NS3 Protease from Hepatitis C Virus: Biophysical Studies on an Intrinsically Disordered Protein Domain

 ,
, 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. NS3 Protease from Hepatitis C Virus
- (1)
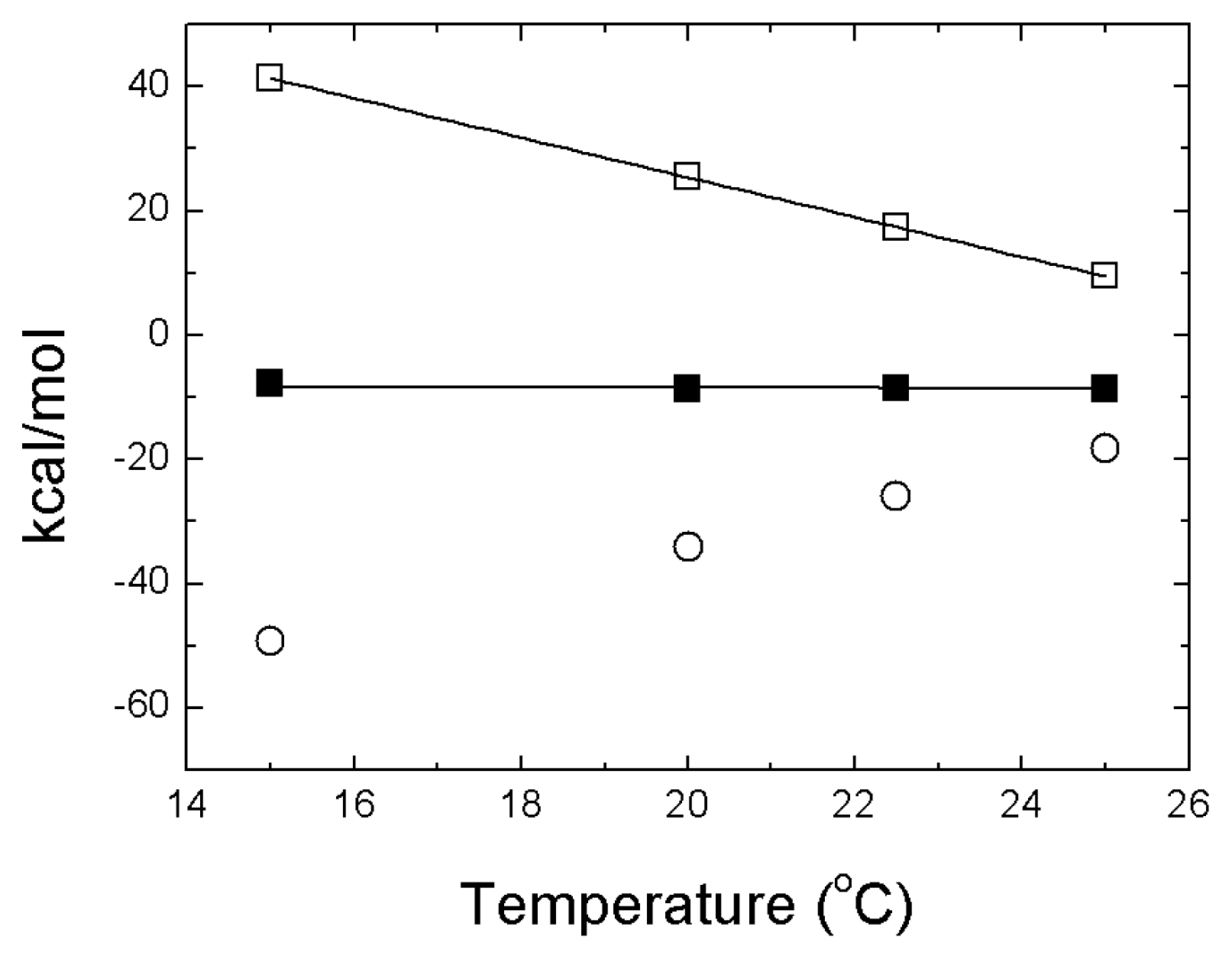
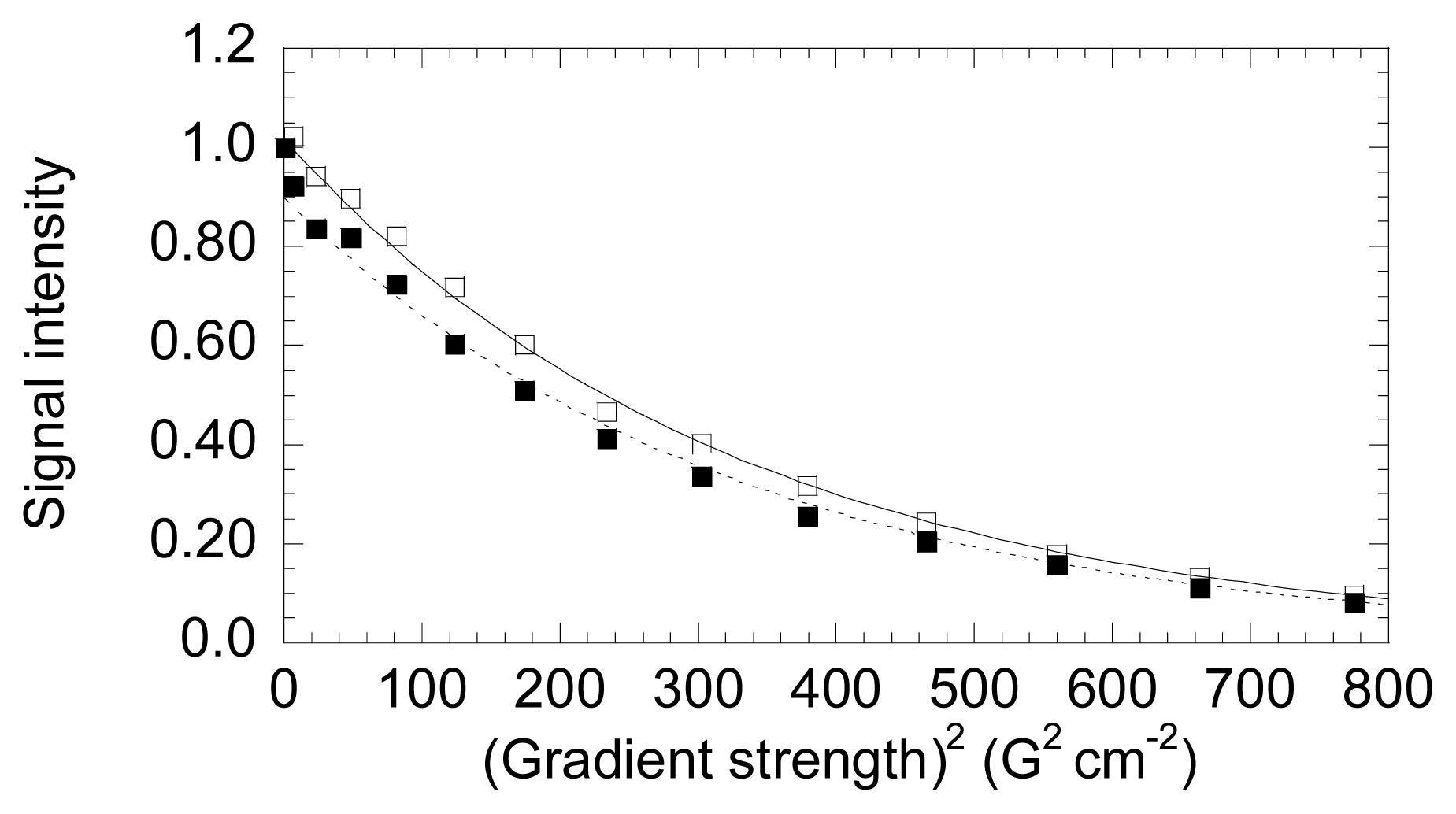
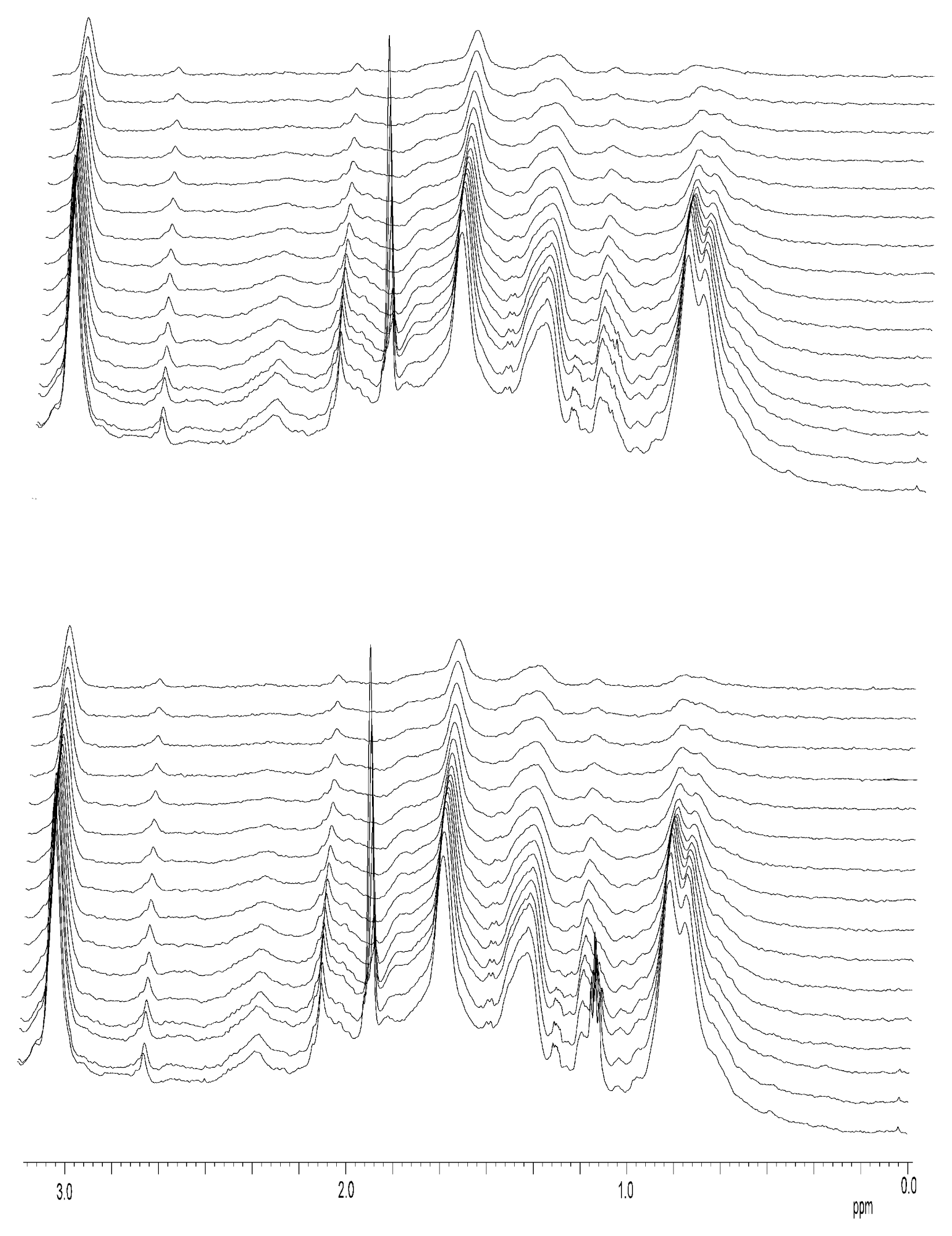
- The energetics of zinc binding to NS3 and its structural implications [15]. Different biophysical techniques have been employed for probing the structural features of the zinc-bound and the zinc-free NS3 conformations. In particular, spectroscopy data (circular dichroism, fluorescence and 1H-1D-NMR) suggested a significant conformational change (in tertiary structure, mainly) associated with zinc interaction [15]. Furthermore, isothermal titration calorimetry (ITC) has been used for determining the energetics of the NS3-zinc interaction, confirming a remarkable conformational change coupled to zinc binding from the determination of the binding heat capacity [15]. Because ITC provides a direct measurement of the binding enthalpy, it is possible to get a reliable estimation of the change in heat capacity upon binding, which is very difficult or simply infeasible using spectroscopic techniques. In addition, here we report translational diffusion experiments and atomic force microscopy data providing further information on the zinc-dependent plasticity of the NS3 protease.
- (2)
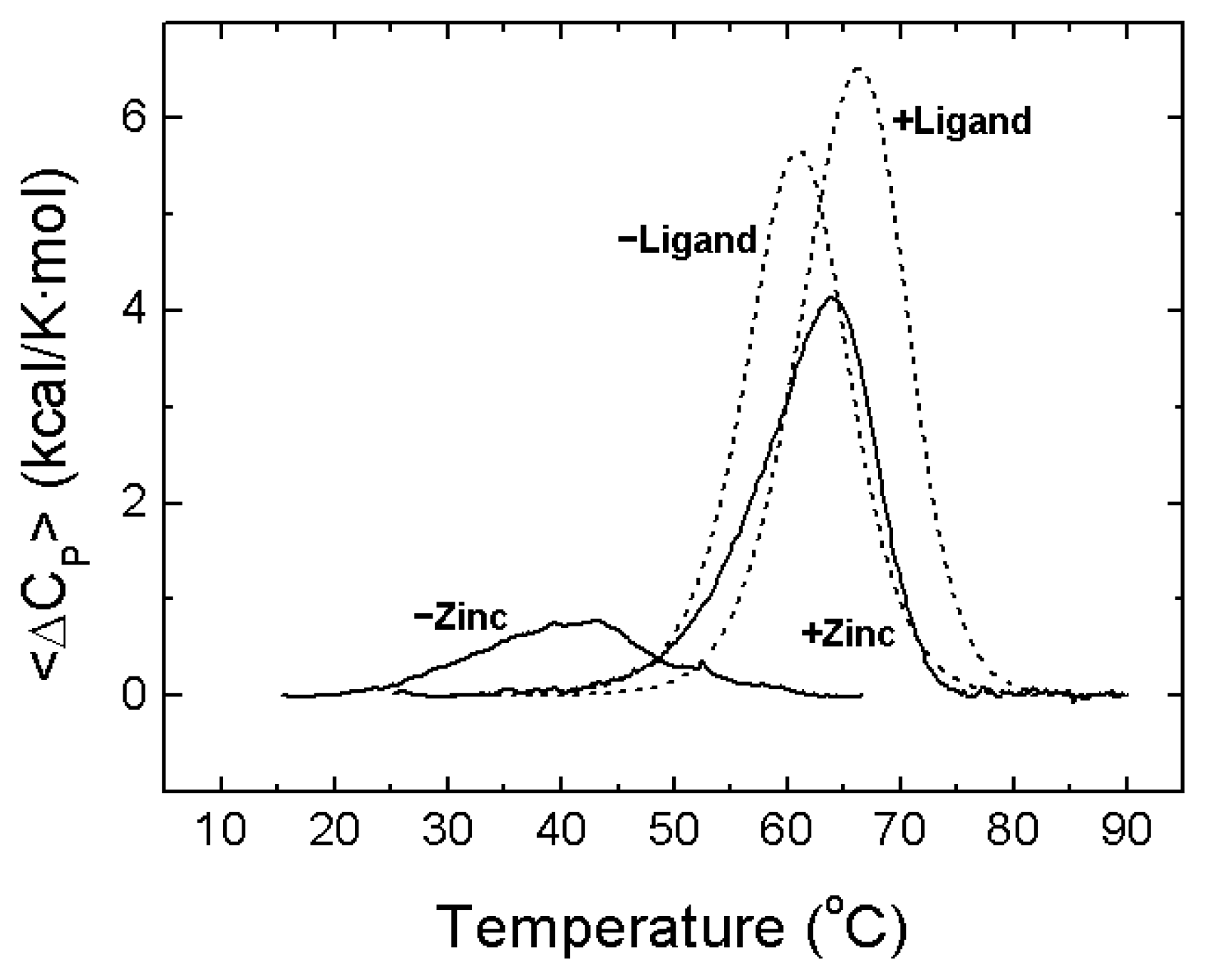
- A description of the conformational landscape of NS3. The structural molecular integrity is dominated by the interaction with zinc. The binding of zinc induces a significant structural global rearrangement of the protein: spectroscopic and calorimetric thermal denaturation assays indicate that zinc-free NS3 protease exhibits very low stability, whereas the zinc-bound NS3 protease is considerably stabilized due to metal binding [16]. Contrary to other homologous zinc-dependent proteases, the zinc-free NS3 protease is not completely unstructured. Thus, the NS3 protease exhibits a fairly complex conformational equilibrium.
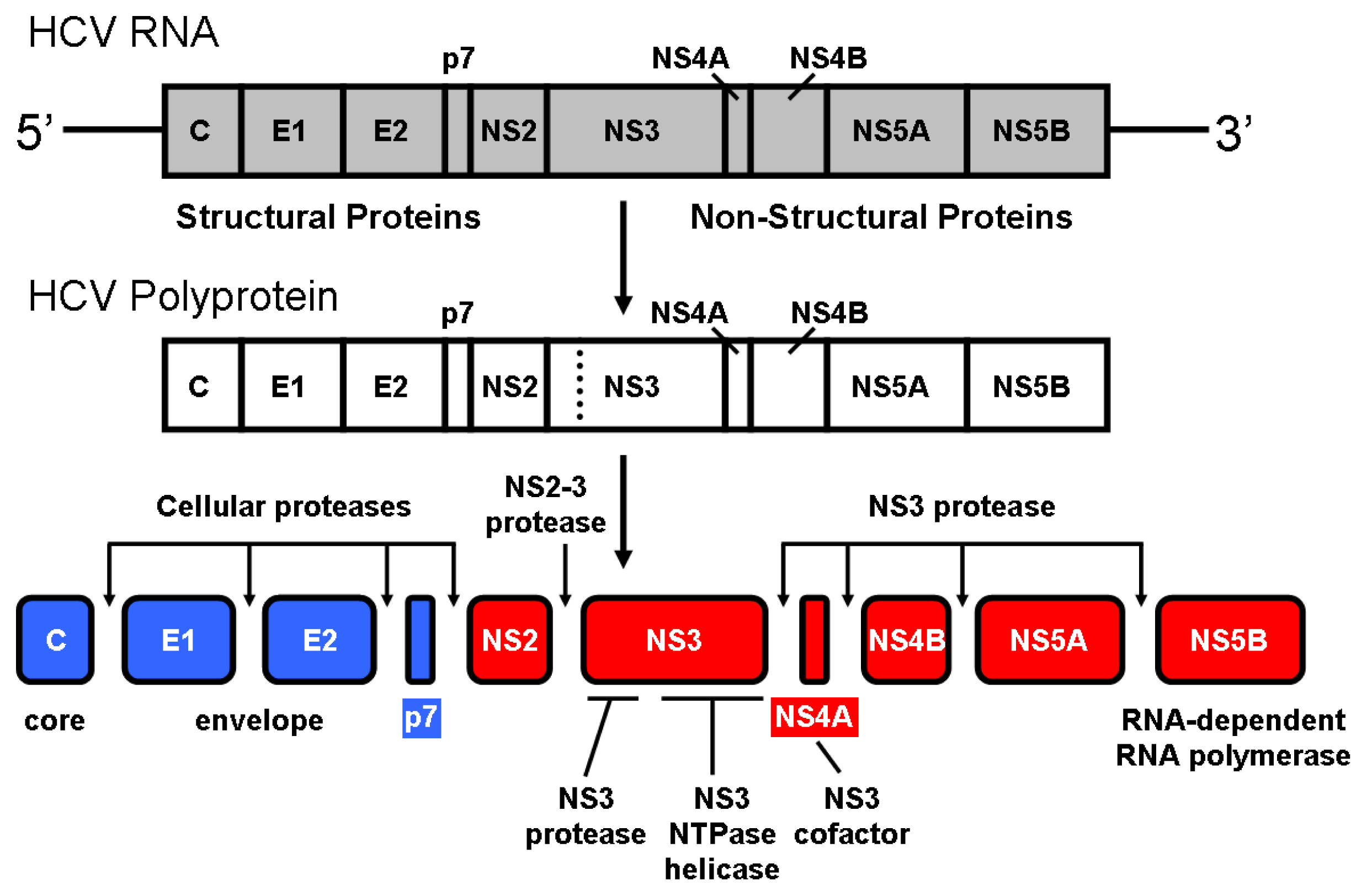
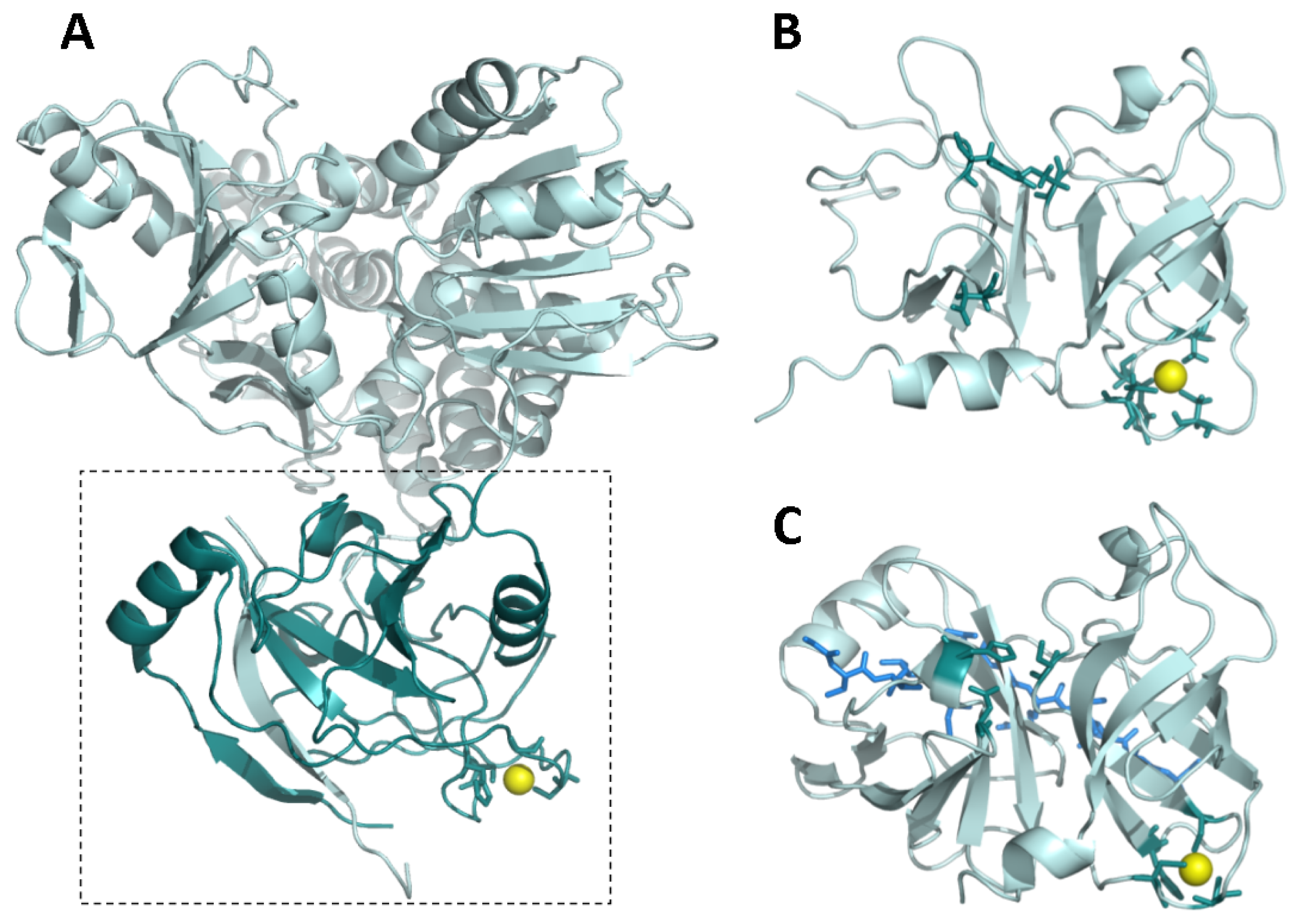
2. NS3 Protease: Structure and Function
3. NS3 Protease: Zinc Interaction
4. NS3 Protease: Structural Stability
5. NS3 Protease: Conformational Landscape
6. Conclusions
Acknowledgments
Appendix
Protein Expression and Purification
Nuclear Magnetic Resonance Spectroscopy
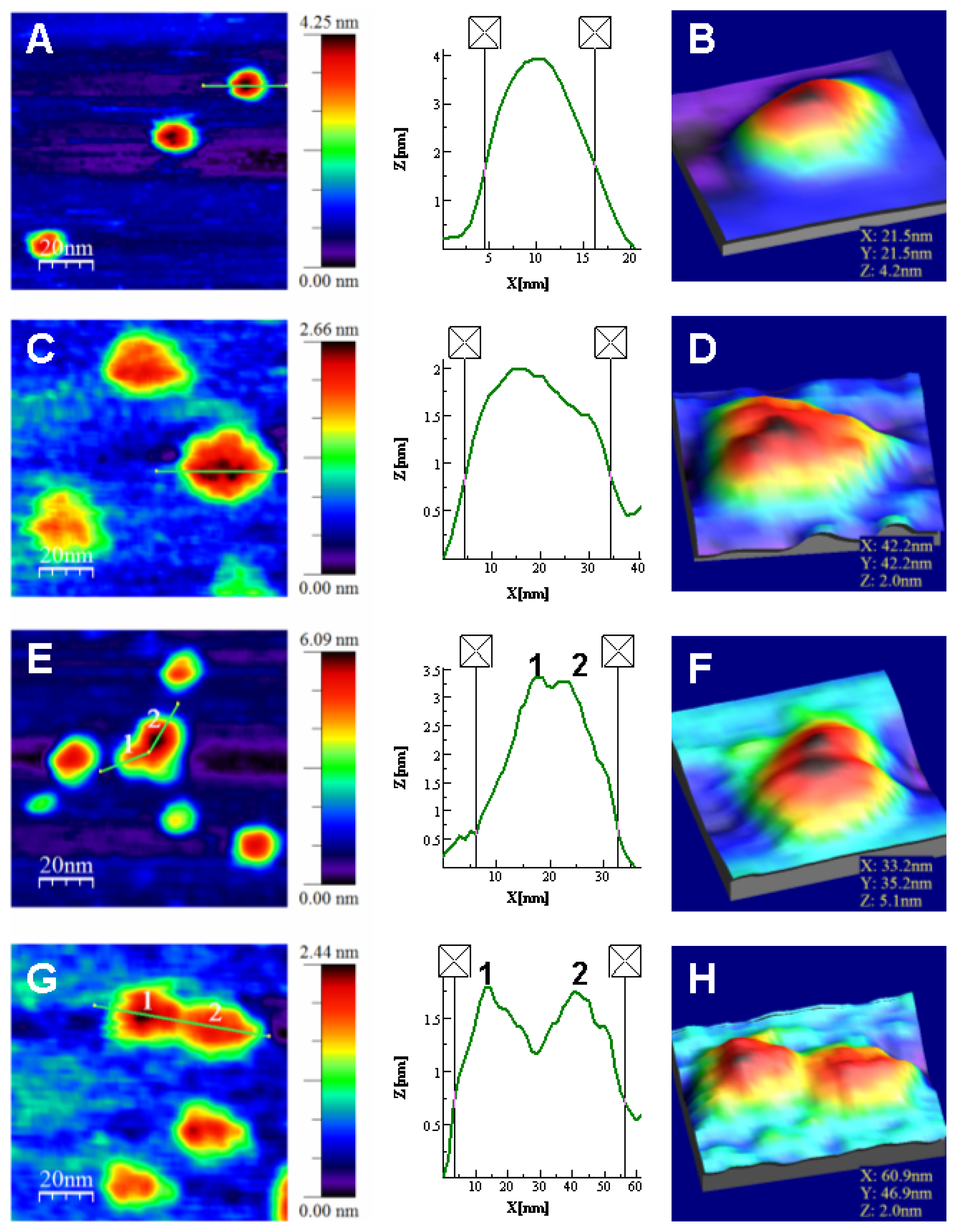
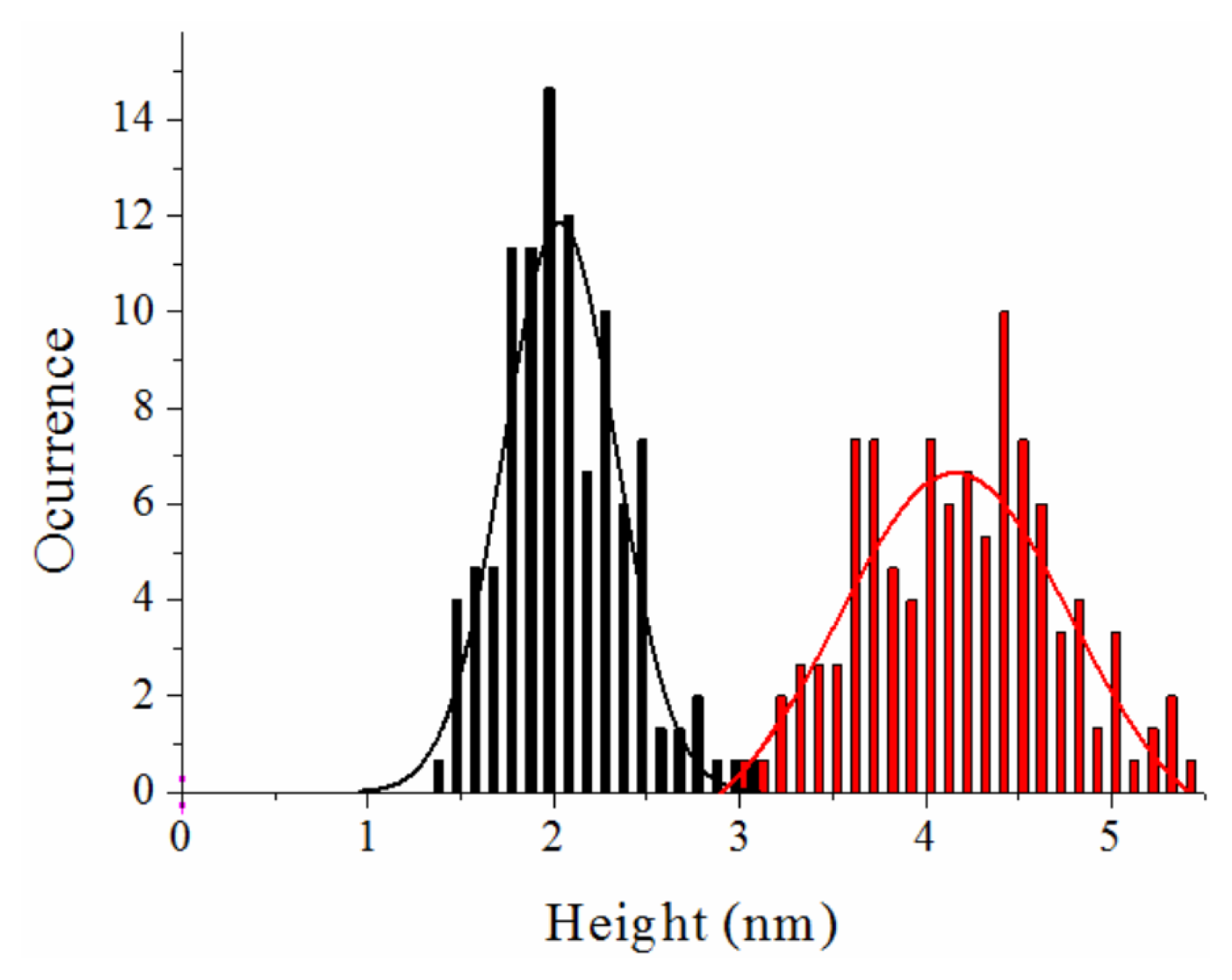
Atomic Force Microscopy Imaging
Differential Scanning Calorimetry

Conflict of Interest
References
- Hijikata, M.; Mizushima, H.; Akagi, T.; Mori, S.; Kakiuchi, N.; Kato, N.; Tanaka, T.; Kimura, K.; Shimotohno, K. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol 1993, 67, 4665–4675. [Google Scholar]
- Grakoui, A.; McCourt, D.W.; Wychowski, C.; Feinstone, S.M.; Rice, C.M. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA 1993, 90, 10583–10587. [Google Scholar]
- Pieroni, L.; Santolini, E.; Fipaldini, C.; Pacini, L.; Migliaccio, G.; la Monica, N. In vitro study of the NS2-3 protease of hepatitis C virus. J. Virol 1997, 71, 6373–6380. [Google Scholar]
- Welbourn, S.; Green, R.; Gamache, I.; Dandache, S.; Lohmann, V.; Bartenschlager, R.; Meerovitch, K.; Pause, A. Hepatitis C virus NS2/3 processing is required for NS3 stability and viral RNA replication. J. Biol. Chem 2005, 280, 29604–29611. [Google Scholar]
- De Francesco, R.; Urbani, A.; Nardi, M.C.; Tomei, L.; Steinkühler, C.; Tramontano, A. A zinc binding site in viral serine proteinases. Biochemistry 1996, 35, 13282–13287. [Google Scholar]
- Love, R.A.; Parge, H.E.; Wickersham, J.A.; Hostomsky, Z.; Habuka, N.; Moomaw, E.W.; Adachi, T.; Hostomska, Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 1996, 87, 331–342. [Google Scholar]
- Kim, J.L.; Morgenstern, K.A.; Lin, C.; Fox, T.; Dwyer, M.D.; Landro, J.A.; Chambers, S.P.; Markland, W.; Lepre, C.A.; O’Malley, E.T.; et al. Crystal-structure of the hepatitis-C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell 1996, 87, 345–355. [Google Scholar]
- Stempniak, M.; Hostomska, Z.; Nodes, B.R.; Hostomsky, Z. The NS3 proteinase domain of hepatitis C virus is a zinc-containing enzyme. J. Virol 1997, 71, 2881–2886. [Google Scholar]
- Kwong, A.D.; Kauffman, R.S.; Hurter, P.; Mueller, P. Discovery and development of telaprevir: An NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol 2011, 29, 993–1003. [Google Scholar]
- Rizza, S.A.; Talwani, R.; Nehra, V.; Temesgen, Z. Boceprevir. Drugs Today 2011, 47, 743–751. [Google Scholar]
- He, Y.; King, M.S.; Kempf, D.J.; Lu, L.; Lim, H.B.; Krishnan, P.; Kati, W.; Middleton, T.; Molla, A. Relative replication capacity and selective advantage profiles of protease inhibitor-resistant hepatitis c virus (HCV) NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob. Agents Chemother 2008, 52, 1101–1110. [Google Scholar]
- Courcambeck, J.; Bouzidi, M.; Perbost, R.; Jouirou, B.; Amrani, N.; Cacoub, P.; Pèpe, G.; Sabatier, J.M.; Halfon, P. Resistance of hepatitis C virus to NS3-4A protease inhibitors: Mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antivir. Ther 2004, 11, 847–855. [Google Scholar]
- Susser, S.; Vermehren, J.; Forestier, N.; Welker, M.W.; Grigorian, N.; Füller, C.; Perner, D.; Zeuzem, S.; Sarrazin, C. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol 2011, 52, 321–327. [Google Scholar]
- Urbani, A.; Bazzo, R.; Nardi, M.C.; Cicero, D.O.; de Francesco, R.; Steinkühler, C.; Barbato, G. The metal binding site of the hepatitis C virus NS3 protease. A spectroscopic investigation. J. Biol. Chem 1998, 273, 18760–18769. [Google Scholar]
- Abian, O.; Neira, J.L.; Velazquez-Campoy, A. Thermodynamics of zinc binding to hepatitis C virus NS3 protease: A folding by binding event. Proteins 2009, 77, 624–636. [Google Scholar]
- Abian, O.; Vega, S.; Neira, J.L.; Velazquez-Campoy, A. Conformational stability of hepatitis C virus NS3 protease. Biophys. J 2010, 99, 3811–3820. [Google Scholar]
- Thibeault, D.; Massariol, M.J.; Zhao, S.; Welchner, E.; Goudreau, N.; Gingras, R.; Llinás-Brunet, M.; White, P.W. Use of the fused NS4A peptide–NS3 protease domain to study the importance of the helicase domain for protease inhibitor binding to hepatitis C virus NS3-NS4A. Biochemistry 2009, 48, 744–753. [Google Scholar]
- Lee, Y.M.; Lim, C. Physical basis of structural and catalytic Zn-binding sites in proteins. J. Mol. Biol 2008, 379, 545–553. [Google Scholar]
- Tedbury, P.R.; Harris, M. Characterisation of the role of zinc in the hepatitis C virus NS2/3 auto-cleavage and NS3 protease activities. J. Mol. Biol 2007, 366, 1652–1660. [Google Scholar]
- Wu, Z.; Yao, N.; Le, H.V.; Weber, P.C. Mechanism of autoproteolysis at the NS2-NS3 junction of the hepatitis C virus polyprotein. Trends Biochem. Sci 1998, 23, 92–94. [Google Scholar]
- Failla, C.; Tomei, L.; de Francesco, R. Both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural proteins. J. Virol 1994, 68, 3753–3760. [Google Scholar]
- Lin, C.; Thomson, J.A.; Rice, C.M. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J. Virol 1995, 69, 4373–4380. [Google Scholar]
- Tanji, Y.; Hijikata, M.; Satoh, S.; Kaneko, T.; Shimotohno, K. Hepatitis C virus-encoded nonstructural protein NS4A has versatile functions in viral protein processing. J. Virol 1995, 69, 1575–1581. [Google Scholar]
- Tomei, L.; Failla, C.; Vitale, R.L.; Bianchi, E.; de Francesco, R. A central hydrophobic domain of the hepatitis C virus NS4A protein is necessary and sufficient for the activation of the NS3 protease. J. Gen. Virol 1996, 77, 1065–1070. [Google Scholar]
- Bianchi, E.; Urbani, A.; Biasiol, G.; Brunetti, M.; Pessi, A.; de Francesco, R.; Steinkühler, C. Complex formation between the hepatitis C virus serine protease and a synthetic NS4A cofactor peptide. Biochemistry 1997, 36, 7890–7897. [Google Scholar]
- Yao, N.; Reichert, P.; Taremi, S.S.; Prosise, W.W.; Weber, P.C. Molecular views of viral polyprotein processing revealed by the crystal structure of the hepatitis C virus bifunctional protease-helicase. Struct. Fold. Des 1999, 7, 1353–1363. [Google Scholar]
- Barbato, G.; Cicero, D.O.; Nardi, M.C.; Steinkuhler, C.; Cortese, R.; de Francesco, R.; Bazzo, R. The solution structure of the N-terminal proteinase domain of the hepatitis C virus (HCV) NS3 protein provides new insights into its activation and catalytic mechanism. J. Mol. Biol 1999, 289, 371–384. [Google Scholar]
- Yan, Y.; Li, Y.; Munshi, S.; Sardana, V.; Cole, J.L.; Sardana, M.; Steinkuehler, C.; Tomei, L.; de Francesco, R.; Kuo, L.C.; et al. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: A 2.2 A resolution structure in a hexagonal crystal form. Protein Sci 1998, 7, 837–847. [Google Scholar]
- Holm, R.H.; Kennepohl, P.; Solomon, E.I. Structural and functional aspects of metal sites in Biology. Chem. Rev 1996, 96, 2239–2314. [Google Scholar]
- Yi, S.; Boys, B.L.; Brickenden, A.; Konermann, L.; Choy, W.Y. Effects of zinc binding on the structure and dynamics of the intrinsically disordered protein prothymosin alpha: Evidence for metalation as an entropic switch. Biochemistry 2007, 46, 13120–13130. [Google Scholar]
- Park, P.S.; Sapra, K.T.; Kolinski, M.; Filipek, S.; Palczewski, K.; Muller, D.J. Stabilizing effect of Zn2+ in native bovine rhodopsin. J. Biol. Chem 2007, 282, 11377–11385. [Google Scholar]
- Colvin, R.; Holmes, W.R.; Fontaine, C.P.; Maret, W. Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis. Metallomics 2010, 2, 306–317. [Google Scholar]
- Binet, M.R.; Poole, R.K. Cd(II); Pb(II) and Zn(II) ions regulate expression of the metal-transporting P-type ATPase ZntA in Escherichia coli. FEBS Lett 2000, 473, 67–70. [Google Scholar]
- Brocklehurst, K.R.; Hobman, J.L.; Lawley, B.; Blank, L.; Marshall, S.J.; Brown, N.L.; Morby, A.P. ZntR is a Zn(II)-responsive MerR-like transcriptional regulator of zntA in Escherichia coli. Mol. Microbiol 1999, 31, 893–902. [Google Scholar]
- Outten, C.E.; O’Halloran, T.V. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 2001, 292, 2488–2492. [Google Scholar]
- Hitomi, Y.; Outten, C.E.; O’Halloran, T.V. Extreme zinc-binding thermodynamics of the metal sensor/regulator protein, ZntR. J. Am. Chem. Soc 2001, 123, 8614–8615. [Google Scholar]
- Robertson, A.D.; Murphy, K.P. Protein structure and the energetics of protein stability. Chem. Rev 1997, 97, 1251–1268. [Google Scholar]
- Fenley, A.T.; Muddana, H.S.; Gilson, M.K. Entropy-enthalpy transduction caused by conformational shifts can obscure the forces driving protein-ligand binding. Proc. Natl. Acad. Sci. USA 2012, 109, 20006–20011. [Google Scholar]
- Eftink, M.; Biltonen, R. Biological Calorimetry; Beezer, A., Ed.; Academic Press: London, UK & New York, NY, USA, 1980; pp. 343–412. [Google Scholar]
- Kouvatsos, N.; Meldrum, J.K.; Searle, M.S.; Thomas, N.R. Coupling ligand recognition to protein folding in an engineered variant of rabbit ileal lipid binding protein. Chem. Commun. (Camb.) 2006, 4623–4625. [Google Scholar]
- Jones, J.A.; Wilkins, D.K.; Smith, L.J.; Dobson, C.M. Characterization of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 1997, 10, 199–203. [Google Scholar]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse filed gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar]
- Casares, S.; Sadqi, M.; Lopez-Mayorga, O.; Conejero-Lara, F.; van Nuland, N.A.J. Detection and characterization of partially unfolded oligomers of the SH3 domain of α-spectrin. Biophys. J 2004, 86, 2403–2413. [Google Scholar]
- Creighton, T.E. Proteins. Structures and Macromolecular Properties, 2nd ed; W.H. Freeman: New York, NY, USA, 1993. [Google Scholar]
- Garcia de la Torre, J.; Huertas, M.L.; Carrasco, B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J 2000, 78, 719–730. [Google Scholar]
- Uversky, V.N. Intrinsically disordered proteins and their environments: Effects of strong denaturants, temperature, pH, counter ions, membranes, binding partners, osmolytes and macromolecular crowding. Protein J 2009, 28, 305–325. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic force microscope. Phys. Rev. Lett 1986, 56, 930–933. [Google Scholar]
- Muller, D.J.; Dufrene, Y.F. Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotechnol 2008, 3, 261–269. [Google Scholar]
- Barrera, N.P.; Betts, J.; You, H.; Henderson, R.M.; Martin, I.L.; Dunn, S.M.; Edwardson, J.M. Atomic force microscopy reveals the stoichiometry and subunit arrangement of the alpha4beta3delta GABA(A) receptor. Mol. Pharmacol 2008, 73, 960–967. [Google Scholar]
- Jensenius, H.; Klein, D.C.; van Hecke, M.; Oosterkamp, T.H.; Schmidt, T.; Jensenius, J.C. Mannan-binding lectin: Structure, oligomerization, and flexibility studied by atomic force microscopy. J. Mol. Biol. 2009, 391, 246–259. [Google Scholar]
- Scheuring, S.; Sturgis, J.N. Atomic force microscopy of the bacterial photosynthetic apparatus: Plain pictures of an elaborate machinery. Photosynth. Res 2009, 102, 197–211. [Google Scholar]
- Martinez-Perez, M.J.; de Miguel, R.; Carbonera, C.; Martinez-Julvez, M.; Lostao, A.; Piquer, C.; Gomez-Moreno, C.; Bartolome, J.; Luis, F. Size-dependent properties of magnetoferritin. Nanotechnology 2010, 21, 465707. [Google Scholar]
- Gomes, S.; Numata, K.; Leonor, I.B.; Mano, J.F.; Reis, R.L.; Kaplan, D.L. AFM Study of morphology and mechanical properties of a chimeric spider silk and bone sialoprotein protein for bone regeneration. Biomacromolecules 2011, 12, 1675–1685. [Google Scholar]
- Lostao, A.; Peleato, M.L.; Gomez-Moreno, C.; Fillat, M.F. Oligomerization properties of FurA from the cyanobacterium Anabaena sp. PCC 7120: Direct visualization by in situ atomic force microscopy under different redox conditions. Biochim. Biophys. Acta 2010, 1804, 1723–1729. [Google Scholar]
- Carnally, S.M.; Edwardson, J.M.; Barrera, N.P. Imaging the spatial orientation of subunits within membrane receptors by atomic force microscopy. Methods Mol. Biol 2012, 736, 47–60. [Google Scholar]
- Marcuello, C.; Arilla-Luna, S.; Medina, M.; Lostao, A. Detection of a quaternary organization into dimer of trimers of Corynebacterium ammoniagenes FAD synthetase at the single-molecule level and at the in cell level. Biochim. Biophys. Acta 2013, 1834, 665–676. [Google Scholar]
- Bustamante, C.; Chemla, Y.R.; Forde, N.R.; Izhaky, D. Mechanical processes in biochemistry. Annu. Rev. Biochem 2004, 73, 705–748. [Google Scholar]
- Meinander, K.; Jensen, T.N.; Simonsen, S.B.; Helveg, S.; Lauritsen, J.V. Quantification of tip-broadening in non-contact atomic force microscopy with carbon nanotube tips. Nanotechnology 2012, 23, 405705. [Google Scholar]
- Horcas, I.; Fernandez, R.; Gomez-Rodriguez, J.M.; Colchero, J.; Gomez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum 2007, 78, 013705. [Google Scholar]
- Voss, T.; Meyer, R.; Sommergruber, W. Spectroscopic characterization of rhinoviral protease 2A: Zn is essential for the structural integrity. Protein Sci 1995, 4, 2526–2531. [Google Scholar]
- Taliani, M.; Bianchi, E.; Narjes, F.; Fossatelli, M.; Urbani, A.; Steinkühler, C.; de Francesco, R.; Pessi, A. A continuous assay of hepatitis C virus protease based on resonance energy transfer depsipeptide substrates. Anal. Biochem 1996, 240, 60–67. [Google Scholar]
- Price, W.S. Pulse-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part I. Basic theory. Conc. Magn. Reson 1997, 9, 299–336. [Google Scholar]
- Price, W.S. Pulse-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part II. Experimental aspects. Conc. Magn. Reson 1998, 10, 197–237. [Google Scholar]
- Lapham, J.; Rife, J.P.; Moore, P.B.; Crothers, D.M. Measurement of diffusion constants for nucleic acids by NMR. J. Biomol. NMR 1997, 10, 255–262. [Google Scholar]
- Hansma, P.K.; Cleveland, J.P.; Radmacher, M.; Walters, D.A.; Hillner, P.E.; Bezanilla, M.; Fritz, M.; Vie, D.; Hansma, H.G.; Prater, C.B.; et al. Tapping mode atomic force microscopy in liquids. Appl. Phys. Lett 1994, 64, 1738–1740. [Google Scholar]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum 1993, 64, 1868–1873. [Google Scholar]








© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vega, S.; Neira, J.L.; Marcuello, C.; Lostao, A.; Abian, O.; Velazquez-Campoy, A. NS3 Protease from Hepatitis C Virus: Biophysical Studies on an Intrinsically Disordered Protein Domain. Int. J. Mol. Sci. 2013, 14, 13282-13306. https://doi.org/10.3390/ijms140713282
Vega S, Neira JL, Marcuello C, Lostao A, Abian O, Velazquez-Campoy A. NS3 Protease from Hepatitis C Virus: Biophysical Studies on an Intrinsically Disordered Protein Domain. International Journal of Molecular Sciences. 2013; 14(7):13282-13306. https://doi.org/10.3390/ijms140713282
Chicago/Turabian StyleVega, Sonia, Jose L. Neira, Carlos Marcuello, Anabel Lostao, Olga Abian, and Adrian Velazquez-Campoy. 2013. "NS3 Protease from Hepatitis C Virus: Biophysical Studies on an Intrinsically Disordered Protein Domain" International Journal of Molecular Sciences 14, no. 7: 13282-13306. https://doi.org/10.3390/ijms140713282
APA StyleVega, S., Neira, J. L., Marcuello, C., Lostao, A., Abian, O., & Velazquez-Campoy, A. (2013). NS3 Protease from Hepatitis C Virus: Biophysical Studies on an Intrinsically Disordered Protein Domain. International Journal of Molecular Sciences, 14(7), 13282-13306. https://doi.org/10.3390/ijms140713282