Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Post-Transcriptional Regulation of Gene Expression in Response to Iron Deficiency
2.1. Cth1 and Cth2 RNA-Binding Proteins Post-Transcriptionally Regulate Iron-Dependent Processes
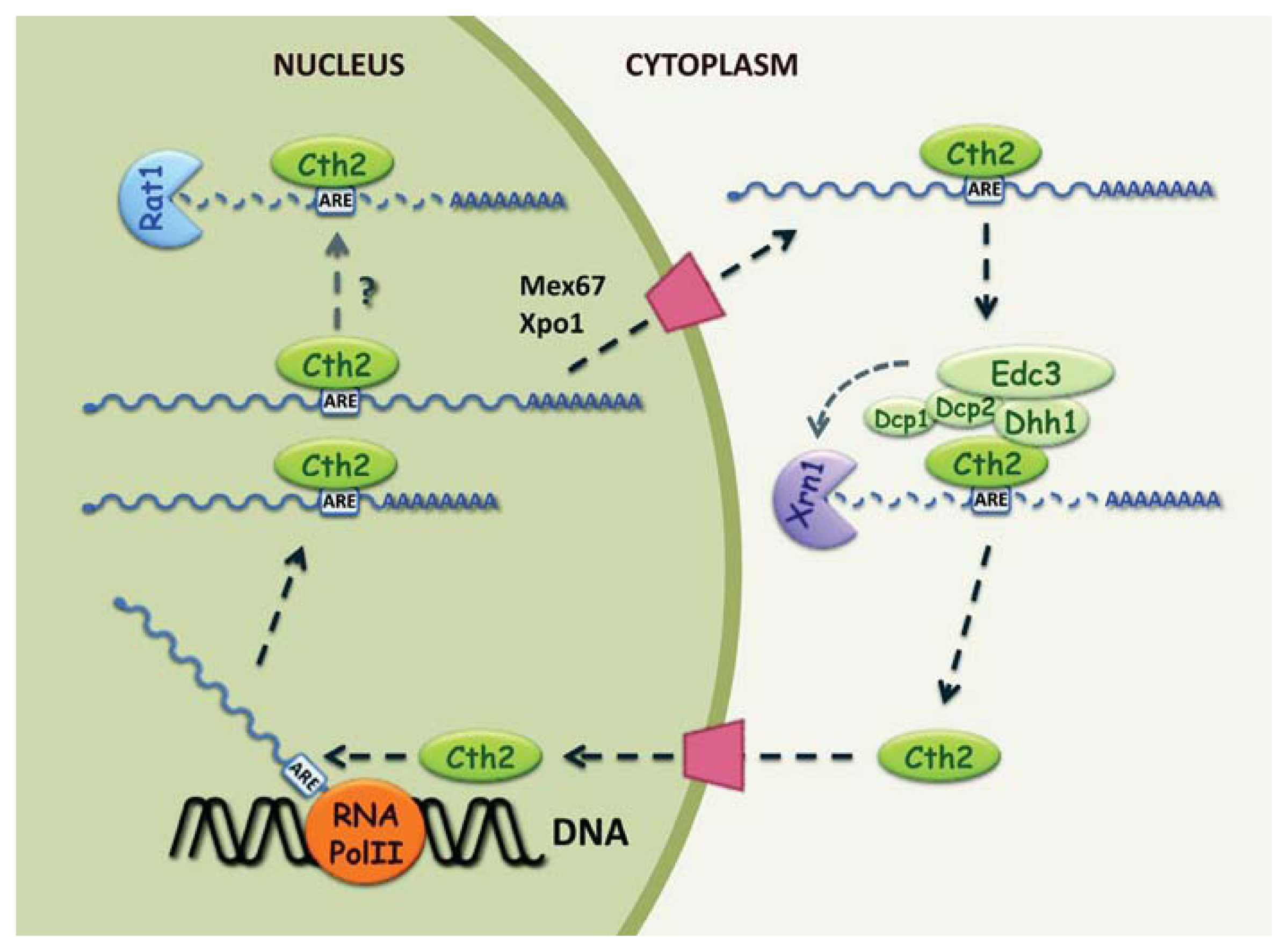
2.2. Cth2 Is a Nucleocytoplasmic Shuttling Protein
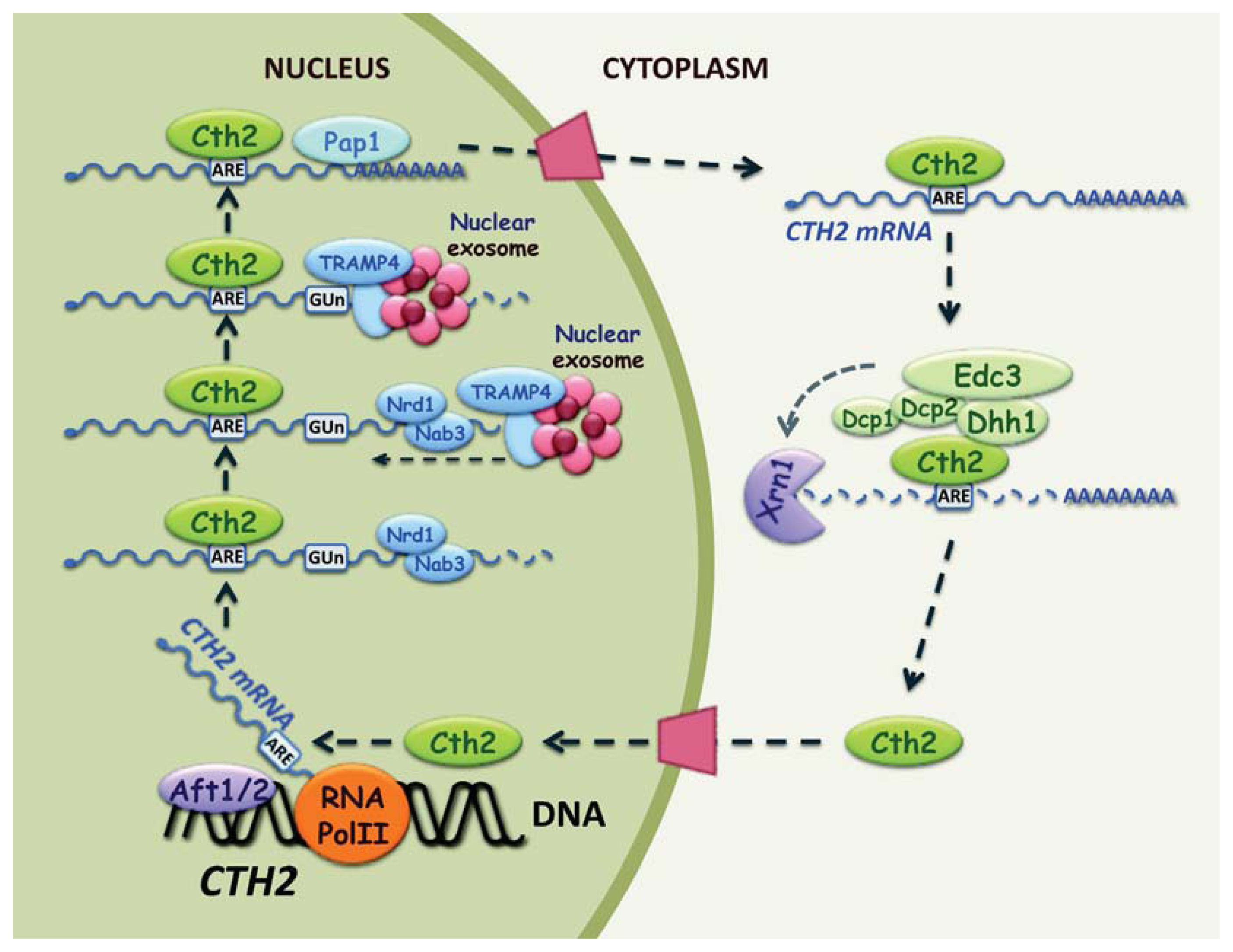
2.3. Cth2 Promotes mRNA Decay by Alternative 3′ End Processing
2.4. Cth2 Stimulates 5′ to 3′ Cytoplasmic Degradation of ARE-Containing mRNAs
2.5. Post-Transcriptional Regulation of CTH2 mRNA
2.6. Multilayered Regulation of Gene Expression by Mammalian TZF-Containing Proteins: Their Contribution to Fe Homeostasis
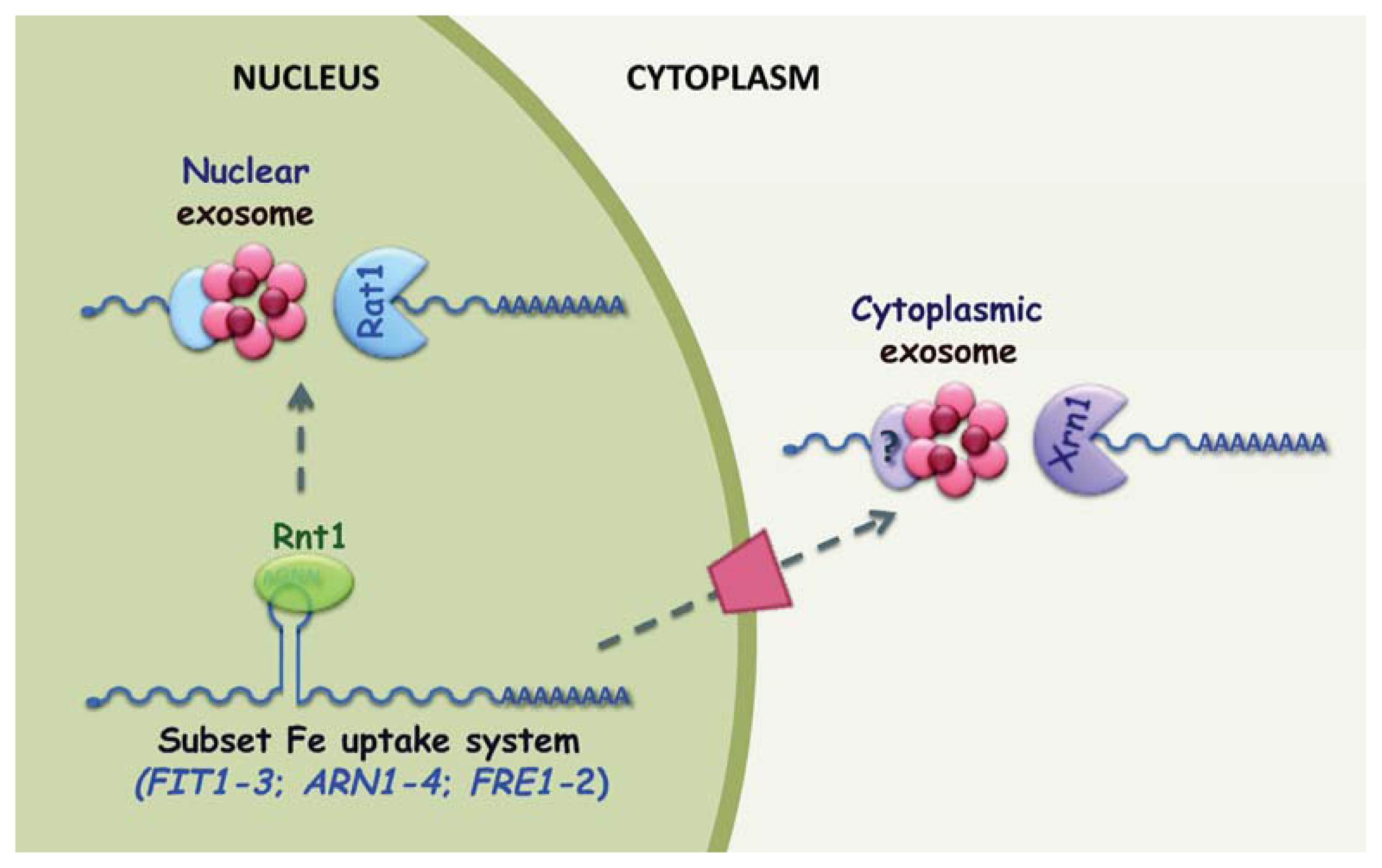
3. Post-Transcriptional Regulation in Response to Iron Excess
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Huma, N.; Salim Ur, R.; Anjum, F.M.; Murtaza, M.A.; Sheikh, M.A. Food fortification strategy-preventing iron deficiency anemia: A review. Crit. Rev. Food Sci. Nutr 2007, 47, 259–265. [Google Scholar]
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem 2005, 12, 1161–1208. [Google Scholar]
- Lee, Y.J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid signaling mediates iron toxicity. Cell. Metab 2012, 16, 90–96. [Google Scholar]
- Lin, H.; Li, L.; Jia, X.; Ward, D.M.; Kaplan, J. Genetic and biochemical analysis of high iron toxicity in yeast: Iron toxicity is due to the accumulation of cytosolic iron and occurs under both aerobic and anaerobic conditions. J. Biol. Chem 2011, 286, 3851–3862. [Google Scholar]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab 2010, 21, 302–314. [Google Scholar]
- Kaplan, J.; Ward, D.M.; de Domenico, I. The molecular basis of iron overload disorders and iron-linked anemias. Int. J. Hematol 2011, 93, 14–20. [Google Scholar]
- Ganz, T.; Nemeth, E. Hepcidin and disorders of iron metabolism. Annu. Rev. Med 2011, 62, 347–360. [Google Scholar]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol 2006, 2, 406–414. [Google Scholar]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr 2008, 28, 197–213. [Google Scholar]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell. Metab 2009, 9, 461–473. [Google Scholar]
- Sanchez, M.; Galy, B.; Schwanhaeusser, B.; Blake, J.; Bahr-Ivacevic, T.; Benes, V.; Selbach, M.; Muckenthaler, M.U.; Hentze, M.W. Iron regulatory protein-1 and -2: Transcriptome-wide definition of binding mRNAs and shaping of the cellular proteome by iron regulatory proteins. Blood 2011, 118, e168–e179. [Google Scholar]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar]
- Vashisht, A.A.; Zumbrennen, K.B.; Huang, X.; Powers, D.N.; Durazo, A.; Sun, D.; Bhaskaran, N.; Persson, A.; Uhlen, M.; Sangfelt, O.; et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009, 326, 718–721. [Google Scholar]
- Chen, O.S.; Crisp, R.J.; Valachovic, M.; Bard, M.; Winge, D.R.; Kaplan, J. Transcription of the yeast iron regulon does not respond directly to iron but rather to iron-sulfur cluster biosynthesis. J. Biol. Chem 2004, 279, 29513–29518. [Google Scholar]
- Li, L.; Miao, R.; Bertram, S.; Jia, X.; Ward, D.M.; Kaplan, J. A role for iron-sulfur clusters in the regulation of transcription factor Yap5-dependent high iron transcriptional responses in yeast. J. Biol. Chem 2012, 287, 35709–35721. [Google Scholar]
- Pujol-Carrion, N.; Belli, G.; Herrero, E.; Nogues, A.; de la Torre-Ruiz, M.A. Glutaredoxins Grx3 and Grx4 regulate nuclear localisation of Aft1 and the oxidative stress response in Saccharomyces cerevisiae. J. Cell. Sci 2006, 119, 4554–4564. [Google Scholar]
- Ojeda, L.; Keller, G.; Muhlenhoff, U.; Rutherford, J.C.; Lill, R.; Winge, D.R. Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces. cerevisiae. J. Biol. Chem 2006, 281, 17661–17669. [Google Scholar]
- Ueta, R.; Fujiwara, N.; Iwai, K.; Yamaguchi-Iwai, Y. Iron-induced dissociation of the Aft1p transcriptional regulator from target gene promoters is an initial event in iron-dependent gene suppression. Mol. Cell. Biol 2012, 32, 4998–5008. [Google Scholar]
- Kaplan, C.D.; Kaplan, J. Iron acquisition and transcriptional regulation. Chem. Rev 2009, 109, 4536–4552. [Google Scholar]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Muhlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar]
- Sanvisens, N.; Puig, S. Causes and Consequences of Nutritional Iron Deficiency in Living Organisms. In Biology of Starvation in Humans and Other Organisms; Merkin, T.C., Ed.; Nova Science Publishers, Inc: Hauppauge, New York, NY, USA, 2011; pp. 245–276. [Google Scholar]
- Kaplan, J.; McVey Ward, D.; Crisp, R.J.; Philpott, C.C. Iron-dependent metabolic remodeling in S. cerevisiae. Biochim. Biophys. Acta 2006, 1763, 646–651. [Google Scholar]
- Philpott, C.C.; Leidgens, S.; Frey, A.G. Metabolic remodeling in iron-deficient fungi. Biochim. Biophys. Acta 2012, 1823, 1509–1520. [Google Scholar]
- Li, L.; Chen, O.S.; McVey Ward, D.; Kaplan, J. CCC1 is a transporter that mediates vacuolar iron storage in yeast. J. Biol. Chem 2001, 276, 29515–29519. [Google Scholar]
- Li, L.; Bagley, D.; Ward, D.M.; Kaplan, J. Yap5 is an iron-responsive transcriptional activator that regulates vacuolar iron storage in yeast. Mol. Cell. Biol 2008, 28, 1326–1337. [Google Scholar]
- Rutherford, J.C.; Jaron, S.; Winge, D.R. Aft1p and Aft2p mediate iron-responsive gene expression in yeast through related promoter elements. J. Biol. Chem 2003, 278, 27636–27643. [Google Scholar]
- Shakoury-Elizeh, M.; Tiedeman, J.; Rashford, J.; Ferea, T.; Demeter, J.; Garcia, E.; Rolfes, R.; Brown, P.O.; Botstein, D.; Philpott, C.C. Transcriptional remodeling in response to iron deprivation in Saccharomyces. cerevisiae. Mol. Biol. Cell 2004, 15, 1233–1243. [Google Scholar]
- Puig, S.; Askeland, E.; Thiele, D.J. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell 2005, 120, 99–110. [Google Scholar]
- Foury, F.; Talibi, D. Mitochondrial control of iron homeostasis. A genome wide analysis of gene expression in a yeast frataxin-deficient strain. J. Biol. Chem 2001, 276, 7762–7768. [Google Scholar]
- SenGupta, D.J.; Zhang, B.; Kraemer, B.; Pochart, P.; Fields, S.; Wickens, M. A three-hybrid system to detect RNA-protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 8496–8501. [Google Scholar]
- Sanvisens, N.; Bano, M.C.; Huang, M.; Puig, S. Regulation of ribonucleotide reductase in response to iron deficiency. Mol. Cell 2011, 44, 759–769. [Google Scholar]
- Thompson, M.J.; Lai, W.S.; Taylor, G.A.; Blackshear, P.J. Cloning and characterization of two yeast genes encoding members of the CCCH class of zinc finger proteins: Zinc finger-mediated impairment of cell growth. Gene 1996, 174, 225–233. [Google Scholar]
- Puig, S.; Vergara, S.V.; Thiele, D.J. Cooperation of two mRNA-binding proteins drives metabolic adaptation to iron deficiency. Cell Metab 2008, 7, 555–564. [Google Scholar]
- Ma, Q.; Herschman, H.R. The yeast homologue YTIS11, of the mammalian TIS11 gene family is a non-essential, glucose repressible gene. Oncogene 1995, 10, 487–494. [Google Scholar]
- Sanvisens, N.; de Llanos, R.; Puig, S. Function and regulation of yeast ribonucleotide reductase: Cell cycle, genotoxic stress, and iron bioavailability. Biomed. J 2013, 36, 51–58. [Google Scholar]
- Cotruvo, J.A.; Stubbe, J. Class I ribonucleotide reductases: Metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem 2011, 80, 733–767. [Google Scholar]
- Ihrig, J.; Hausmann, A.; Hain, A.; Richter, N.; Hamza, I.; Lill, R.; Muhlenhoff, U. Iron regulation through the back door: Iron-dependent metabolite levels contribute to transcriptional adaptation to iron deprivation in Saccharomyces. cerevisiae. Eukaryot. Cell 2010, 9, 460–471. [Google Scholar]
- Pedro-Segura, E.; Vergara, S.V.; Rodriguez-Navarro, S.; Parker, R.; Thiele, D.J.; Puig, S. The Cth2 ARE-binding protein recruits the Dhh1 helicase to promote the decay of succinate dehydrogenase SDH4 mRNA in response to iron deficiency. J. Biol. Chem 2008, 283, 28527–28535. [Google Scholar]
- Vergara, S.V.; Puig, S.; Thiele, D.J. Early recruitment of AU-rich element-containing mRNAs determines their cytosolic fate during iron deficiency. Mol. Cell. Biol 2011, 31, 417–429. [Google Scholar]
- Millevoi, S.; Vagner, S. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res 2010, 38, 2757–2774. [Google Scholar]
- Mandel, C.R.; Bai, Y.; Tong, L. Protein factors in pre-mRNA 3′-end processing. Cell. Mol. Life Sci 2008, 65, 1099–1122. [Google Scholar]
- Mischo, H.E.; Proudfoot, N.J. Disengaging polymerase: Terminating RNA polymerase II transcription in budding yeast. Biochim. Biophys. Acta 2013, 1829, 174–185. [Google Scholar]
- Waern, K.; Snyder, M. Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 2013, 3, 343–352. [Google Scholar]
- Pelechano, V.; Wei, W.; Steinmetz, L.M. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 2013, 497, 127–131. [Google Scholar]
- Ozsolak, F.; Kapranov, P.; Foissac, S.; Kim, S.W.; Fishilevich, E.; Monaghan, A.P.; John, B.; Milos, P.M. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 2010, 143, 1018–1029. [Google Scholar]
- Yoon, O.K.; Brem, R.B. Noncanonical transcript forms in yeast and their regulation during environmental stress. RNA 2010, 16, 1256–1267. [Google Scholar]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar]
- Miura, F.; Kawaguchi, N.; Sese, J.; Toyoda, A.; Hattori, M.; Morishita, S.; Ito, T. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc. Natl. Acad. Sci. USA 2006, 103, 17846–17851. [Google Scholar]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 2005, 33, 201–212. [Google Scholar]
- Kim Guisbert, K.S.; Li, H.; Guthrie, C. Alternative 3′ pre-mRNA processing in Saccharomyces. cerevisiae is modulated by Nab4/Hrp1 in vivo. PLoS Biol 2007, 5, e6. [Google Scholar]
- Perez-Canadillas, J.M. Grabbing the message: Structural basis of mRNA 3′UTR recognition by Hrp1. EMBO J 2006, 25, 3167–3178. [Google Scholar]
- Minvielle-Sebastia, L.; Beyer, K.; Krecic, A.M.; Hector, R.E.; Swanson, M.S.; Keller, W. Control of cleavage site selection during mRNA 3′ end formation by a yeast hnRNP. EMBO J 1998, 17, 7454–7468. [Google Scholar]
- Kessler, M.M.; Henry, M.F.; Shen, E.; Zhao, J.; Gross, S.; Silver, P.A.; Moore, C.L. Hrp1, a sequence-specific RNA-binding protein that shuttles between the nucleus and the cytoplasm, is required for mRNA 3′-end formation in yeast. Genes Dev 1997, 11, 2545–2556. [Google Scholar]
- Gonzalez, C.I.; Ruiz-Echevarria, M.J.; Vasudevan, S.; Henry, M.F.; Peltz, S.W. The yeast hnRNP-like protein Hrp1/Nab4 marks a transcript for nonsense-mediated mRNA decay. Mol. Cell 2000, 5, 489–499. [Google Scholar]
- Prouteau, M.; Daugeron, M.C.; Seraphin, B. Regulation of ARE transcript 3′ end processing by the yeast Cth2 mRNA decay factor. EMBO J 2008, 27, 2966–2976. [Google Scholar]
- Aitchison, J.D.; Blobel, G.; Rout, M.P. Kap104p: A karyopherin involved in the nuclear transport of messenger RNA binding proteins. Science 1996, 274, 624–627. [Google Scholar]
- Milligan, L.; Torchet, C.; Allmang, C.; Shipman, T.; Tollervey, D. A nuclear surveillance pathway for mRNAs with defective polyadenylation. Mol. Cell. Biol 2005, 25, 9996–10004. [Google Scholar]
- Parker, R. RNA degradation in Saccharomyces. cerevisiae. Genetics 2012, 191, 671–702. [Google Scholar]
- Balagopal, V.; Fluch, L.; Nissan, T. Ways and means of eukaryotic mRNA decay. Biochim. Biophys. Acta 2012, 1819, 593–603. [Google Scholar]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar]
- Coller, J.; Parker, R. General translational repression by activators of mRNA decapping. Cell 2005, 122, 875–886. [Google Scholar]
- Nissan, T.; Rajyaguru, P.; She, M.; Song, H.; Parker, R. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol. Cell 2010, 39, 773–783. [Google Scholar]
- Carroll, J.S.; Munchel, S.E.; Weis, K. The DExD/H box ATPase Dhh1 functions in translational repression, mRNA decay, and processing body dynamics. J. Cell. Biol 2011, 194, 527–537. [Google Scholar]
- Decker, C.J.; Parker, R. P-bodies and stress granules: Possible roles in the control of translation and mRNA degradation. Cold Spring Harb. Perspect. Biol 2012, 4, a012286. [Google Scholar]
- Erickson, S.L.; Lykke-Andersen, J. Cytoplasmic mRNP granules at a glance. J. Cell. Sci 2011, 124, 293–297. [Google Scholar]
- Sheth, U.; Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 2003, 300, 805–808. [Google Scholar]
- Teixeira, D.; Sheth, U.; Valencia-Sanchez, M.A.; Brengues, M.; Parker, R. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 2005, 11, 371–382. [Google Scholar]
- Teixeira, D.; Parker, R. Analysis of P-body assembly in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 2274–2287. [Google Scholar]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell. Biol 2007, 8, 9–22. [Google Scholar]
- Ciais, D.; Bohnsack, M.T.; Tollervey, D. The mRNA encoding the yeast ARE-binding protein Cth2 is generated by a novel 3′ processing pathway. Nucleic Acids Res. 2008. [Google Scholar] [CrossRef]
- Roth, K.M.; Byam, J.; Fang, F.; Butler, J.S. Regulation of NAB2 mRNA 3′-end formation requires the core exosome and the Trf4p component of the TRAMP complex. RNA 2009, 15, 1045–1058. [Google Scholar]
- Roth, K.M.; Wolf, M.K.; Rossi, M.; Butler, J.S. The nuclear exosome contributes to autogenous control of NAB2 mRNA levels. Mol. Cell. Biol 2005, 25, 1577–1585. [Google Scholar]
- Torchet, C.; Bousquet-Antonelli, C.; Milligan, L.; Thompson, E.; Kufel, J.; Tollervey, D. Processing of 3′-extended read-through transcripts by the exosome can generate functional mRNAs. Mol. Cell 2002, 9, 1285–1296. [Google Scholar]
- Lykke-Andersen, S.; Tomecki, R.; Jensen, T.H.; Dziembowski, A. The eukaryotic RNA exosome: Same scaffold but variable catalytic subunits. RNA Biol 2011, 8, 61–66. [Google Scholar]
- Schmid, M.; Jensen, T.H. The exosome: A multipurpose RNA-decay machine. Trends Biochem. Sci 2008, 33, 501–510. [Google Scholar]
- Houseley, J.; LaCava, J.; Tollervey, D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell. Biol 2006, 7, 529–539. [Google Scholar]
- Houalla, R.; Devaux, F.; Fatica, A.; Kufel, J.; Barrass, D.; Torchet, C.; Tollervey, D. Microarray detection of novel nuclear RNA substrates for the exosome. Yeast 2006, 23, 439–454. [Google Scholar]
- Wlotzka, W.; Kudla, G.; Granneman, S.; Tollervey, D. The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J 2011, 30, 1790–1803. [Google Scholar]
- Jamonnak, N.; Creamer, T.J.; Darby, M.M.; Schaughency, P.; Wheelan, S.J.; Corden, J.L. Yeast Nrd1, Nab3, and Sen1 transcriptome-wide binding maps suggest multiple roles in post-transcriptional RNA processing. RNA 2011, 17, 2011–2025. [Google Scholar]
- Martinez-Pastor, M.; Vergara, S.V.; Puig, S.; Thiele, D.J. Negative feedback regulation of the yeast Cth1 and Cth2 mRNA binding proteins is required for adaptation to iron deficiency and iron supplementation. Mol. Cell. Biol 2013, 33, 2178–2187. [Google Scholar]
- Perez-Ortin, J.E.; Alepuz, P.M.; Moreno, J. Genomics and gene transcription kinetics in yeast. Trends Genet 2007, 23, 250–257. [Google Scholar]
- Ciais, D.; Cherradi, N.; Feige, J.J. Multiple functions of tristetraprolin/TIS11 RNA-binding proteins in the regulation of mRNA biogenesis and degradation. Cell. Mol. Life Sci 2012, 70, 2031–2044. [Google Scholar]
- Brooks, S.A.; Blackshear, P.J. Tristetraprolin (TTP): Interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim. Biophys. Acta 2013, 1829, 666–679. [Google Scholar]
- Baou, M.; Jewell, A.; Murphy, J.J. TIS11 family proteins and their roles in posttranscriptional gene regulation. J. Biomed. Biotechnol 2009, 2009, 634520. [Google Scholar]
- Tiedje, C.; Ronkina, N.; Tehrani, M.; Dhamija, S.; Laass, K.; Holtmann, H.; Kotlyarov, A.; Gaestel, M. The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet 2012, 8, e1002977. [Google Scholar]
- Pfeiffer, J.R.; Brooks, S.A. Cullin 4B is recruited to tristetraprolin-containing messenger ribonucleoproteins and regulates TNF-alpha mRNA polysome loading. J. Immunol 2012, 188, 1828–1839. [Google Scholar]
- Qi, M.Y.; Wang, Z.Z.; Zhang, Z.; Shao, Q.; Zeng, A.; Li, X.Q.; Li, W.Q.; Wang, C.; Tian, F.J.; Li, Q.; et al. AU-rich-element-dependent translation repression requires the cooperation of tristetraprolin and RCK/P54. Mol. Cell. Biol 2012, 32, 913–928. [Google Scholar]
- Murata, T.; Yoshino, Y.; Morita, N.; Kaneda, N. Identification of nuclear import and export signals within the structure of the zinc finger protein TIS11. Biochem. Biophys. Res. Commun 2002, 293, 1242–1247. [Google Scholar]
- Phillips, R.S.; Ramos, S.B.; Blackshear, P.J. Members of the tristetraprolin family of tandem CCCH zinc finger proteins exhibit CRM1-dependent nucleocytoplasmic shuttling. J. Biol. Chem 2002, 277, 11606–11613. [Google Scholar]
- Desroches-Castan, A.; Cherradi, N.; Feige, J.J.; Ciais, D. A novel function of Tis11b/BRF1 as a regulator of Dll4 mRNA 3′-end processing. Mol. Biol. Cell 2011, 22, 3625–3633. [Google Scholar]
- Brooks, S.A.; Connolly, J.E.; Rigby, W.F. The role of mRNA turnover in the regulation of tristetraprolin expression: Evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol 2004, 172, 7263–7271. [Google Scholar]
- Tchen, C.R.; Brook, M.; Saklatvala, J.; Clark, A.R. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem 2004, 279, 32393–32400. [Google Scholar]
- Clement, S.L.; Scheckel, C.; Stoecklin, G.; Lykke-Andersen, J. Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing deadenylase recruitment. Mol. Cell. Biol 2011, 31, 256–266. [Google Scholar]
- Marchese, F.P.; Aubareda, A.; Tudor, C.; Saklatvala, J.; Clark, A.R.; Dean, J.L. MAPKAP kinase 2 blocks tristetraprolin-directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J. Biol. Chem 2010, 285, 27590–27600. [Google Scholar]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar]
- Bayeva, M.; Khechaduri, A.; Puig, S.; Chang, H.C.; Patial, S.; Blackshear, P.J.; Ardehali, H. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell. Metab 2012, 16, 645–657. [Google Scholar]
- Felice, M.R.; de Domenico, I.; Li, L.; Ward, D.M.; Bartok, B.; Musci, G.; Kaplan, J. Post-transcriptional regulation of the yeast high affinity iron transport system. J. Biol. Chem 2005, 280, 22181–22190. [Google Scholar]
- Lee, A.; Henras, A.K.; Chanfreau, G. Multiple RNA surveillance pathways limit aberrant expression of iron uptake mRNAs and prevent iron toxicity in S. cerevisiae. Mol. Cell 2005, 19, 39–51. [Google Scholar]
- Hartman, E.; Wang, Z.; Zhang, Q.; Roy, K.; Chanfreau, G.; Feigon, J. Intrinsic dynamics of an extended hydrophobic core in the S. cerevisiae RNase III dsRBD contributes to recognition of specific RNA binding sites. J. Mol. Biol 2013, 425, 546–562. [Google Scholar]
- Wang, Z.; Hartman, E.; Roy, K.; Chanfreau, G.; Feigon, J. Structure of a yeast RNase III dsRBD complex with a noncanonical RNA substrate provides new insights into binding specificity of dsRBDs. Structure 2011, 19, 999–1010. [Google Scholar]
- Chanfreau, G.; Buckle, M.; Jacquier, A. Recognition of a conserved class of RNA tetraloops by Saccharomyces. cerevisiae RNase III. Proc. Natl. Acad. Sci. USA 2000, 97, 3142–3147. [Google Scholar]
- Nagel, R.; Ares, M., Jr. Substrate recognition by a eukaryotic RNase III: The double-stranded RNA-binding domain of Rnt1p selectively binds RNA containing a 5′-AGNN-3′ tetraloop. RNA 2000, 6, 1142–1156. [Google Scholar]
- Leulliot, N.; Quevillon-Cheruel, S.; Graille, M.; van Tilbeurgh, H.; Leeper, T.C.; Godin, K.S.; Edwards, T.E.; Sigurdsson, S.T.; Rozenkrants, N.; Nagel, R.J.; et al. A new alpha-helical extension promotes RNA binding by the dsRBD of Rnt1p RNAse III. EMBO J 2004, 23, 2468–2477. [Google Scholar]
- Wu, H.; Henras, A.; Chanfreau, G.; Feigon, J. Structural basis for recognition of the AGNN tetraloop RNA fold by the double-stranded RNA-binding domain of Rnt1p RNase III. Proc. Natl. Acad. Sci. USA 2004, 101, 8307–8312. [Google Scholar]
- Meaux, S.; Lavoie, M.; Gagnon, J.; Abou Elela, S.; van Hoof, A. Reporter mRNAs cleaved by Rnt1p are exported and degraded in the cytoplasm. Nucleic Acids Res 2011, 39, 9357–9367. [Google Scholar]
- Ghazal, G.; Gagnon, J.; Jacques, P.E.; Landry, J.R.; Robert, F.; Elela, S.A. Yeast RNase III triggers polyadenylation-independent transcription termination. Mol. Cell 2009, 36, 99–109. [Google Scholar]
- Rondon, A.G.; Mischo, H.E.; Kawauchi, J.; Proudfoot, N.J. Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol. Cell 2009, 36, 88–98. [Google Scholar]
- Ge, D.; Lamontagne, B.; Elela, S.A. RNase III-mediated silencing of a glucose-dependent repressor in yeast. Curr. Biol 2005, 15, 140–145. [Google Scholar]
- Zer, C.; Chanfreau, G. Regulation and surveillance of normal and 3′-extended forms of the yeast aci-reductone dioxygenase mRNA by RNase III cleavage and exonucleolytic degradation. J. Biol. Chem 2005, 280, 28997–29003. [Google Scholar]
- Larose, S.; Laterreur, N.; Ghazal, G.; Gagnon, J.; Wellinger, R.J.; Elela, S.A. RNase III-dependent regulation of yeast telomerase. J. Biol. Chem 2007, 282, 4373–4381. [Google Scholar]
- Catala, M.; Aksouh, L.; Abou Elela, S. RNA-dependent regulation of the cell wall stress response. Nucleic Acids Res 2012, 40, 7507–7517. [Google Scholar]
- Catala, M.; Lamontagne, B.; Larose, S.; Ghazal, G.; Elela, S.A. Cell cycle-dependent nuclear localization of yeast RNase III is required for efficient cell division. Mol. Biol. Cell 2004, 15, 3015–3030. [Google Scholar]



© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez-Pastor, M.T.; Llanos, R.D.; Romero, A.M.; Puig, S. Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2013, 14, 15785-15809. https://doi.org/10.3390/ijms140815785
Martínez-Pastor MT, Llanos RD, Romero AM, Puig S. Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2013; 14(8):15785-15809. https://doi.org/10.3390/ijms140815785
Chicago/Turabian StyleMartínez-Pastor, María Teresa, Rosa De Llanos, Antonia María Romero, and Sergi Puig. 2013. "Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae" International Journal of Molecular Sciences 14, no. 8: 15785-15809. https://doi.org/10.3390/ijms140815785
APA StyleMartínez-Pastor, M. T., Llanos, R. D., Romero, A. M., & Puig, S. (2013). Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae. International Journal of Molecular Sciences, 14(8), 15785-15809. https://doi.org/10.3390/ijms140815785