L. Melatonin reduces acute lung injury in endotoxemic rats. Chin. Med. J. (Engl. ) 2009, 122, 1388–1393. [Google Scholar]
- Veneroso, C.; Tunon, M.J.; Gonzalez-Gallego, J.; Collado, P.S. Melatonin reduces cardiac inflammatory injury induced by acute exercise. J. Pineal Res 2009, 47, 184–191. [Google Scholar]
- Sasaki, M.; Jordan, P.; Joh, T.; Itoh, M.; Jenkins, M.; Pavlick, K.; Minagar, A.; Alexander, S.J. Melatonin reduces TNF-a induced expression of MAdCAM-1 via inhibition of NF-kappaB. BMC Gastroenterol 2002, 2, 9. [Google Scholar]
- Li, J.H.; Yu, J.P.; Yu, H.G.; Xu, X.M.; Yu, L.L.; Liu, J.; Luo, H.S. Melatonin reduces inflammatory injury through inhibiting NF-kappaB activation in rats with colitis. Mediators Inflamm 2005, 2005, 185–193. [Google Scholar]
- Ganguly, K.; Swarnakar, S. Chronic gastric ulceration causes matrix metalloproteinases-9 and −3 augmentation: Alleviation by melatonin. Biochimie 2012, 94, 2687–2698. [Google Scholar]
- Negi, G.; Kumar, A.; Sharma, S.S. Melatonin modulates neuroinflammation and oxidative stress in experimental diabetic neuropathy: Effects on NF-kappaB and Nrf2 cascades. J. Pineal Res 2011, 50, 124–131. [Google Scholar]
- Esposito, E.; Paterniti, I.; Mazzon, E.; Bramanti, P.; Cuzzocrea, S. Melatonin reduces hyperalgesia associated with inflammation. J. Pineal Res 2010, 49, 321–331. [Google Scholar]
- Genovese, T.; Mazzon, E.; Muia, C.; Bramanti, P.; de Sarro, A.; Cuzzocrea, S. Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J. Pineal Res 2005, 38, 198–208. [Google Scholar]
- Jung, K.H.; Hong, S.W.; Zheng, H.M.; Lee, D.H.; Hong, S.S. Melatonin downregulates nuclear erythroid 2-related factor 2 and nuclear factor-kappaB during prevention of oxidative liver injury in a dimethylnitrosamine model. J. Pineal Res 2009, 47, 173–183. [Google Scholar]
- Cai, C.; Zhang, H.Y.; Le, J.J.; Dong, J.C.; Cui, Y.; Xu, C.Q.; Liu, B.J.; Wu, J.F.; Duan, X.H.; Cao, Y.X. Inflammatory airway features and hypothalamic-pituitary-adrenal axis function in asthmatic rats combined with chronic obstructive pulmonary disease. Chin. Med. J. (Engl. ) 2010, 123, 1720–1726. [Google Scholar]
- Huang, S.H.; Cao, X.J.; Wei, W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-kappa B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J. Pineal Res 2008, 45, 93–100. [Google Scholar]
- Cristofanon, S.; Uguccioni, F.; Cerella, C.; Radogna, F.; Dicato, M.; Ghibelli, L.; Diederich, M. Intracellular prooxidant activity of melatonin induces a survival pathway involving NF-kappaB activation. Ann. N. Y. Acad. Sci 2009, 1171, 472–478. [Google Scholar]
- Radogna, F.; Nuccitelli, S.; Mengoni, F.; Ghibelli, L. Neuroprotection by melatonin on astrocytoma cell death. Ann. N. Y. Acad. Sci 2009, 1171, 509–513. [Google Scholar]
- Soybir, G.; Topuzlu, C.; Odabas, O.; Dolay, K.; Bilir, A.; Koksoy, F. The effects of melatonin on angiogenesis and wound healing. Surg. Today 2003, 33, 896–901. [Google Scholar]
- Pugazhenthi, K.; Kapoor, M.; Clarkson, A.N.; Hall, I.; Appleton, I. Melatonin accelerates the process of wound repair in full-thickness incisional wounds. J. Pineal Res 2008, 44, 387–396. [Google Scholar]
- Klein, D.C. Arylalkylamine N-acetyltransferase: “the Timezyme”. J. Biol. Chem 2007, 282, 4233–4237. [Google Scholar]
- Ekstrom, P.; Meissl, H. Evolution of photosensory pineal organs in new light: The fate of neuroendocrine photoreceptors. Philos. Trans. R. Soc. Lond. B 2003, 358, 1679–1700. [Google Scholar]
- Ferreira, Z.S.; Fernandes, P.A.; Duma, D.; Assreuy, J.; Avellar, M.C.; Markus, R.P. Corticosterone modulates noradrenaline-induced melatonin synthesis through inhibition of nuclear factor kappa B. J. Pineal Res 2005, 38, 182–188. [Google Scholar]
- Cecon, E.; Fernandes, P.A.; Pinato, L.; Ferreira, Z.S.; Markus, R.P. Daily variation of constitutively activated nuclear factor kappa B (NFKB) in rat pineal gland. Chronobiol. Int 2010, 27, 52–67. [Google Scholar]
- de Freitas, M.S.; Spohr, T.C.; Benedito, A.B.; Caetano, M.S.; Margulis, B.; Lopes, U.G.; Moura-Neto, V. Neurite outgrowth is impaired on HSP70-positive astrocytes through a mechanism that requires NF-kappaB activation. Brain Res 2002, 958, 359–370. [Google Scholar]
- Luningschror, P.; Stocker, B.; Kaltschmidt, B.; Kaltschmidt, C. miR-290 cluster modulates pluripotency by repressing canonical NF-kappaB signaling. Stem Cells 2012, 30, 655–664. [Google Scholar]
- Imielski, Y.; Schwamborn, J.C.; Luningschror, P.; Heimann, P.; Holzberg, M.; Werner, H.; Leske, O.; Puschel, A.W.; Memet, S.; Heumann, R.; et al. Regrowing the adult brain: NF-kappaB controls functional circuit formation and tissue homeostasis in the dentate gyrus. PLoS One 2012, 7, e30838. [Google Scholar]
- Lee, I.C.; Kim, S.H.; Lee, S.M.; Baek, H.S.; Moon, C.; Kim, S.H.; Park, S.C.; Kim, H.C.; Kim, J.C. Melatonin attenuates gentamicin-induced nephrotoxicity and oxidative stress in rats. Arch. Toxicol 2012, 86, 1527–1536. [Google Scholar]
- Tamura, E.K.; Cecon, E.; Monteiro, A.W.; Silva, C.L.; Markus, R.P. Melatonin inhibits LPS-induced NO production in rat endothelial cells. J. Pineal Res 2009, 46, 268–274. [Google Scholar]
- Lopes, C.; Mariano, M.; Markus, R.P. Interaction between the adrenal and the pineal gland in chronic experimental inflammation induced by BCG in mice. Inflamm. Res 2001, 50, 6–11. [Google Scholar]
- Carvalho-Sousa, C.E.; da Silveira Cruz-Machado, S.; Tamura, E.K.; Fernandes, P.A.; Pinato, L.; Muxel, S.M.; Cecon, E.; Markus, R.P. Molecular basis for defining the pineal gland and pinealocytes as targets for tumor necrosis factor. Front. Endocrinol. (Lausanne) 2011, 2, 10. [Google Scholar]
- Pontes, G.N.; Cardoso, E.C.; Carneiro-Sampaio, M.M.; Markus, R.P. Pineal melatonin and the innate immune response: The TNF-alpha increase after cesarean section suppresses nocturnal melatonin production. J. Pineal Res 2007, 43, 365–371. [Google Scholar]
- Tatsch-Dias, M.O.; Levandovski, R.M.; Rocha, M.G.; Fernandes, P.A.; Torres, I.S.L.; Hidalgo, M.P.L.; Markus, R.P.; Caumo, W. Evaluating the immune-pineal axis in patients undergoing abdominal hysterectomy. Neuroimmunomodulation 2013, 20, 205–212. [Google Scholar]
- Brugger, P.; Marktl, W.; Herold, M. Impaired nocturnal secretion of melatonin in coronary heart disease. Lancet 1995, 345, 1408. [Google Scholar]
- Fiorina, P.; Lattuada, G.; Silvestrini, C.; Ponari, O.; Dall’Aglio, P. Disruption of nocturnal melatonin rhythm and immunological involvement in ischaemic stroke patients. Scand. J. Immunol 1999, 50, 228–231. [Google Scholar]
- Carvalho, L.A.; Gorenstein, C.; Moreno, R.A.; Markus, R.P. Melatonin levels in drug-free patients with major depression from the southern hemisphere. Psychoneuroendocrinology 2006, 31, 761–768. [Google Scholar]
- Anderson, G.; Maes, M.; Berk, M. Inflammation-related disorders in the tryptophan catabolite pathway in depression and somatization. Adv. Protein Chem. Struct. Biol 2012, 88, 27–48. [Google Scholar]
- Antonioli, M.; Rybka, J.; Carvalho, L.A. Neuroimmune endocrine effects of antidepressants. Neuropsychiatr. Dis. Treat 2012, 8, 65–83. [Google Scholar]
- Liu, R.Y.; Zhou, J.N.; van Heerikhuize, J.; Hofman, M.A.; Swaab, D.F. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J. Clin. Endocrinol. Metab 1999, 84, 323–327. [Google Scholar]
- Lahiri, D.K.; Chen, D.M.; Lahiri, P.; Bondy, S.; Greig, N.H. Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases. Ann. N. Y. Acad. Sci 2005, 1056, 430–449. [Google Scholar]
- Cecon, E.; Markus, R.P. Relevance of the chronobiological and non-chronobiological actions of melatonin for enhancing therapeutic efficacy in neurodegenerative disorders. Recent Pat. Endocr. Metab. Immune Drug Discov 2011, 5, 91–99. [Google Scholar]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Furio, A.M.; Brusco, L.I. Therapeutic application of melatonin in mild cognitive impairment. Am. J. Neurodegener. Dis 2012, 1, 280–291. [Google Scholar]
- Tosini, G.; Baba, K.; Hwang, C.K.; Iuvone, P.M. Melatonin: An underappreciated player in retinal physiology and pathophysiology. Exp. Eye Res 2012, 103, 82–89. [Google Scholar]
- Jimenez-Jorge, S.; Guerrero, J.M.; Jimenez-Caliani, A.J.; Naranjo, M.C.; Lardone, P.J.; Carrillo-Vico, A.; Osuna, C.; Molinero, P. Evidence for melatonin synthesis in the rat brain during development. J. Pineal Res 2007, 42, 240–246. [Google Scholar]
- Bubenik, G.A. Thirty four years since the discovery of gastrointestinal melatonin. J. Physiol. Pharmacol 2008, 59, 33–51. [Google Scholar]
- Konturek, S.J.; Konturek, P.C.; Brzozowski, T.; Bubenik, G.A. Role of melatonin in upper gastrointestinal tract. J. Physiol. Pharmacol 2007, 58, 23–52. [Google Scholar]
- Conti, A.; Conconi, S.; Hertens, E.; Skwarlo-Sonta, K.; Markowska, M.; Maestroni, J.M. Evidence for melatonin synthesis in mouse and human bone marrow cells. J. Pineal Res 2000, 28, 193–202. [Google Scholar]
- Carrillo-Vico, A.; Calvo, J.R.; Abreu, P.; Lardone, P.J.; Garcia-Maurino, S.; Reiter, R.J.; Guerrero, J.M. Evidence of melatonin synthesis by human lymphocytes and its physiological significance: Possible role as intracrine, autocrine, and/or paracrine substance. FASEB J 2004, 18, 537–539. [Google Scholar]
- Martins, E., Jr; Ferreira, A.C.; Skorupa, A.L.; Afeche, S.C.; Cipolla-Neto, J.; Costa Rosa, L.F. Tryptophan consumption and indoleamines production by peritoneal cavity macrophages. J. Leukoc. Biol. 2004, 75, 1116–1121. [Google Scholar]
- Liu, Y.J.; Zhuang, J.; Zhu, H.Y.; Shen, Y.X.; Tan, Z.L.; Zhou, J.N. Cultured rat cortical astrocytes synthesize melatonin: Absence of a diurnal rhythm. J. Pineal Res 2007, 43, 232–238. [Google Scholar]
- Markus, R.P.; Tamura, E.K. G protein-Coupled Receptors and Other Mechanisms that Translate Melatonin Effects. In G Protein-Coupled Receptors in Vertebrates: Comparative Perspectives; Research Signpost: Kerala, Índia, 2009; Volume 2, pp. 93–111. [Google Scholar]
- Luchetti, F.; Canonico, B.; Betti, M.; Arcangeletti, M.; Pilolli, F.; Piroddi, M.; Canesi, L.; Papa, S.; Galli, F. Melatonin signaling and cell protection function. FASEB J 2010, 24, 3603–3624. [Google Scholar]
- Tan, D.X.; Manchester, L.C.; Sanchez-Barcelo, E.; Mediavilla, M.D.; Reiter, R.J. Significance of high levels of endogenous melatonin in Mammalian cerebrospinal fluid and in the central nervous system. Curr. Neuropharmacol 2010, 8, 162–167. [Google Scholar]
- Silva, S.O.; Rodrigues, M.R.; Ximenes, V.F.; Bueno-da-Silva, A.E.; Amarante-Mendes, G.P.; Campa, A. Neutrophils as a specific target for melatonin and kynuramines: Effects on cytokine release. J. Neuroimmunol 2004, 156, 146–152. [Google Scholar]
- Pires-Lapa, M.A.; Tamura, E.K.; Salustiano, E.M.A; Markus, R.P. Local melatonin synthesis enhances dectin-1-mediated phagocytosis by human colostrum mononuclear cells. J. Pineal Res. 2013, in press. [Google Scholar]
- Carrillo-Vico, A.; Garcia-Maurino, S.; Calvo, J.R.; Guerrero, J.M. Melatonin counteracts the inhibitory effect of PGE2 on IL-2 production in human lymphocytes via its mt1 membrane receptor. FASEB J 2003, 17, 755–757. [Google Scholar]
- Brunner, T.; Mogil, R.J.; LaFace, D.; Yoo, N.J.; Mahboubi, A.; Echeverri, F.; Martin, S.J.; Force, W.R.; Lynch, D.H.; Ware, C.F.; et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature 1995, 373, 441–444. [Google Scholar]
- Lissoni, P.; Brivio, F.; Barni, S.; Tancini, G.; Cattaneo, G.; Archili, C.; Conti, A.; Maestroni, G.J. Neuroimmunotherapy of human cancer with interleukin-2 and the neurohormone melatonin: Its efficacy in preventing hypotension. Anticancer Res 1990, 10, 1759–1761. [Google Scholar]
- Srinivasan, V.; Spence, D.W.; Trakht, I.; Pandi-Perumal, S.R.; Cardinali, D.P.; Maestroni, G.J. Immunomodulation by melatonin: Its significance for seasonally occurring diseases. Neuroimmunomodulation 2008, 15, 93–101. [Google Scholar]
- Ban, J.Y.; Kim, B.S.; Kim, S.C.; Kim, D.H.; Chung, J.H. Microarray analysis of gene expression profiles in response to treatment with melatonin in lipopolysaccharide activated raw 264.7 cells. Korean J. Physiol. Pharmacol 2011, 15, 23–29. [Google Scholar]
- Xia, M.Z.; Liang, Y.L.; Wang, H.; Chen, X.; Huang, Y.Y.; Zhang, Z.H.; Chen, Y.H.; Zhang, C.; Zhao, M.; Xu, D.X.; et al. Melatonin modulates TLR4-mediated inflammatory genes through MyD88- and TRIF-dependent signaling pathways in lipopolysaccharide-stimulated RAW264.7 cells. J. Pineal Res 2012, 53, 325–334. [Google Scholar]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef]
- Tamura, E.K.; Fernandes, P.A.; Marcola, M.; da Silveira Cruz-Machado, S.; Markus, R.P. Long-lasting priming of endothelial cells by plasma melatonin levels. PLoS One 2010, 5, e13958. [Google Scholar]
- Marcola, M.; da Silveira Cruz-Machado, S.; Fernandes, P.A.; Monteiro, A.W.; Markus, R.P.; Tamura, E.K. Endothelial cell adhesiveness is a function of environmental lighting and melatonin level. J. Pineal Res 2013, 54, 162–169. [Google Scholar]
- Skwarlo-Sonta, K.; Majewski, P.; Markowska, M.; Oblap, R.; Olszanska, B. Bidirectional communication between the pineal gland and the immune system. Can. J. Physiol. Pharmacol 2003, 81, 342–349. [Google Scholar]
- Fernandes, P.A.; Markus, R.P. Melatonin and Inflammation—The Role of the Immune–Pineal Axis and the Sympathetic Tonus. In Melatonin in the Promotion of Health, 2nd ed.; Watson, R.R., Ed.; CRC Press: Tucson, AZ, USA, 2011; pp. 435–450. [Google Scholar]
- Mauriz, J.L.; Collado, P.S.; Veneroso, C.; Reiter, R.J.; Gonzalez-Gallego, J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: Recent insights and new perspectives. J. Pineal Res. 2012. [Google Scholar] [CrossRef]
- Fernandes, P.A.; Bothorel, B.; Clesse, D.; Monteiro, A.W.; Calgari, C.; Raison, S.; Simonneaux, V.; Markus, R.P. Local corticosterone infusion enhances nocturnal pineal melatonin production in vivo. J. Neuroendocrinol 2009, 21, 90–97. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Markus, R.P.; Cecon, E.; Pires-Lapa, M.A. Immune-Pineal Axis: Nuclear Factor κB (NF-kB) Mediates the Shift in the Melatonin Source from Pinealocytes to Immune Competent Cells. Int. J. Mol. Sci. 2013, 14, 10979-10997. https://doi.org/10.3390/ijms140610979
Markus RP, Cecon E, Pires-Lapa MA. Immune-Pineal Axis: Nuclear Factor κB (NF-kB) Mediates the Shift in the Melatonin Source from Pinealocytes to Immune Competent Cells. International Journal of Molecular Sciences. 2013; 14(6):10979-10997. https://doi.org/10.3390/ijms140610979
Chicago/Turabian StyleMarkus, Regina P., Erika Cecon, and Marco Antonio Pires-Lapa. 2013. "Immune-Pineal Axis: Nuclear Factor κB (NF-kB) Mediates the Shift in the Melatonin Source from Pinealocytes to Immune Competent Cells" International Journal of Molecular Sciences 14, no. 6: 10979-10997. https://doi.org/10.3390/ijms140610979
APA StyleMarkus, R. P., Cecon, E., & Pires-Lapa, M. A. (2013). Immune-Pineal Axis: Nuclear Factor κB (NF-kB) Mediates the Shift in the Melatonin Source from Pinealocytes to Immune Competent Cells. International Journal of Molecular Sciences, 14(6), 10979-10997. https://doi.org/10.3390/ijms140610979