Effect of δ-Opioid Receptor Activation on BDNF-TrkB vs. TNF-α in the Mouse Cortex Exposed to Prolonged Hypoxia

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
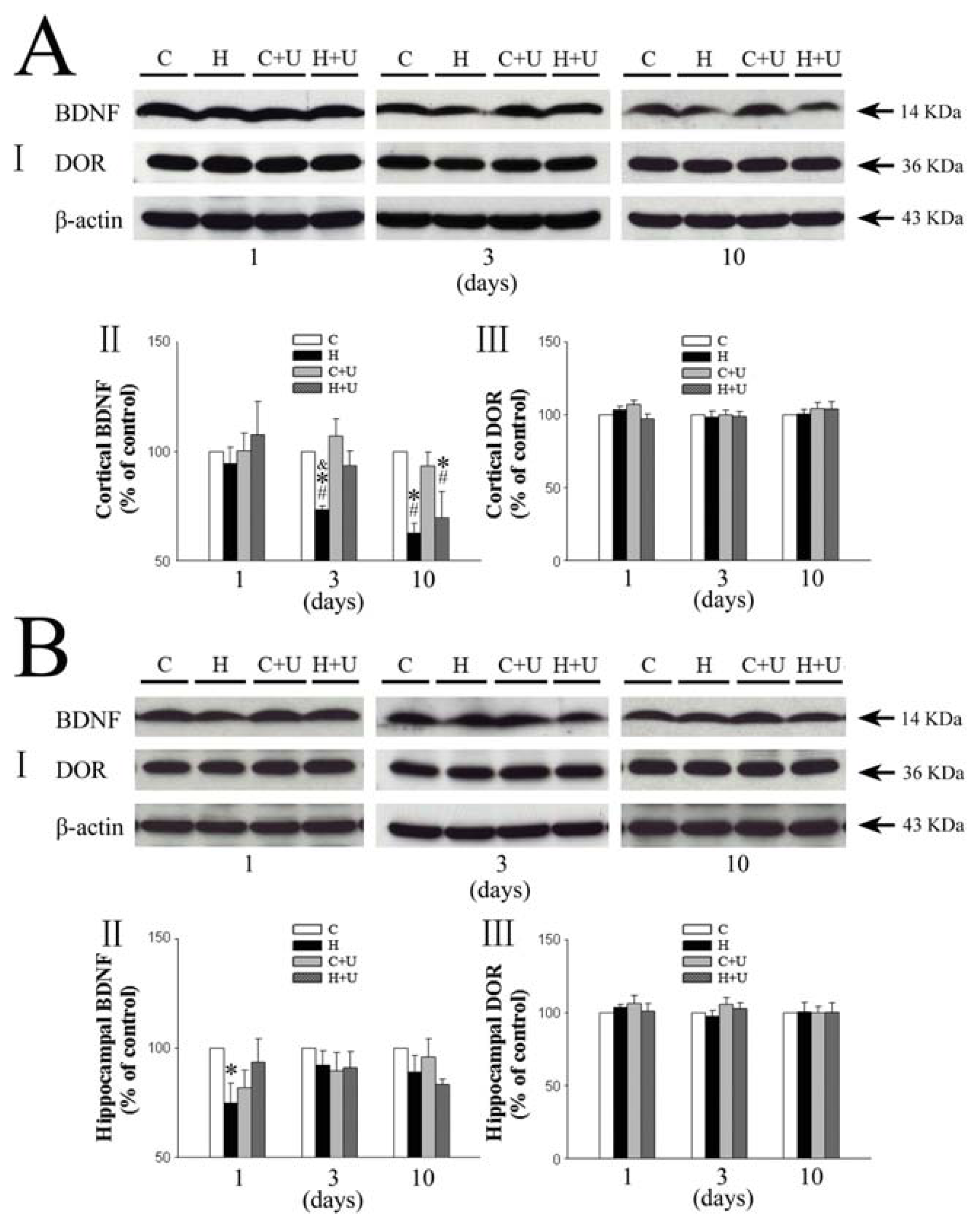
2.1. Effect of DOR Activation on Cortical BDNF under Hypoxia
2.2. Effect of DOR Activation on Hippocampal BDNF Expression under Hypoxia
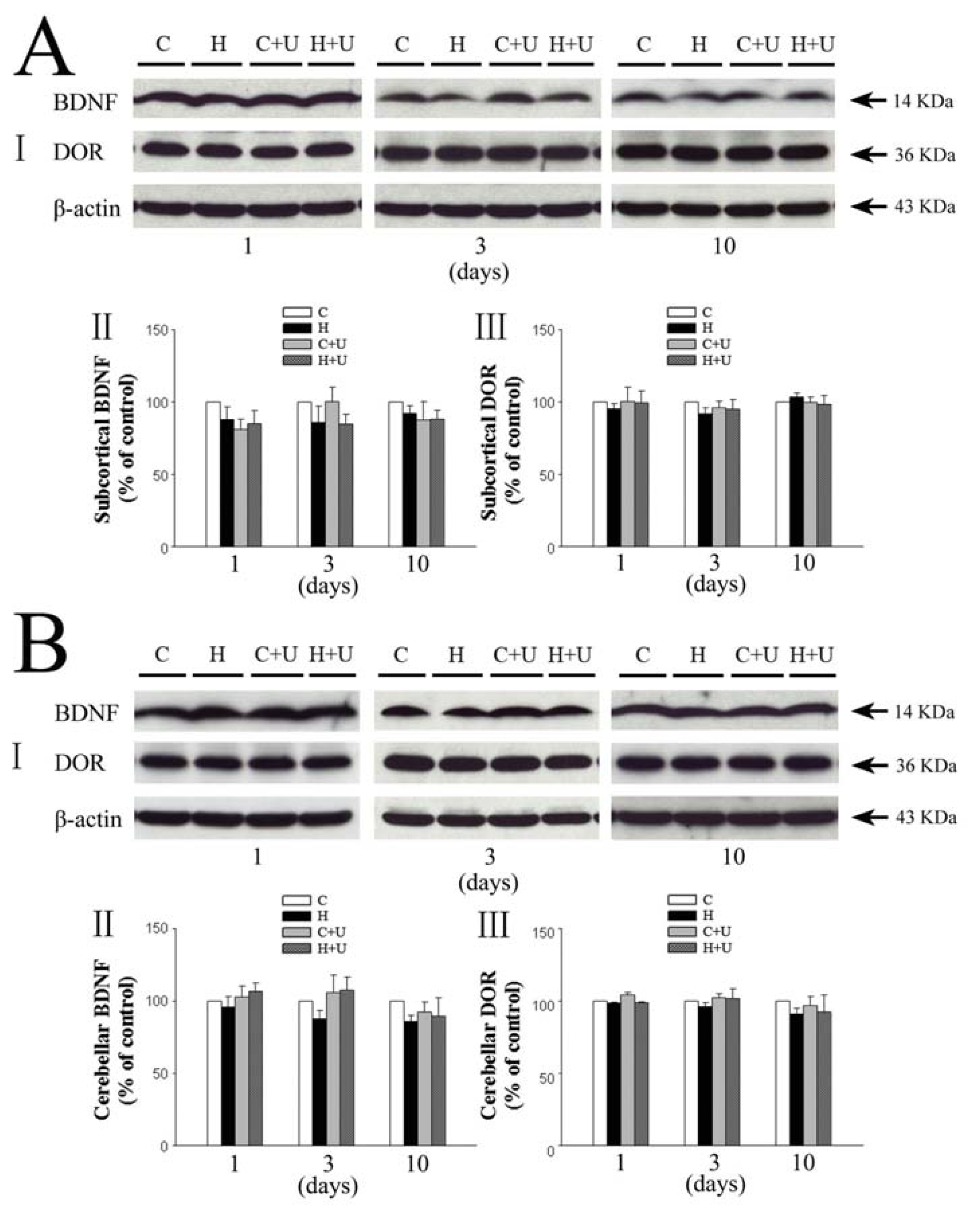
2.3. Effect of DOR Activation on Subcortical and Cerebellar BDNF in Hypoxia
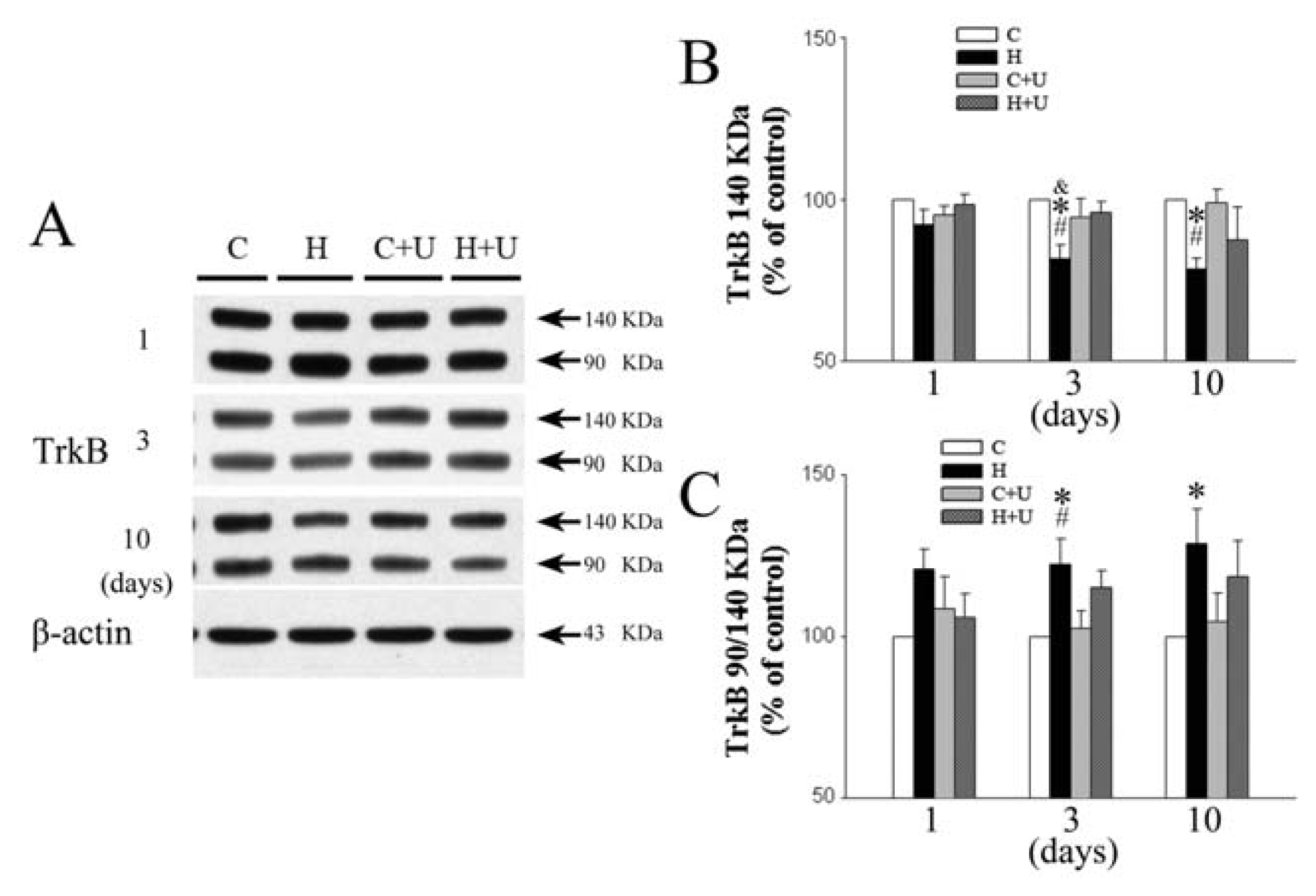
2.4. Effect of DOR Activation on Cortical TrkB Protein Expression
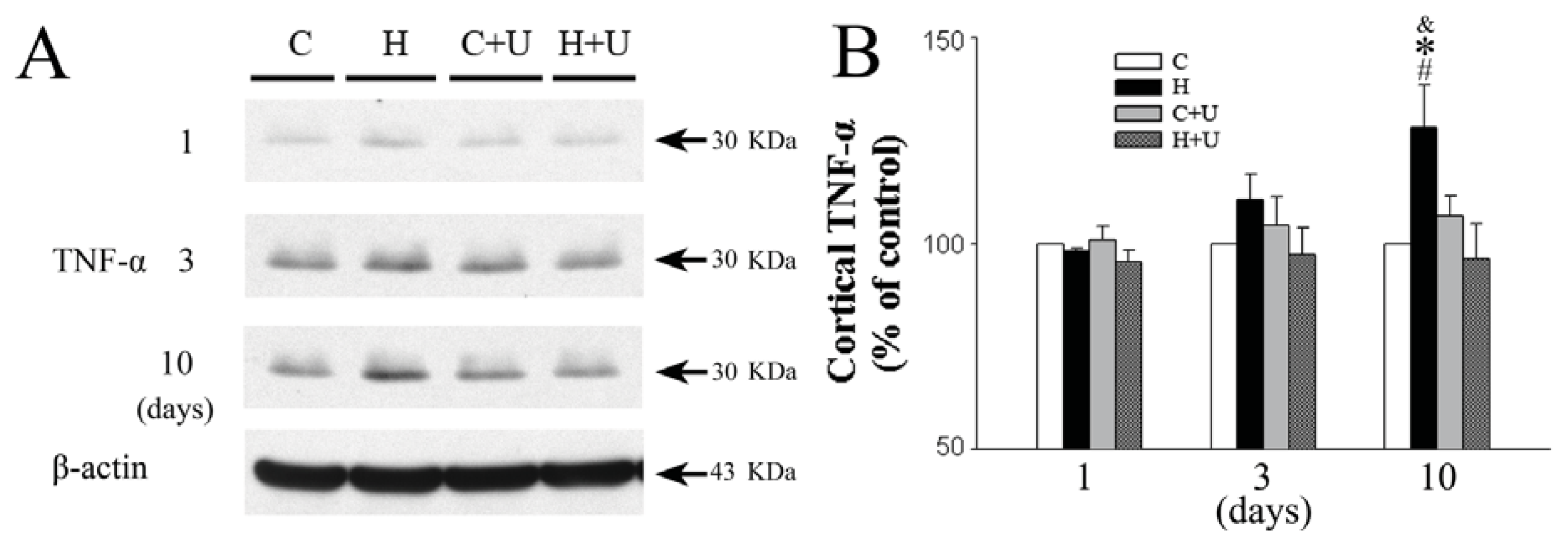
2.5. Effect of DOR Activation on Cortical TNF-α Protein Expression
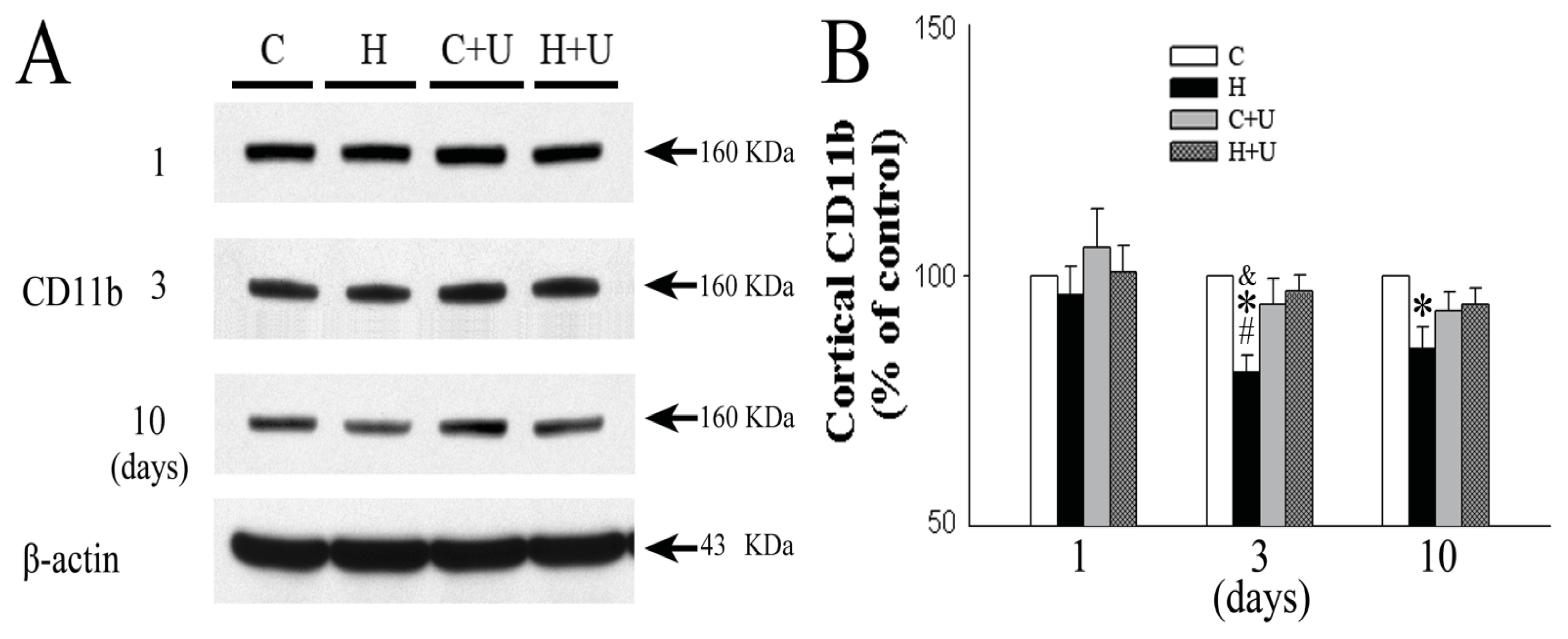
2.6. Effect of DOR Activation on Cortical CD11b Protein Expression
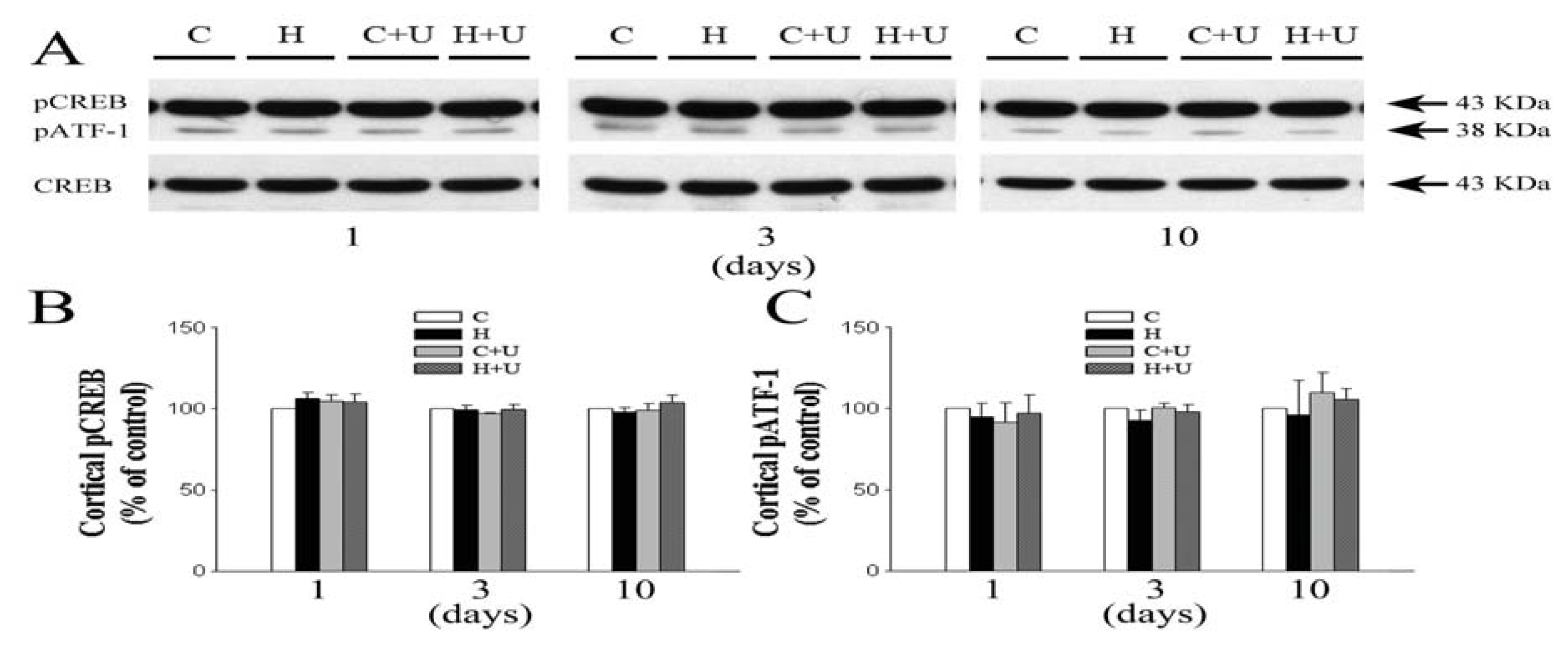
2.7. Effect of DOR Activation on Cortical pATF-1 and pCREB/CREB Expression
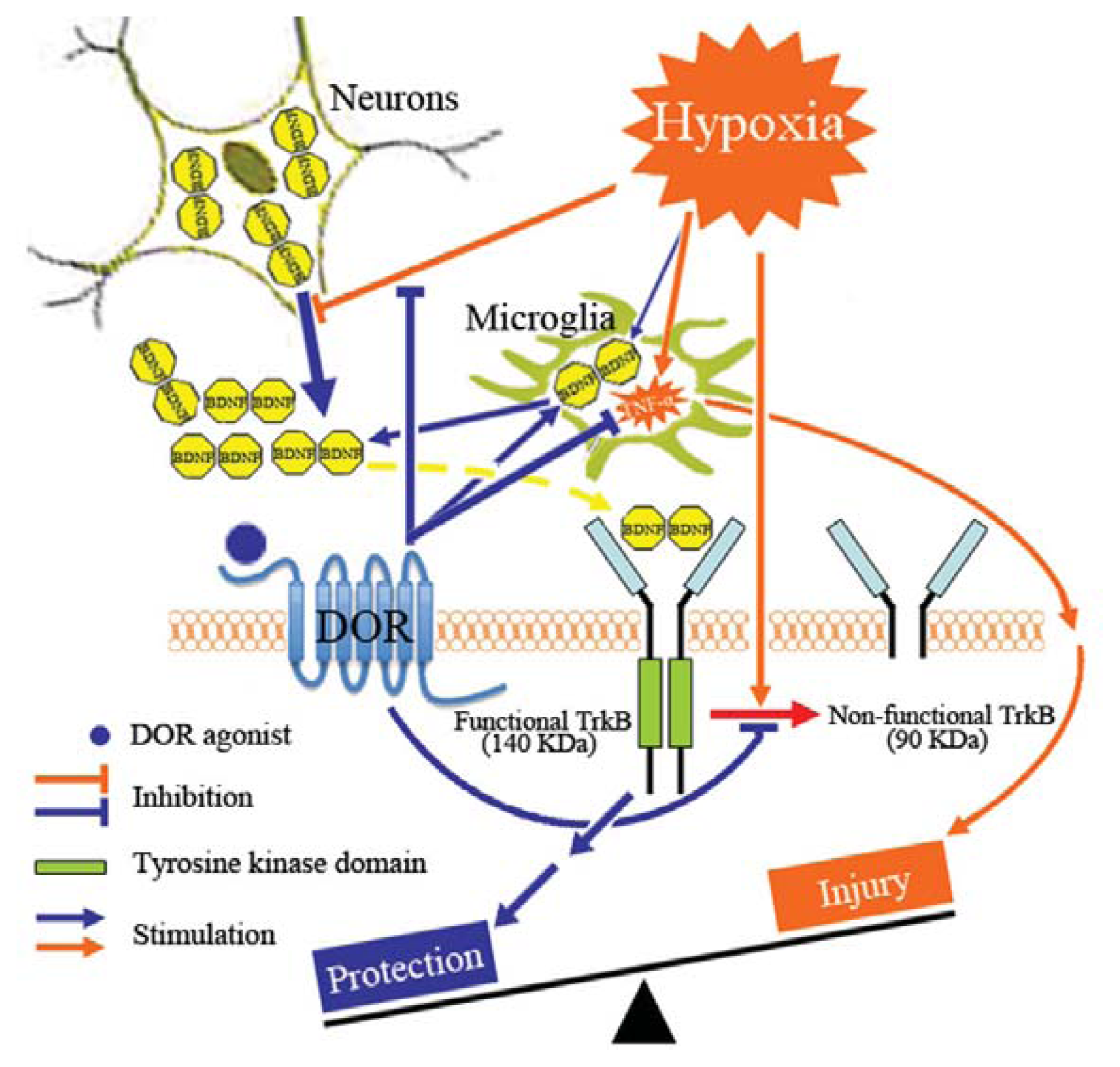
3. Discussion
4. Experimental Section
4.1. Animals and Hypoxic Exposure
4.2. Chemicals and Reagents
4.3. Western Blot Analysis
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
Abbreviations
| DOR | delta-opioid receptor |
| BDNF | brain-derived neurotrophic factor |
| TrkB | tyrosine kinase B |
| TNF-α | tumor necrosis factor α |
| CREB | cyclic AMP response element binding protein |
| p-CREB | CREB phosphorylation |
References
- Zhang, J.; Haddad, G.G.; Xia, Y. Delta-, but not mu- and kappa-, opioid receptor activation protects neocortical neurons from glutamate-induced excitotoxic injury. Brain Res 2000, 885, 143–153. [Google Scholar]
- Tian, X.S.; Zhou, F.; Yang, R.; Xia, Y.; Wu, G.C.; Guo, J.C. Effects of intracerebroventricular injection of delta-opioid receptor agonist TAN-67 or antagonist naltrindole on acute cerebral ischemia in rat. Sheng Li Xue Bao 2008, 60, 475–484. [Google Scholar]
- He, X.; Sandhu, H.K.; Yang, Y.; Hua, F.; Belser, N.; Kim, D.H.; Xia, Y. Neuroprotection against hypoxia/ischemia: Delta-opioid receptor-mediated cellular/molecular events. Cell Mol. Life Sci 2013, 70, 2291–2303. [Google Scholar]
- Zhao, P.; Huang, Y.; Zuo, Z. Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J. Neuropathol. Exp. Neurol 2006, 65, 945–952. [Google Scholar]
- Gwak, M.S.; Li, L.; Zuo, Z. Morphine preconditioning reduces lipopolysaccharide and interferon-gamma-induced mouse microglial cell injury via delta 1 opioid receptor activation. Neuroscience 2010, 167, 256–260. [Google Scholar]
- Borlongan, C.V.; Wang, Y.; Su, T.P. Delta opioid peptide (D-Ala 2, D-Leu 5) enkephalin: Linking hibernation and neuroprotection. Front. Biosci 2004, 9, 3392–3398. [Google Scholar]
- Borlongan, C.V.; Hayashi, T.; Oeltgen, P.R.; Su, T.P.; Wang, Y. Hibernation-like state induced by an opioid peptide protects against experimental stroke. BMC Biol 2009, 7, 31. [Google Scholar]
- Sun, X.; Zhou, H.; Luo, X.; Li, S.; Yu, D.; Hua, J.; Mu, D.; Mao, M. Neuroprotection of brain-derived neurotrophic factor against hypoxic injury in vitro requires activation of extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. Int. J. Dev. Neurosci 2008, 26, 363–370. [Google Scholar]
- Narumiya, S.; Ohno, M.; Tanaka, N.; Yamano, T.; Shimada, M. Enhanced expression of full-length TrkB receptors in young rat brain with hypoxic/ischemic injury. Brain Res 1998, 797, 278–286. [Google Scholar]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci 2001, 24, 677–736. [Google Scholar]
- Frisen, J.; Verge, V.M.; Fried, K.; Risling, M.; Persson, H.; Trotter, J.; Hökfelt, T.; Lindholm, D. Characterization of glial trkB receptors: Differential response to injury in the central and peripheral nervous systems. Proc. Natl. Acad. Sci. USA 1993, 90, 4971–4975. [Google Scholar]
- Wong, J.; Higgins, M.; Halliday, G.; Garner, B. Amyloid beta selectively modulates neuronal TrkB alternative transcript expression with implications for Alzheimer’s disease. Neuroscience 2012, 210, 363–374. [Google Scholar]
- Wong, J.; Rothmond, D.A.; Webster, M.J.; Weickert, C.S. Increases in two truncated TrkB isoforms in the prefrontal cortex of people with schizophrenia. Schizophr. Bull 2013, 39, 130–140. [Google Scholar]
- Torregrossa, M.M.; Folk, J.E.; Rice, K.C.; Watson, S.J.; Woods, J.H. Chronic administration of the delta opioid receptor agonist (+)BW373U86 and antidepressants on behavior in the forced swim test and BDNF mRNA expression in rats. Psychopharmacology (Berl. ) 2005, 183, 31–40. [Google Scholar]
- Torregrossa, M.M.; Isgor, C.; Folk, J.E.; Rice, K.C.; Watson, S.J.; Woods, J.H. The delta-opioid receptor agonist (+)BW373U86 regulates BDNF mRNA expression in rats. Neuropsychopharmacology 2004, 29, 649–659. [Google Scholar]
- Torregrossa, M.M.; Jutkiewicz, E.M.; Mosberg, H.I.; Balboni, G.; Watson, S.J.; Woods, J.H. Peptidic delta opioid receptor agonists produce antidepressant-like effects in the forced swim test and regulate BDNF mRNA expression in rats. Brain Res 2006, 1069, 172–181. [Google Scholar]
- Zhang, H.; Torregrossa, M.M.; Jutkiewicz, E.M.; Shi, Y.G.; Rice, K.C.; Woods, J.H.; Watson, S.J.; Ko, M.C. Endogenous opioids upregulate brain-derived neurotrophic factor mRNA through deltaand micro-opioid receptors independent of antidepressant-like effects. Eur. J. Neurosci 2006, 23, 984–994. [Google Scholar]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm 2008, 5, 45. [Google Scholar]
- Makar, T.K.; Bever, C.T.; Singh, I.S.; Royal, W.; Sahu, S.N.; Sura, T.P.; Sultana, S.; Sura, K.T.; Patel, N.; Dhib-Jalbut, S.; et al. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J. Neuroimmunol 2009, 210, 40–51. [Google Scholar]
- Fujino, H.; Kitaoka, Y.; Hayashi, Y.; Munemasa, Y.; Takeda, H.; Kumai, T.; Kobayashi, S.; Ueno, S. Axonal protection by brain-derived neurotrophic factor associated with CREB phosphorylation in tumor necrosis factor-alpha-induced optic nerve degeneration. Acta Neuropathol 2009, 117, 75–84. [Google Scholar]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar]
- Finkbeiner, S.; Tavazoie, S.F.; Maloratsky, A.; Jacobs, K.M.; Harris, K.M.; Greenberg, M.E. CREB: A major mediator of neuronal neurotrophin responses. Neuron 1997, 19, 1031–1047. [Google Scholar]
- Bilecki, W.; Hollt, V.; Przewlocki, R. Acute delta-opioid receptor activation induces CREB phosphorylation in NG108–15 cells. Eur. J. Pharmacol 2000, 390, 1–6. [Google Scholar]
- Zhang, J.; Xia, Y.; Haddad, G.G. Activation of δ-opioid receptors protects cortical neurons from glutamate excitotoxic injury. Soc. Neurosci 1999, 28, 736. [Google Scholar]
- Gao, C.J.; Niu, L.; Ren, P.C.; Wang, W.; Zhu, C.; Li, Y.Q.; Chai, W.; Sun, X.D. Hypoxic preconditioning attenuates global cerebral ischemic injury following asphyxial cardiac arrest through regulation of delta opioid receptor system. Neuroscience 2011, 202, 352–362. [Google Scholar]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov 2011, 10, 209–219. [Google Scholar]
- Xie, Z. Neuronal vulnerability to anesthesia neurotoxicity depends on age of neurons. Ann. Neurol 2013, 73, 686–687. [Google Scholar]
- Chao, D.; Xia, Y. Ionic storm in hypoxic/ischemic stress: Can opioid receptors subside it? Prog. Neurobiol 2010, 90, 439–470. [Google Scholar]
- Yan, Q.; Rosenfeld, R.D.; Matheson, C.R.; Hawkins, N.; Lopez, O.T.; Bennett, L.; Welcher, A.A. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience 1997, 78, 431–448. [Google Scholar]
- Dieni, S.; Rees, S. BDNF and TrkB protein expression is altered in the fetal hippocampus but not cerebellum after chronic prenatal compromise. Exp. Neurol 2005, 192, 265–273. [Google Scholar]
- Altar, C.A.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997, 389, 856–860. [Google Scholar]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol 2007, 81, 294–330. [Google Scholar]
- Narantuya, D.; Nagai, A.; Sheikh, A.M.; Masuda, J.; Kobayashi, S.; Yamaguchi, S.; Kim, S.U. Human microglia transplanted in rat focal ischemia brain induce neuroprotection and behavioral improvement. PLoS One 2010, 5, e11746. [Google Scholar]
- Nolan, S.M.; Mathew, E.C.; Scarth, S.L.; Al-Shamkhani, A.; Law, S.K. The effects of cysteine to alanine mutations of CD18 on the expression and adhesion of the CD11/CD18 integrins. FEBS Lett 2000, 486, 89–92. [Google Scholar]
- Fan, L.W.; Lin, S.; Pang, Y.; Rhodes, P.G.; Cai, Z. Minocycline attenuates hypoxia-ischemia-induced neurological dysfunction and brain injury in the juvenile rat. Eur. J. Neurosci 2006, 24, 341–350. [Google Scholar]
- Choi, B.R.; Kwon, K.J.; Park, S.H.; Jeon, W.K.; Han, S.H.; Kim, H.Y.; Han, J.S. Alternations of septal-hippocampal system in the adult wistar rat with spatial memory impairments induced by chronic cerebral hypoperfusion. Exp. Neurobiol 2011, 20, 92–99. [Google Scholar]
- Zhang, J.; Geula, C.; Lu, C.; Koziel, H.; Hatcher, L.M.; Roisen, F.J. Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp. Neurol 2003, 183, 469–481. [Google Scholar]
- Hallenbeck, J.M. The many faces of tumor necrosis factor in stroke. Nat. Med 2002, 8, 1363–1368. [Google Scholar]
- Kaur, C.; Rathnasamy, G.; Ling, E.A. Roles of activated microglia in hypoxia induced neuroinflammation in the developing brain and the retina. J. Neuroimmune. Pharmacol 2013, 8, 66–78. [Google Scholar]
- Wang, Y.; Cao, M.; Liu, A.; Di, W.; Zhao, F.; Tian, Y.; Jia, J. Changes of inflammatory cytokines and neurotrophins emphasized their roles in hypoxic-ischemic brain damage. Int. J. Neurosci 2013, 123, 191–195. [Google Scholar]
- Jiang, Y.; Wei, N.; Lu, T.; Zhu, J.; Xu, G.; Liu, X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neuroscience 2011, 172, 398–405. [Google Scholar]
- Lu, B. Pro-region of neurotrophins: role in synaptic modulation. Neuron 2003, 39, 735–738. [Google Scholar]
- Ginty, D.D.; Kornhauser, J.M.; Thompson, M.A.; Bading, H.; Mayo, K.E.; Takahashi, J.S.; Greenberg, M.E. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 1993, 260, 238–241. [Google Scholar]
- Balboni, G.; Salvadori, S.; Guerrini, R.; Negri, L.; Giannini, E.; Jinsmaa, Y.; Bryant, S.D.; Lazarus, L.H. Potent delta-opioid receptor agonists containing the Dmt-Tic pharmacophore. J. Med. Chem 2002, 45, 5556–5563. [Google Scholar]
- Vergura, R.; Balboni, G.; Spagnolo, B.; Gavioli, E.; Lambert, D.G.; McDonald, J.; Trapella, C.; Lazarus, L.H.; Regoli, D.; Guerrini, R.; et al. Anxiolytic- and antidepressant-like activities of H-Dmt-Tic-NH-CH(CH2-COOH)-Bid (UFP-512), a novel selective delta opioid receptor agonist. Peptides 2008, 29, 93–103. [Google Scholar]
- Aguila, B.; Coulbault, L.; Boulouard, M.; Léveillé, F.; Davis, A.; Tóth, G.; Borsodi, A.; Balboni, G.; Salvadori, S.; Jauzac, P.; et al. In vitro and in vivo pharmacological profile of UFP-512, a novel selective delta-opioid receptor agonist; correlations between desensitization and tolerance. Br. J. Pharmacol 2007, 152, 1312–1324. [Google Scholar]
- Yang, Y.; Zhi, F.; He, X.; Moore, M.L.; Kang, X.; Chao, D.; Wang, R.; Kim, D.H.; Xia, Y. Delta-opioid receptor activation and microRNA expression of the rat cortex in hypoxia. PLoS One 2012, 7, e51524. [Google Scholar]
- He, X.; Yang, Y.; Zhi, F.; Moore, M.L.; Kang, X.; Chao, D.; Wang, R.; Balboni, G.; Salvadori, S.; Kim, D.H.; et al. Delta-Opioid receptor activation modified microRNA expression in the rat kidney under prolonged hypoxia. PLoS One 2013, 8, e61080. [Google Scholar]







© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, X.; Hua, F.; Sandhu, H.K.; Chao, D.; Balboni, G.; Salvadori, S.; He, X.; Xia, Y. Effect of δ-Opioid Receptor Activation on BDNF-TrkB vs. TNF-α in the Mouse Cortex Exposed to Prolonged Hypoxia. Int. J. Mol. Sci. 2013, 14, 15959-15976. https://doi.org/10.3390/ijms140815959
Tian X, Hua F, Sandhu HK, Chao D, Balboni G, Salvadori S, He X, Xia Y. Effect of δ-Opioid Receptor Activation on BDNF-TrkB vs. TNF-α in the Mouse Cortex Exposed to Prolonged Hypoxia. International Journal of Molecular Sciences. 2013; 14(8):15959-15976. https://doi.org/10.3390/ijms140815959
Chicago/Turabian StyleTian, Xuesong, Fei Hua, Harleen K Sandhu, Dongman Chao, Gianfranco Balboni, Severo Salvadori, Xiaozhou He, and Ying Xia. 2013. "Effect of δ-Opioid Receptor Activation on BDNF-TrkB vs. TNF-α in the Mouse Cortex Exposed to Prolonged Hypoxia" International Journal of Molecular Sciences 14, no. 8: 15959-15976. https://doi.org/10.3390/ijms140815959
APA StyleTian, X., Hua, F., Sandhu, H. K., Chao, D., Balboni, G., Salvadori, S., He, X., & Xia, Y. (2013). Effect of δ-Opioid Receptor Activation on BDNF-TrkB vs. TNF-α in the Mouse Cortex Exposed to Prolonged Hypoxia. International Journal of Molecular Sciences, 14(8), 15959-15976. https://doi.org/10.3390/ijms140815959